
Abstract
Background
Telethoninopathy or TCAP-gene related Limb Girdle Muscular Dystrophy is a rare genetic disease that was first described in Brazil. There are around 100 families reported worldwide. Due to its rarity, detailed information on muscle biopsy light and electron microscopic features are lacking.
Cases presentation
Retrospective study of consecutive muscle biopsies performed in patients from a Neuromuscular Outpatient Clinic between 2011 and 2023. Inclusion criteria: telethoninopathy diagnosed by both immunohistochemistry and molecular studies. Seven patients (0.7% or 7/953) were found: five male and two female, admitted from 6 to 54 years old. Detailed light and electron microscopy findings are illustrated. Muscle imaging is presented. A dystrophic pattern on muscle biopsy was found in 57% (4/7) of the patients. Other 43% (3/7) presented myopathic features such as variation in fibre calibre, nuclear internalization, rimmed vacuoles, and oxidative irregularities. Morphometry disclosed type 1 lobulated fibres that were 34%, 52%, and 57% smaller than type 2 fibres, respectively, in three patients, without type 1 fibre predominance. Electron microscopy demonstrated nuclear pseudoinclusions, pyknosis, multifocal loss of the sarcolemma, and 17 nm intrasarcoplasmic filamentous inclusions. All patients presented: (1) complete absence of the immunohistochemical expression of telethonin, and (2) the homozygous c.157C > T, p.(Gln53*) pathogenic variant in exon 2 of the TCAP gene.
Conclusion
Anti-telethonin immunohistochemistry may be helpful in unsolved cases with nonspecific myopathic abnormalities, specially with small type 1 lobulated fibres. Appropriate diagnosis is important for adequate genetic counselling.
Similar content being viewed by others

