
Abstract
Background
Parinaud’s syndrome, also known as dorsal midbrain syndrome, is characterized by a combination of signs, including upgaze palsy, convergence–retraction nystagmus, eyelid retraction and pupillary dissociation. It is caused by pretectal or tegmental lesions of upper brainstem. Miller Fisher syndrome, characterized by the triad of ataxia, areflexia and ophthalmoplegia, has rarely been reported to present as Parinaud’s syndrome in adults. To our knowledge, this clinical manifestation has not been previously described in children.
Case presentation
A previously healthy 13-year-old girl presented with blurred vision and diplopia, 10 days after a viral infection. Initial examination revealed incomplete Parinaud’s syndrome, while rest of neurological examination was normal. Brain imaging (MRI, MRA) did not reveal any abnormal findings and CSF findings were also normal. During the first days after admission she gradually deteriorated, showing complete external ophthalmoplegia, unsteady gait, and absent deep tendon reflexes of lower limbs with normal muscle power. With the clinical suspicion of Miller Fisher syndrome IVIG was administered, leading to subsequent resolution of her symptoms. AntiGQ1b and antiGD1b antibodies came back positive confirming diagnosis. On 1 month follow-up, neurological examination revealed diplopia in left gaze, and a second dose of IVIG was administered with good response. She remains asymptomatic 1 year from disease onset.
Conclusions
Miller Fisher syndrome can rarely present as Parinaud’s syndrome in adults. Herein we described the first pediatric patient with similar clinical presentation. As the typical semiology of ataxia and areflexia may not be present initially, high index of suspicion is required to recognise and treat those patients promptly. Serological detection of anti-ganglioside antibodies, such as anti-GQ1b and anti-GD1b, may help confirm diagnosis.
Similar content being viewed by others


Background
Miller Fisher syndrome (MFS) is an immune-mediated neuropathy, typically presenting with ophthalmoplegia associated with areflexia and ataxia. Besides these key clinical features, various neuro-ophthalmic manifestations have been reported. Here we present a case of a child presenting with incomplete Parinaud’s syndrome as the initial manifestation of MFS.
Case presentation
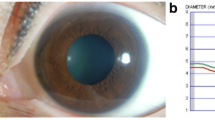
A previously healthy 13-year-old girl presented with complaints of blurred vision and diplopia in the 24 h preceding the initial examination. Her symptoms were not persistent and fluctuated during the day. Past medical history was not significant and she was not on any medication, while recent history revealed a non-febrile upper respiratory infection 10 days prior to the onset of the presenting symptoms. Family history was not contributory. On examination she was alert with normal mental status examination. Neurological examination revealed upgaze palsy, as well as eyelid retraction on attempted upgaze and convergence. Pupils were equal and reactive to both light and accommodation. No other abnormal signs were noted. Laboratory testing including full blood count, comprehensive metabolic panel, markers of infection and thyroid function was normal (Fig. 1). Written consent has been obtained by parient’s parent for publication.

Patient with upgaze palsy and eyelid retraction at presentation
Since the patient’s presentation was suggestive of incomplete Parinaud’s syndrome, brain MRI and MRA was performed with particular attention on the tectal region. However, imaging was negative for midbrain pathology or other abnormal findings.
Two days after admission neurological examination revealed areflexia, first affecting patellar deep tendon reflexes and subsequently the Achilles, normal muscle strength, and unsteady gait. With the diagnosis of Miller Fisher syndrome suspected, a lumbar puncture was performed, which demonstrated normal cell count and protein concentration. Thereafter the patient received intravenous immune globulin (IVIG), in a dose of 2gr/kg in 2 days, to treat for possible MFS.
During the following days the patient gradually deteriorated, reaching the nadir of her symptoms by the fifth hospital day, when she developed complete external ophthalmoplegia. She began to improve on the sixth hospital day and 2 days later she was discharged, with near-complete resolution of her signs and symptoms. Serum antibody test results came back positive for GQ1b-IgG and GD1b-IgG, confirming the diagnosis of MFS. One month later, on outpatient follow-up, left gaze diplopia was noted on examination, and a second course of IVIG was administered in a dose of 2gr/kg in 5 days, followed by complete resolution of symptoms. One year from disease onset, she remains asymptomatic (Fig. 2).

Full recovery after treatment
Discussion
Parinaud’s syndrome, also known as dorsal midbrain or sylvian aqueduct syndrome, was first described by Henri Parinaud in the late 1800s, and originally included upgaze palsy and convergence paralysis [1]. However, the above definition has now been expanded to include the combination of upgaze palsy, convergence retraction nystagmus and pupillary light-near dissociation. This classic triad is seen in about 65% of patients [2]. Collier’s eyelid retraction sign may also be present, as well as loss of convergence. Fragments of the syndrome are common, such being the case with our patient.
The syndrome is caused by compression of the rostral midbrain and pretectum. Damage of the vertical gaze centers, mainly the interstitial nucleus of Cajal (INC) and the rostral interstitial nuclei of the medial longitudinal fasciculus (riMLF), results to the characteristic limited conjugate upgaze [1, 3]. The most common causes are tumors (mainly located in the midbrain or the pineal gland), stroke, obstructive hydrocephalus, demyelination and infection. Tumors are more common in children and young adults, while vascular causes are more common among older patients. [2]
MFS is a rare variant of Guillain–Barre syndrome. It was first described by James Collier in 1932 as a triad of symptoms including ophthalmoplegia, ataxia and areflexia [4]. MFS most commonly affects male adults, although cases have been reported in children. A preceding infectious illness is reported in the majority of MFS cases, most common being upper respiratory infection followed by gastrointestinal illness [5, 6]. The pathogenesis has been shown to involve molecular mimicry triggered by antecedent infection [7]. Anti-ganglioside antibodies GQ1b-IgG were suggested in 1992 as a diagnostic marker of MFS [8]. They are present in over 90% of the affected patients and are highly specific for MFS [9]. They target epitopes located both in peripheral and central nervous system, leading to variable clinical presentations. Those epitopes are found in high concentrations in the cranial nerves III, IV and VI, resulting to the characteristic ophthalmoplegia of MFS [6]. Moreover, they seem to peak early in the course of the disease, making them a more useful diagnostic marker comparing to CSF albuminocytological dissociation [10]. Other antibodies against gangliosides are also rarely detected, such as against GM1b, GD1a, GD1b and GT1a, corresponding to a variety of phenotypes.
There are only a few reports in the literature of MFS presenting as Parinaud syndrome in adults, and to our knowledge none in the pediatric population. Mallia et al. and Magun et al. reported a 25-year-old and a 29-year-old male, respectively, with preceding history of diarrhea, presenting as Parinaud’s Syndrome. Both were diagnosed with Miller Fisher syndrome, on the grounds of concurrent ataxia and areflexia, normal brain imaging and positive anti-GQ1b antibodies. They both responded to intravenous immunoglobulin with full resolution of their symptoms and signs [11, 12]. In the review of al-Din et al. investigating the neuro-ophthalmic manifestations in Miller Fisher syndrome, 109 reports were included, describing 243 cases. Parinaud's syndrome was described in 2 cases. It is of note, though, that while the usual finding regarding lid involvement was ptosis, active lid retraction on attempted upgaze was described in 22 cases, and was observed early in the course of the disease, indicating brainstem involvement in MFS [13]. Apart from the ophthalmological hallmarks of MFS (relatively symmetric bilateral ophthalmoplegia, ptosis, and facial palsies), other neuro-ophthalmic manifestations reported include internuclear ophthalmoplegia, convergence spasm, horizontal, vertical or rotatory nystagmus, ocular flutter, pupillary dysfunction, and optic neuritis. [13, 14] Considering the above, after the main causes of Parinaud’s syndrome are ruled out, high index of suspicion is required to include MFS in differential diagnosis, as early treatment may result to a favourable outcome. Serological detection of anti-ganglioside antibodies, such as anti-GQ1b and anti-GD1b, may help confirm diagnosis.
Conclusions
MFS is primarily a clinical diagnosis based on the key clinical presentation of ataxia, areflexia and external ophthalmoplegia. However, the typical semiology may not be present initially, leading to a more challenging diagnostic work up. In our case, the patient presented with Parinaud’s syndrome and gradually developed areflexia, ataxia and complete ophthalmoplegia. Considering the above, this case highlights that after exclusion of structural pathology of dorsal midbrain, MFS should be considered in the differential diagnosis of Parinaud’s syndrome.
Availability of data and materials
Data sharing is not applicable to this article as no data sets were generated or analysed during the current study. Patient’s file is available on request.