Background
Limb Girdle Muscular Dystrophy (LGMD) is a heterogeneous group of genetic progressive muscle diseases characterized by proximal limbs, scapular, and pelvic girdle muscle weakness. Morphologically they are characterized by dystrophic features on muscle biopsy: distorted architecture, endomysial fibrosis, muscle fat replacement, severe variation in muscle fibre calibre, atrophy, hypertrophy, fibre splitting, necrosis, phagocytosis, and regeneration (Cotta et al. 2014; Dubowitz et al. 2020). They have been classified in five dominant and 28 recessive subtypes according to the chronological order of discovery of the related genes (Straub et al. 2018; Benarroch et al. 2024).
Limb Girdle Muscular Dystrophy recessive seven (LGMDR7), previously classified as LGMD2G, is related to the telethonin or titin-cap (TCAP) gene located in the 17q12 (Straub et al. 2018; Benarroch et al. 2024). It was the seventh described recessive Limb Girdle Muscular Dystrophy and it was first reported in Brazilian patients (Moreira et al. 1997, 2000). Later descriptions of Brazilian patients and other countries were published (Almeida et al. 2012; Barresi et al. 2015; Blanco-Palmero et al. 2019; Brusa et al. 2018; Chakravorty et al. 2020; Chamova et al. 2017; Chamova et al. 2018; Chen et al. 2020; Chen et al. 2023; Cotta et al. 2014; de Fuenmayor-Fernández de la Hoz et al. 2016; Ferreiro et al. 2011; Fichna et al. 2018; Francis et al. 2014; Huang et al. 2022; Hudson et al. 2017; Ikenberg et al. 2017; Liang et al. 2020; Lima et al. 2005; Lin et al. 2023; Lorenzoni et al. 2023; Lv et al. 2021; Lv et al. 2023; Magri et al. 2017; Moutinho-Pereira et al. 2022; Nalini et al. 2015; Negrão et al. 2010; Olivé et al. 2008; Paim et al. 2013; Savarese et al. 2016; Seong et al. 2016; Tian et al. 2015; Vainzof et al. 2002; Waddell et al. 2012; Yee et al. 2007; Zatz et al. 2000).
Telethoninopathy is considered an extremely rare subtype of Limb Girdle Muscular Dystrophy. There are around 100 families reported worldwide, with an estimated prevalence of 0.004/100,000 in the gnomAD database (Liu et al. 2019). In Brazil, this rarity was explained by the discovery of a rare allele A of rs1053651 SNP (the ancestral allele is C) in Brazilian and Portuguese patients that shared the c.157C > T (Q53X) or p.(Gln53*) homozygous nonsense mutation in the TCAP gene (Almeida et al. 2013). The frequency of this allele in the European population was estimated in only 28% (Almeida et al. 2013).
Besides the Limb Girdle Muscular Dystrophy phenotype, the TCAP gene has been related to: (1) congenital muscular dystrophy (Ferreiro et al. 2011; Almeida et al. 2012), (2) distal myopathy (Blanco-Palmero et al. 2019; Chen et al. 2020; Lv et al. 2021), and (3) cardiomyopathy of the subtypes hypertrophic, and dilated (Benarroch et al. 2024; Hayashi et al. 2004).
Telethonin is the gene product of the TCAP gene. It was discovered in 1997 as one of the most abundant skeletal muscle sarcomeric proteins (Valle et al. 1997). That group was supported by the Italian Telethon (tele- for television, and -thon for marathon), hence the name telethonin (Valle et al. 1997). Telethonin binds to the giant protein titin and it is involved in T tubule organization, sarcomere assembly and development, sarcomere-membrane interaction, and signalling (Murphy and Straub 2015; Lv et al. 2021). Therefore, the commercially available antibodies anti-telethonin present strong intrasarcoplasmic reaction as they react with a sarcomeric protein. This information is important for pathologists because in routine muscle biopsy immunohistochemistry several antibodies are reactive at the sarcolemmal membrane such as dystrophin, dysferlin, caveolin, and the sarcoglycans (alpha, beta, gamma, and delta) (Cotta et al. 2021).
Methods
Design of the study
A retrospective study of all consecutive muscle biopsies performed in patients from the SARAH Network of Rehabilitation Hospitals, Neuromuscular Outpatient Clinic in Belo Horizonte, Brazil, between 2011 and 2023.
Muscle imaging
Patients were submitted either to axial T1-weighted Magnetic Resonance Imaging (MRI), or to Computed Tomography of the pelvis, thighs, and legs during their diagnostic work-up.
Muscle biopsy light microscopy
Muscle biopsies were performed at the SARAH Network of Rehabilitation Hospitals, Belo Horizonte, Brazil, according to standard protocols (Udd et al. 2019; Cotta et al. 2021). Histochemical reactions and stains included were: hematoxylin and eosin (HE), modified Gomori trichrome, PAS (Periodic acid Schiff) with and without diastase, Oil-red-O, SDH (succinate dehydrogenase), COX (cytochrome c oxidase), NADH (nicotinamide adenine dinucleotide), myosinic ATPase pH9.4, pH4.6, and pH4.3, acid phosphatase, and nonspecific esterase. Morphometric studies were performed on photographs of myosinic ATPase slides captured with Axiocam 105 color with Zeiss Zen software and analysed using Image J software. Muscle immunohistochemistry was performed with anti-telethonin antibody (G-11 sc-25327) (Santa Cruz, biotechnology, Inc.), at 1:50 dilution. Telethonin complete deficiency was observed when no reaction was noted in the sarcoplasm of the patient compared to a normal control sample.
Muscle biopsy transmission electron microscopy
Electron microscopy was performed in muscle fragments fixed in glutaraldehyde, postfixed in osmium tetroxide, dehydrated in ethanol/ propylene oxide, and embedded in Epon resin. Semithin sections were stained with toluidine blue. Ultrathin sections were stained by uranyl acetate/ lead citrate and analyzed in a JEM 1010 transmission electron microscope at 80 kV.
Genetic analysis
Genetic analysis was performed at The SARAH Network of Rehabilitation Hospitals, Brasilia, DF, Brazil (Patients 1, 2, 3, 4, and 6), and at the Human Genome Centre from the University of São Paulo USP, Brazil (Patients 5 and 7), with the search for point mutations in the telethonin (titin-cap, TCAP) gene (NM_003673.3) through direct sequencing in the ABI 3130XL (Applied Biosystems) with analysis in the softwares Sequencing Analysis (v. 5.2) and SeqScape (v. 2.5), and the search for mutations in the Leiden mutation database (http://www.dmd.nl/index.html), ClinVar (https://www.ncbi.nlm.nih.gov/clinvar), and dbSNP (http://www.ncbi.nlm.nih.gov/projects/SNP).
Case series
A total of 953 muscle biopsies were found in the period and their diagnosis were reviewed. Inclusion criteria for this study was a diagnosis of telethoninopathy that had been confirmed by both immunohistochemistry, and molecular studies. Seven patients fulfilled inclusion criteria, five were male, and two were female. The age of admission varied from 6 to 54 years old. They represented 0.7% (7/953) of the muscle biopsy diagnosis in the period. Light and electron microscopy findings of two patients had been partially described by our group: Patient 5 (Cotta et al. 2014), and Patient 7 (Paim et al. 2013). Patients 2, 3, 5, 6, and 7 had been included in a Table with no description of light or electron microscopic findings (Winckler et al. 2019). Patient 1 and Patient 4 were not included in previous studies. Clinical and laboratorial findings of all patients are summarized in Table 1 (Paim et al. 2013; Cotta et al. 2014; Winckler et al. 2019; current paper). A detailed description of clinical, neurophysiological, muscle serum enzymes, muscle imaging, and morphological, and molecular findings are presented in detail below.
Case 1
Patient 1 was admitted at the age of 14 years. When he was 2 years old, he started abnormal gait with difficulties running. He was born of non-consanguineous parents. Physical examination demonstrated long face, high arched palate, divergent strabismus on the right eye with slow extraocular muscle movements, distal joint laxity, scoliosis, winged scapula on the left side, flexion contractures of the elbows, slight flexion of the knees and ankles, generalized muscle atrophy, specially in the thighs and arms, calf volume increase, and absent lower and upper limbs reflexes. He presented abnormal gait with increased lumbar lordosis, and toe walking.
Total creatine kinase was 694 IU/L (< 190) (3.7x), and aldolase of 11.6 U/L (< 15.2). Electromyogram was suggestive of a myopathic process. Muscle imaging (Fig. 1) presented slight right glutei atrophy. Right triceps brachialis muscle biopsy (Fig. 2) demonstrated severe variation in fibre calibre with numerous atrophic fibres, 52% of fibre type disproportion: mean diameter of Type 1 fibres = 210 (± 69) micrometre, and mean diameter of Type 2 fibres = 435 (± 140) micrometre, with no fibre type predominance (Type 1 = 47.4%, and Type 2 = 52.6%). No sarcomeric telethonin reaction was observed on immunohistochemistry. Electron microscopy (Fig. 3) demonstrated atrophic fibres, nuclear internalization, and intrasarcoplasmic glycogen accumulation. Molecular studies demonstrated a homozygous pathogenic variant c.157C > T, p.(Gln53*) or (Q53X) in exon 2 of the titin-cap (TCAP or telethonin) gene.