Abbreviations
- MFS:
-
Miller Fisher Syndrome
- MRI:
-
Magnetic Resonance Imaging
- MRA:
-
Magnetic Resonance Angiography
- CSF:
-
Cerebrospinal fluid
- IVIG:
-
Intravenous Immunoglobulin
- AntiGQ1b:
-
Ganglioside Q1b antibody
- AntiGD1b:
-
Ganglioside D1b antibody
- IgG:
-
Immunoglobulin G
- INC:
-
Interstitial nucleus of Cajal
- riMLF:
-
Rostral interstitial nuclei of the medial longitudinal fasciculus
- GM1b:
-
Ganglioside M1b
- GD1a:
-
Ganglioside D1a
- GT1a:
-
Ganglioside T1a
References
Roper-Hall G. Historical vignette: Henri Parinaud (1844–1905): French ophthalmologist and pioneer in neuroophthalmology. Am Orthopt J. 2014;64:126–33. https://doi.org/10.3368/aoj.64.1.126.
Shields M, Sinkar S, Chan W, Crompton J. Parinaud syndrome: a 25-year (1991–2016) review of 40 consecutive adult cases. Acta Ophthalmol. 2017;95(8):e792–3. https://doi.org/10.1111/aos.13283.
Pierrot-Deseilligny CH, Chain F, Gray F, Serdaru M, Escourolle R, Lhermitte F. Parinaud’s syndrome: electro-oculographic and anatomical analyses of six vascular cases with deductions about vertical gaze organization in the premotor structures. Brain. 1982;105(Pt 4):667–96.
Fisher M. An unusual variant of acute idiopathic polyneuritis (syndrome of ophthalmoplegia, ataxia and areflexia). N Engl J Med. 1956;255(2):57–65. https://doi.org/10.1056/NEJM195607122550201.
Bukhari S, Taboada J. A Case of Miller Fisher Syndrome and Literature Review. Cureus. 2017;9(2): e1048. https://doi.org/10.7759/cureus.1048.
Snyder LA, Rismondo V, Miller NR. The Fisher variant of Guillain-Barré syndrome (Fisher syndrome). J Neuroophthalmol. 2009;29(4):312–24. https://doi.org/10.1097/WNO.0b013e3181c2514b.
Willison HJ, O’Hanlon GM. The Immunopathogenesis of Miller Fisher Syndrome. J Neuroimmunol. 1999;100:3–12.
Chiba A, Kusunoki S, Shimizu T, Kanazawa I. Serum IgG antibody to ganglioside GQ1b is a possible marker of Miller Fisher syndrome. Ann Neurol. 1992;31(6):677–9.
Kusunoki S. Diagnosis, pathogenesis and treatment of Miller Fisher syndrome and related disorders: clinical significance of antiGQ1b IgG antibody. Expert Rev Neurother. 2003;3(1):133–40. https://doi.org/10.1586/14737175.3.1.133.
Nishimoto Y, Odaka M, Hirata K, Yuki N. Usefulness of anti-GQ1b IgG antibody testing in Fisher syndrome compared with cerebrospinal fluid examination. J Neuroimmunol. 2004;148(1–2):200–5. https://doi.org/10.1016/j.jneuroim.2003.11.017.
Magun R, Silva J. This is a novel case a variant of miller-fisher syndrome with a positve anti-GQ1b antibody presenting as Parinaud’s Syndrome (P5038). Neurology. 2014;82(10 Supplement):P5.038.
Mallia M, Chircop Ch, Aquilina J. An unusual case of Parinaud’s syndrome. BMJ Case Reports. 2012. https://doi.org/10.1136/bcr-2012-006722.
Al-Din SN, Anderson M, Eeg-Olofsson O, Trontelj JV. Neuro-ophthalmic manifestations of the syndrome of ophthalmoplegia, ataxia and areflexia: a review. Acta Neurol Scand. 1994;89(3):157–63. https://doi.org/10.1111/j.1600-0404.1994.tb01654.x.
Saul RF. Neuro-ophthalmology and the Anti-GQ1b antibody syndromes. Curr Neurol Neurosci Rep. 2009;9(5):379–83. https://doi.org/10.1007/s11910-009-0055-0.
Acknowledgements
Not applicable.
Funding
All authors have no financial relationships relevant to this article to disclose.
Author information
Authors and Affiliations
Contributions
AK was involved in patient’s clinical care, analyzed and interpreted the patient data, wrote the initial draft; D.M. provided critical editing and revision to the final drafts; AM and AS were involved in patient’s clinical care, contributed to the manuscript writing, S.M. performed the literature reviews and revision to the final drafts, GV was involved in patient’s clinical care, provided supervision, critical editing and revision to the final drafts and the discussion of the available literature; all authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Written consent obtained from patient’s parent.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Korona, A., Maritsi, D., Markante, A. et al. Eyes wide open—an atypical presentation of Miller Fisher syndrome (MFS): case report. Egypt J Neurol Psychiatry Neurosurg 58, 14 (2022). https://doi.org/10.1186/s41983-022-00451-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s41983-022-00451-3