Muscle imaging in a series of TCAP gene-related limb girdle muscular dystrophy patients: schematic diagram (a, b, and c) and muscle imaging (d, e, f, g, h, i, j, k, l, m, n, o, p, q, r, s, t, u, v, w, and x). Schematic diagram of the pelvis (a), thighs (b), and legs (c) representing a summary of muscle imaging pattern of telethoninopathy in this series from complete muscle fat replacement (white) to normal muscle (dark gray): calf volume increase, early gluteus maximus (Gmax) muscle fat replacement, followed by gluteus medius (Gmed), and gluteus minimus (Gmin), asymmetric (worse in the right side); tibialis anterior (Ta), adductor magnus (AdM), adductor longus (AdL), semitendinosus (St), biceps femoris (Bf), semimembranosus (Sm), vastus lateralis (VL), rectus femoris (Rf), vastus medialis (Vmed), vastus intermedius (Vi), soleus (So), peroneus group (P), extensor group (E), tibialis posterior (Tp), gastronemius lateralis (Gl), asymmetric gastronemius medialis (Gm), with compensatory hypertrophy of sartorius (S) and gracilis (G). Muscle imaging of the pelvis (d, g, j, m, p, s, and v), thighs (e, h, k, n, q, t, and w) and legs (f, i, l, o, r, u, and x) of patient 1 (d, e, and f), patient 2 (g, h, and i), patient 3 (j, k, and l), patient 4 (m, n, and o), patient 5 (p, q, and r), patient 6 (s, t, and u), and patient 7 (v, w, and x). Patient 1 (d, e, and f) at 14 years old, with 14 years duration of disease, presented only slight right glutei atrophy (d). Patient 2 (g, h, and i) at 6 years old, with 3 years duration of disease, presented subtle sparse areas of muscle fat replacement. Patient 3 (j, k, and l) at 22 years old, with 7 years of duration of disease, presented moderate asymmetry of the glutei, smaller on the right side (arrow in j), and severe atrophy of vastus lateralis (arrow head in k). Patient 4 (m, n, and o) at 24 years old, with 7 years of duration of disease presented asymmetric muscle fat replacement of semitendinosus severe at the right side and slight at the left side (arrow in n), and atrophy and muscle fat replacement of the left gastrocnemius medialis (arrow head in o), and peroneus muscles fat replacement severe in the left side (* in o). Patient 5 (p, q, and r) at 32 years old, with 12 years of duration of disease, presented moderate gastrocnemius medialis fat replacement severe in the right side, slight in the left side (arrow head in r), and peroneus fat replacement severe in the left side (* in r). Patient 6 (s, t, and u) at 35 years old, with 20 years of duration of disease, presented asymmetric preservation of the right tibialis posterior (arrow head in u), and left peroneus (* in u). Patient 7 (v, w, and x) at 54 years old, with 46 years duration of disease, presented asymmetric muscle fat replacement of gastrocnemius lateralis, less severe at the left side (arrow head in x). Patient 1 (d, e, and f), patient 3 (j, k, and l), patient 4 (m, n, and o), and patient 6 (s, t, and u) were submitted to Axial T1-weighted magnetic resonance imaging (MRI) of the pelvis, thighs and legs. Patient 2 (g, h, and i), patient 5 (p, q, and r), and patient 7 (v, w, and x) were submitted to computed tomography (CT); yo: years old

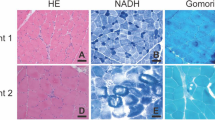
Light microscopy findings of right triceps brachialis muscle biopsy of Patient 1. Muscle with severe variation in fibre calibre with numerous atrophic fibres (arrows in a, b, c, e, f, g, h, i, and j), with mild increase in intrasarcoplasmic lipid content (d), with 52% of fibre type disproportion (e) (mean diameter of Type 1 fibres = 210 (± 69) micrometre and mean diameter of Type 2 fibres = 435 (± 140) micrometre, with no fibre type predominance (Type 1 = 47.4% and Type 2 = 52.6%) (e), immunohistochemistry with negative sarcomeric telethonin reaction (arrow in k) compared to the normal control (arrow in l) (a HE 400x, b Modified Gomori trichrome 200x, c PAS 200x original optic magnification (x 4.5195), d Oil-red-O 400x, e ATPase pH4.6 100x, f SDH 200x, g COX-SDH 100x, h NADH 20x, i Acid phosphatase 200x, j Nonspecific esterase 200x, Patient (k), and Control (l), Immunoperoxidase Antibody anti-telethonin (G-11 sc-25327, Santa Cruz Biotechnology, Inc. at 1:50 dilution) (k) 200x, and (l) 400x)

Electron microscopy findings of right triceps brachialis muscle biopsy of Patient 1. Muscle with atrophic fibres (arrows in a, and b), nuclear internalization (arrow in c), intrasarcoplasmic glycogen accumulation (red * in e and red * f), and absent intrasarcoplasmic, intranuclear, or endothelial (d) inclusions (Transmission electron microscopy a. 2,500x, b. 2,500x, c. 3,000x, d. 5,000x, e. 5,000x, f. 6,000x)
Case 2
Patient 2 was admitted at 6 years old. When he was 3 years old, he started with difficulties climbing stairs, frequent falls, and myalgia in the lower limbs. He was referred to our service with a clinical suspicion of Duchenne muscular dystrophy. He was born on non-consanguineous parents. Physical examination disclosed slight bilateral eyelid ptosis, proximal lower limb weakness with decreased lower limb reflexes, slightly increased calf volume, and abnormal tiptoe gait.
Serum creatine kinase was 981 IU/L (< 225) (4.4x), and aldolase 30.2 U/L (< 15.2). Muscle imaging (Fig. 1) demonstrated subtle sparse areas of muscle fat replacement. Right vastus lateralis muscle biopsy (Fig. 4) demonstrated internal nuclei, regeneration, irregular basophilic intrasarcoplasmic areas, necrosis, phagocytosis, slight type 1 fibre predominance of 57.9%; irregular areas of myofibrillar disorganization, oxidative irregularities, increased acid phosphatase activity, negative sarcomeric telethonin reaction, and ectopic dystrophin sarcoplasmic deposits. Electron microscopy (Fig. 5) demonstrated atrophy, ring fibres, nuclear pyknosis, invaginations, and pseudoinclusions. Molecular studies demonstrated a homozygous pathogenic variant c.157C > T, p.(Gln53*) or (Q53X) in exon 2 of the titin-cap (TCAP or telethonin) gene.

Light microscopy findings of right vastus lateralis muscle biopsy of Patient 2. Muscle with internal nuclei that are single (blue arrow in a) or multiple (green arrow in a), regeneration (yellow * in a), and irregular basophilic intrasarcoplasmic areas (yellow arrow in a), necrosis and phagocytosis (yellow arrow in b). Slight type 1 (yellow * in c) fibre predominance of 57.9%; irregular areas of myofibrillar disorganization with increased reaction (yellow arrows in d, e, f, and h; green arrow in k), and oxidative irregularities (green arrow in e). Increased acid phosphatase activity (yellow arrow in g). Immunohistochemistry with negative sarcomeric telethonin reaction (arrow in i) compared to the normal control (arrow in j). Abnormal dystrophin reactive ectopic sarcoplasmic deposits in k, compared to normal control in l. (a HE 400x, b Modified Gomori trichrome 400x, c ATPase pH4.6 100x, d SDH 200x, e COX-SDH 400x, f NADH 400x, g Acid phosphatase 200x, h Nonspecific esterase 200x, Immunoperoxidase Antibody anti-telethonin (G-11 sc-25327, Santa Cruz Biotechnology, Inc. at 1:50 dilution) 200x, patient (i), 400x control (j). Immunoperoxidase Antibody anti-dystrophin carboxy-terminal (NCL-DYS2, Novocastra, Leica) 200x (k) patient, and 200x (l) control)

Electron microscopy findings of right vastus lateralis muscle biopsy of Patient 2. Muscle with atrophic fibres (yellow arrows in a, and b), ring fibres (red arrow in c), nuclear internalization (yellow arrow in c), intrasarcoplasmic lipid accumulation (arrow in d), nuclear irregularities including pyknosis (arrow in c), invaginations (arrow in e), and pseudoinclusions (red arrow in f) (Transmission electron microscopy a 2,500x, b 2,000x, c 3,000x, d 5,000x, e 6,000x, f 15,000x)
Case 3
Patient 3 was admitted at 22 years old. At the age of 15 years, he began difficulties running and climbing irregular terrains followed by weakness of the upper limbs. He was born of consanguineous parents. Physical examination demonstrated orbicularis oris paresis, left foot drop, limited abduction of the eyes, predominant proximal lower limb weakness, winged scapula, bilateral thigh atrophy, and calf volume increase.
Total serum creatine kinase was 218 IU/L (< 190) (1.1x), and aldolase of 7.4 U/L (< 15.2). Electromyogram was suggestive of a myopathic process. Muscle imaging (Fig. 1) demonstrated moderate asymmetry of the glutei, smaller on the right side, and severe atrophy of vastus lateralis. Right biceps brachialis muscle biopsy (Fig. 6) demonstrated atrophy, irregular intrassarcoplasmic basophilic deposits, internal nuclei, fibre type disproportion with Type 1 fibres 57% smaller than type 2 fibres: Type 1 = 30 (± 10) micrometre, Type 2 = 71 (± 11) micrometre, with no fibre type predominance: 47% Type 1 and 53% Type 2 (fibre type predominance criteria in superficial biceps brachialis Type 1 > 62,3%, and Type 2 > 77.7%), small lobulated fibers, and negative sarcomeric telethonin reaction. Electron microscopy (Fig. 7) demonstrated ring fibres, myofibrillar loss with glycogen accumulation, nuclear indentations, and irregularities of the nuclear membrane, subsarcolemmal accumulation of mitochondriae, membrane irregularities, and intrasarcoplasmic filamentous inclusions from 15 to 19 nm diameter. Molecular studies demonstrated a homozygous pathogenic variant c.157C > T, p.(Gln53*) or (Q53X) in exon 2 of the titin-cap (TCAP or telethonin) gene.

Light microscopy findings of right biceps brachialis muscle biopsy of Patient 3. Muscle with atrophic fibres (yellow arrows in a, b, c, d, e, f, g, h, i, and j), irregular intrassarcoplasmic basophilic deposits (white arrow in b), internal nuclei (green arrow in b), fibre type disproportion with Type 1 (dark fibre in yellow asterisk * in f) fibres 57% smaller than type 2 (light fibre at yellow arrow in f) fibres, Type 1 = 30 (± 10) micrometre, Type 2 = 71 (± 11) micrometre, with no fibre type predominance: 47% Type 1 and 53% Type 2 (fibre type predominance criteria in superficial biceps brachialis Type 1 > 62,3% and Type 2 > 77.7%). Small fibres (yellow * in g) with subsarcolemmal and intrasarcoplasmic mitochondrial proliferation with some characteristics of lobulated fibers, except for the irregularity of the sarcolemmal membrane (red arrow in g). Oxidative irregularities with moth eaten aspect (green arrow in h). Immunohistochemistry with completely negative sarcomeric telethonin reaction (arrow in k), compared to the normal control (arrow in l). (a HE 400x, b HE 200x. c Modified Gomori trichrome 200x, d PAS 200x original optic magnification (x 4.5195), e Oil-red-O 400x, f ATPase pH4.6 100x, g SDH 200x, h COX-SDH 100x, i NADH 20x, j Nonspecific esterase 200x, Immunoperoxidase Antibody anti-telethonin (G-11 sc-25327, Santa Cruz Biotechnology, Inc. at 1:50 dilution) Patient (k) 200x, and Control (l) 400x)

Electron microscopy findings of right biceps brachialis muscle biopsy of Patient 3. Muscle with ring fibres (yellow * in a, yellow arrow in d), characterized by a rim of perpendicularly oriented myofibrils around the fibre. Myofibrillar loss with glycogen accumulation (yellow * in b), nuclear abnormalities with indentations, and irregularities (yellow arrows in c, f, and g) of the nuclear membrane. Subsarcolemmal accumulation of mitochondriae (red arrow in c), membrane irregularities (yellow arrow in e), intrasarcoplasmic filamentous inclusions from 15 to 19 nm diameter (inset in h, and yellow arrow in i) (i is the inset area in h) (Transmission electron microscopy a 2,500x, b 3,000x, c 4,000x, d 4,000x, e 8,000x, f 10,000x, g 12,000x, h 10,000x, and i 60,000x)
Case 4
Patient 4 was admitted when she was 24 years old. She had presented difficulties to walk, and frequent falls in childhood. At the age of 17 years old, she started difficulties standing up, and climbing stairs. She was born of non-consanguineous parents with one symptomatic cousin, and one possible symptomatic distant relative. Physical examination demonstrated proximal lower limbs weakness, right foot drop, waddling gait, and decreased lower limbs reflexes.
Electromyogram was myopathic. Total creatine kinase was 1320 IU/L (< 165) (8x), and aldolase was 6.3 U/L (< 7.6). Muscle imaging (Fig. 1) demonstrated moderate asymmetric muscle fat replacement of semitendinosus, gastrocnemius medialis, and peroneus muscles. Right extensor digitorum longus muscle biopsy (Fig. 8) demonstrated variation in fibre calibre, nuclear internalization, fibre splitting, degeneration, rimmed vacuoles, small type 1 fibres, core-like oxidative irregular reactions, intrasarcoplasmic acid phosphatase activity, and negative intrasarcoplasmic immunohistochemical telethonin reaction. Electron microscopy (Fig. 9) demonstrated areas of subsarcolemmal loss of thick filaments with remnants of Z disks, myofibrillar degeneration, autophagic vacuoles with myelinoid figures, abnormal mitochondriae with variation in size and shape, dilated and ruptured cristae, paracrystalline inclusions, nuclear pyknosis, pseudoinclusions, i.e., invaginations of the sarcoplasm inside the irregular nucleus, and multiple focal areas of loss of the sarcolemmal membrane. Molecular studies demonstrated a homozygous pathogenic variant c.157C > T, p.(Gln53*) or (Q53X) in exon 2 of the titin-cap (TCAP or telethonin) gene.

Light microscopy findings of extensor digitorum longus muscle biopsy in the right leg of Patient 4. Variation in fibre calibre, nuclear internalization (white arrow in a), fibre splitting (yellow arrows in a, and b), multiple splitting in degenerating fibre (white arrow in b), rimmed vacuole (yellow arrow in c), small dark type 1 fibres (yellow * in d), core-like oxidative irregular reactions (yellow arrows in e, f, and g), and intrasarcoplasmic acid phosphatase activity (yellow arrow in h). Immunohistochemistry with complete negative intrassarcoplasmic telethonin reaction (arrow in i, compared to the normal control (inset arrow in i). Serial sections of a non-necrotic muscle fibre (same fibre marked with yellow * in j, k, and l) with focal secondary reduction in membrane dysferlin expression (yellow arrow in k) in the sarcolemmal membrane, compared to strong sarcolemmal membrane reaction of the adjacent fibres, and compared to normal dystrophin reaction in serial sections of the same fibre (yellow arrow in l) as proof of membrane integrity. (a HE 200x, b HE 400x. c HE 400x, d ATPase pH4.6 100x, e SDH 200x, f COX-SDH 200x, g NADH 400x, h Acid phosphatase 200x, Immunoperoxidase Antibody anti-telethonin (G-11 sc-25327, Santa Cruz Biotechnology, Inc. at 1:50 dilution) 200x, Patient (i), and 400x Control (inset in i), j. HE 400x, k. Immunoperoxidase Antibody anti-dysferlin (HAM1/7B6, Vector Laboratories) 400x, and l. Immunoperoxidase Antibody anti-dystrophin carboxy-terminal (NCL-DYS2, Novocastra, Leica) 400x)

Electron microscopy findings of extensor digitorum longus muscle biopsy in the right leg of Patient 4. Muscle with areas of subsarcolemmal loss of thick filaments with remnants of Z disks (yellow arrow in a), and myofibrillar degeneration (red arrow in e). Autophagic vacuoles with myelinoid figures (yellow arrows in b, c, and e), abnormal mitochondriae with variation in size and shape, dilated and ruptured cristae (yellow arrows in d, h, and i). Mitochondrial paracrystalline inclusions (arrow in g). Nuclear irregularities including pyknosis and nuclear pseudoinclusions, i.e., invaginations of the sarcoplasm inside the irregular nucleus (yellow arrow in f), multiple focal areas of loss of the sarcolemmal membrane (yellow * in h and i) (Transmission electron microscopy a 5,000x, b 6,000x, c 6,000x, d 8,000x, e 8,000x, f 12,000x, g 15,000x, h 40,000x, and i 50,000x)
Case 5
Patient 5 was admitted in our service when he was 29 years old. He presented frequent falls since he had acquired independent gait. He had started progressive gait disturbance, and weakness in the lower limbs when he was 20 years old. At the same time, he had noticed also tremor on the hands. He was born of non-consanguineous parents with no similar cases in the family. Physical examination disclosed increased calves, difficulties performing extraocular eye movement, and lower limbs proximal weakness.
Electromyogram disclosed myopathic motor unit potentials (MUPs) in all muscles studied but in the upper limbs a mixed myopathic and neurogenic pattern with both low amplitude and/or short duration, and high amplitude and/or long duration MUP. Total creatine kinase level was 1438 IU/L (< 190 IU/L), and aldolase was 10.1 U/L (< 7.6 U/L). Muscle imaging (Fig. 1) demonstrated moderate asymmetric gastrocnemius medialis, and peroneus fat replacement. Left quadriceps femoris muscle biopsy (Fig. 10) demonstrated 14.9% of the fibres with rimmed vacuoles, nuclear clumps, endomysial fibrosis, atrophy, hypertrophy, fibre splitting, type 2 fibre predominance with 97.5% of type 2 fibres, and 2.5% of type 1 fibres, and negative immunohistochemical telethonin reaction. Electron microscopy (Fig. 11) demonstrated autophagic vacuoles, atrophic fibres with invaginations of the sarcoplasmic membrane, atrophy with remnants of Z disks, intrasarcoplasmic degeneration, subsarcolemal accumulation of mitochondria, and myofibrillar disorganization with perpendicularly oriented sarcomeres. Molecular studies demonstrated a homozygous pathogenic variant c.157C > T, p.(Gln53*) or (Q53X) in exon 2 of the titin-cap (TCAP or telethonin) gene.

Light microscopy findings of left quadriceps femoris muscle biopsy of Patient 5. Muscle biopsy with 14.9% of the fibres presenting rimmed vacuoles (yellow arrows in a, b, c, d, e, and f; green arrow in b). Nuclear clumps (white arrows in b, f, and g), endomysial fibrosis (yellow * in h), atrophy (blue arrow in h, yellow arrow in j), hypertrophy with muscle fibre splitting (yellow arrow in h), type 2 fibre predominance of 97.5% (dark fibre with yellow * in i), with 2.5% of type 1 fibres (light fibre with white * in i). Immunohistochemistry with negative intrasarcoplasmic telethonin reaction (arrow in k), compared to the normal control (arrow in l). (a HE 400x, b HE 400x, c HE 400x, d HE 400x, e HE 200x, f HE 400x, g HE 400x, h HE 200x, i ATPase pH9.4 100x, j NADH 200x. Immunoperoxidase Antibody anti-telethonin (G-11 sc-25327, Santa Cruz Biotechnology, Inc. at 1:50 dilution) Patient (k) 100x, and Control (l) 200x)

Electron microscopy findings of left quadriceps femoris muscle biopsy of Patient 5. Muscle with autophagic vacuoles (yellow arrow in a, c, f, g, and i), atrophic fibres with invaginations of the sarcoplasmic membrane (green arrow in a), atrophy with remnants of Z disks (blue arrows in b, e, and h), intrasarcoplasmic degeneration (yellow * in c), and subsarcolemal accumulation of mitochondria (brown arrow in d). Myofibrillar disorganization with perpendicularly oriented sarcomeres (red arrow in d) (Transmission electron microscopy a 2,500x, b 2,500x, c 4,000x, d 4,000x, e 5,000x, f 5,000x, g 5,000x, h 12,000x, and i 15,000x)
Case 6
Patient 6 was admitted at 34 years of age. He had started at the age of 15, weakness in the left lower limb. He described the perception of pronounced worsening of his condition at the age of 25, with impossibility to stand up, and necessity to use a wheelchair. Simultaneously, at the age of 25, he began difficulties raising his arms, and he noticed limb atrophy. He had not been evaluated in our service between 15 and 25 years old. Therefore, the precise rate of gait impairment could not be documented in our outpatient clinic. He was born of non-consanguineous parents. Physical examination, at the age of 34 years, disclosed left divergent strabismus that he reported that had started at the age of 25 years, tongue atrophy, global muscular atrophy, proximal and distal upper limbs and lower limbs weakness, absent upper, and lower limb reflexes.
Electromyogram demonstrated myopathic and some neurogenic motor unit potentials. Total creatine kinase levels were 358 IU/L (< 190) (1.9x). Muscle imaging (Fig. 1) demonstrated diffuse muscle fat replacement with asymmetric involvement of tibialis posterior, and peroneus muscles. Right triceps brachialis muscle biopsy (Fig. 12) demonstrated variation in fibre calibre, isolated atrophic fibres, predominance of 95.1% type 1 fibres with 4.9% of type 2 fibres, lobulated-like fibres with irregular subsarcolemmal mitocondrial proliferation but without the irregular sarcolemmal membrane characteristic lobulated-trabeculated fibres, focal activity of acid phosphatase, and negative immunohistochemical telethonin reaction. Electron microscopy (Fig. 13) demonstrated autophagic vacuoles with granular and membranous material, myelinoid figures, Z disk streaming, disorganization of the Z disk, myofibrillar degeneration, remnants of myofilaments, invaginations of the sarcolemmal membrane, and proliferation of dense mitochondria. Molecular studies demonstrated a homozygous pathogenic variant c.157C > T, p.(Gln53*) or (Q53X) in exon 2 of the titin-cap (TCAP or telethonin) gene.

Light microscopy findings of right triceps brachialis muscle biopsy of Patient 6. Muscle with variation in fibre calibre, isolated atrophic fibres (yellow arrows in a, b, c, and j), slight increase in intrasarcoplasmic lipid content (yellow arrow in d), predominance of 95.1% type 1 fibres (dark fibre with yellow * in e) with 4.9% type 2 fibres (light fibre with white * in e), lobulated-like fibres with irregular subsarcolemmal mitocondrial proliferation but without the irregular sarcolemmal membrane of characteristic lobulated-trabeculated fibres (yellow arrows in f, g, and h), acid phosphatase activity (yellow arrow in i), immunohistochemistry with negative intrasarcoplasmic telethonin reaction (yellow arrow in k), compared to the normal control (yellow arrow in l) (a HE 400x, b Modified Gomori trichrome 400x, c PAS 400x, d Oil-red-O 200x, e ATPase pH4.6 100x, f SDH 200x, g COX-SDH 100x, h NADH 20x, i Acid phosphatase 200x, j Nonspecific esterase 200x, Immunoperoxidase Antibody anti-telethonin (G-11 sc-25327, Santa Cruz Biotechnology, Inc. at 1:50 dilution) 200x Patient (k), and 200x (l) Control)

Electron microscopy findings of right triceps brachialis muscle biopsy of Patient 6. Autophagic vacuoles with granular and membranous material and myelinoid figures (yellow * in a, e, f, and g), Z disk streaming (blue arrow in b), disorganization of the Z disk (red arrow in d), myofibrillar degeneration (red * in h) in atrophic fibre with nucleus with a prominent nucleolus (white arrow in h). Remnants of myofilaments (green arrow in f). Invaginations of the sarcolemmal membrane (brown arrow in i), mitochondrial proliferation (red arrow in c with dense mitochondria (green arrow in i) (Transmission electron microscopy a 4,000x, b 5,000x, c 5,000x, d 5,000x, e 6,000x, f 15,000x, g 6,000x, h 6,000x, and i 8,000x)
Case 7
Patient 7 was admitted at the age of 54. She had started tiptoe walking at 8 years old. She reported frequent falls due to a foot deformity in the left lower limb. She was born of non-consanguineous parents. She was wheelchair bound when she was 41 years old. Physical examination disclosed scapula alata, atrophy of the pelvic girdle, feet deformity in dorsiflexion, proximal upper limbs weakness, proximal and distal lower limb weakness, and decreased lower limbs reflexes.
Electromyogram was myopathic. Total creatine kinase was 269 IU/L (< 165) (1.6x), and aldolase was 5.8 U/L (< 7.6). Muscle imaging (Fig. 1) demonstrated severe almost complete muscle fat replacement with asymmetric involvement of the gastrocnemius lateralis. Right deltoid muscle biopsy (Fig. 14) demonstrated variation in fibre calibre, atrophy, hypertrophy, muscle fat replacement, internal nuclei, ragged-red fibres, necrosis, phagocytosis, nemaline rods, type 1 fibre predominance of 90.3% with 9.7% type 2 fibres, type 1 fibre disproportion of 34%, mean diameter of type 1 = 43 (± 13) micrometre, mean diameter of type 2 = 67 (± 18) micrometre, 37.3% of lobulated or trabeculated fibres, and negative immunohistochemical telethonin reaction. Electron microscopy (Fig. 15) demonstrated nemaline bodies, areas of myofibrillar disruption with accumulation of mitochondria, subsarcolemal mitochondrial proliferation, and abnormal mitochondrial paracrystalline inclusions. Molecular studies demonstrated a homozygous pathogenic variant c.157C > T, p.(Gln53*) or (Q53X) in exon 2 of the titin-cap (TCAP or telethonin) gene.

Light microscopy findings of right deltoid muscle biopsy of Patient 7. Muscle with variation in fibre calibre, atrophy, hypertrophy, replacement of muscle tissue by adipose tissue (blue arrow in a), internal nuclei (yellow arrow in a, and b), subsarcolemal basophilic accumulation (green arrow in b) that corresponds to red subsarcolemal proliferation (green arrow in e), and to ragged-red equivalent/ ragged blue fibres (green arrow in i). Necrosis with phagocytosis (yellow arrow in c), nemaline rods (arrow in d), subtle glycogen accumulation (yellow arrow in f), and intrasarcoplasmic lipid accumulation (yellow arrow in g). Type 1 fibre predominance of 90.3% (light fibre with yellow * in h) with 9.7% type 2 fibres (dark fibre with white * in h), and type 1 fibre disproportion of 34% (mean diameter of type 1 = 43 (± 13) micrometre, mean diameter of type 2 = 67 (± 18) micrometre, and 37.3% of lobulated or trabeculated fibres (yellow arrow in j). Immunohistochemistry with negative intrasarcoplasmic telethonin reaction (yellow arrow in k), compared to the normal control (yellow arrow in l) (a HE 100x original optic magnification (x 9.039), b HE 200x, c HE 400x, d Gomori modified trichrome 100x, e Gomori modified trichrome 400x, f PAS 200x original optic magnification (x 4.5195), g Oil-red-O 200x, h ATPase pH9.4 100x, i SDH 400x, j COX-SDH 200x, Immunoperoxidase Antibody anti-telethonin (G-11 sc-25327, Santa Cruz Biotechnology, Inc. at 1:50 dilution) Patient (k) 200x, and (l) Control 200x)

Electron microscopy findings of right deltoid muscle biopsy of Patient 7. Muscle with areas of myofibrillar disruption with accumulation of mitochondria (yellow * in a, and b), subsarcolemal mitochondrial proliferation (yellow * in c, yellow arrow in d, and e) with abnormal mitochondrial paracrystalline inclusions (yellow arrow in f), nemaline bodies (red arrows in g, h, and i) (Transmission electron microscopy a 2,500x, b 2,000x, c 12,000x, d 5,000x, e 20,000x, f 50,000x, g 6,000x, h 10,000x, and i 100,000x)
Imaging findings in this series
Muscle imaging studies of the patients in this series were organized in Fig. 1 from the least to the most affected. In Patients 2, 3, 4, 5, 6, and 7, it was possible to observe a progressive muscle fat replacement that was directly proportional to the disease duration. In childhood, muscle imaging abnormalities were subtle, but a rapid progression was observed after the age of 24 years old in this series of patients.
In these patients, the order of progression among muscles was: gluteus maximus, followed by gluteus medius, gluteus minimus, tibialis anterior, adductor magnus, adductor longus, semitendinosus, biceps femoris, semimembranosus, vastus lateralis, rectus femoris, vastus medialis, soleus, peroneus group (peroneus longus and peroneus brevis), extensor digitorum longus, tibialis posterior, gastronemius lateralis, and gastronemius medialis. A compensatory hypertrophy of sartorius and gracilis muscles was observed between 7 and 12 years of duration of disease (Fig. 1k, n, and q).
Muscle fat replacement was moderately asymmetric in three patients: Patient 3 (j, k, and l in Fig. 1), Patient 4 (m, n, and o in Fig. 1), and Patient 5 (p, q, and r in Fig. 1). Muscle involvement was slightly asymmetric in three patients: Patients 1 (d, e, and f in Fig. 1), Patient 6 (s, t, and u, in Fig. 1), and Patient 7 (v, w, and x in Fig. 1). Muscle involvement was symmetric in Patient 2 (g, h, and i in Fig. 1).
Morphological findings in this series
Some morphological findings were recurrent in this series (Table 1). Lobulated (trabeculated) fibres were observed in 57% (4/7): Patient 1 (Fig. 2f), Patient 3 (Fig. 6g, h, and i), Patient 6 (Fig. 12f, g, and h), and Patient 7 (Fig. 14i). Rimmed vacuoles were found in 43% (3/7): Patient 4 (Fig. 8c), Patient 5 (Fig. 10a, b, c, d, e, f, and g), and Patient 6 (Fig. 12i). Rimmed vacuoles and ultrastructural autophagic vacuoles were observed in the same patients that corresponded to 43% (3/7): Patient 4 (Fig. 9b, c, and e), Patient 5 (Fig. 11f, g, and i), and Patient 6 (Fig. 13a, e, f, and g).
Other morphological findings (Table 1) were: 17 nm intrasarcoplasmic filamentous inclusions in 14% (1/7), Patient 3 (Fig. 7h, and i); multifocal loss of the sarcolemmal membrane in 14% (1/7), Patient 4 (Fig. 9h, and i); mitochondrial paracrystalline inclusions in 29% (2/7), Patient 4 (Fig. 9g), and Patient 7 (Fig. 15f); and severe nuclear morphological abnormalities in 29% (2/7), Patient 2 (Fig. 5c, e, and f), and Patient 4 (Fig. 9f) characterized by pyknosis, nuclear membrane invaginations, and intranuclear sarcoplasmic pseudo-inclusions. Nemaline bodies were observed in 14% (1/7), Patient 7 (Fig. 15g, h, and i). Core-like areas were observed in 14% (1/7), Patient 4 (Fig. 8e, f, and g) patient in histochemical reactions.
Abnormal smallness of type fibres was observed without type 1 fibre predominance in 43% (3/7) of the patients. Small type 1 fibres were 34%, 52% and 57% smaller than type 2 fibres, respectively, in Patient 7 (Fig. 14h), Patient 1 (Fig. 2e), and Patient 3 (Fig. 6f) (Table 1). These fibres presented the same diameter as the lobulated (trabeculated) fibres with irregular subsarcolemmal and intermyofibrillary distribution of mitochondriae that could be noticed in ultrastructural studies in Patient 3 (Fig. 7c, and h), and Patient 7 (Fig. 15a, b, c, d, and e). Serial sections revealed that most type 1 atrophic fibres corresponded to lobulated or trabeculated fibres on oxidative stains (SDH, NADH, and COX) in Patient 1 (Fig. 2f, and g); Patient 3 (Fig. 6g, h, and i), and Patient 7 (Fig. 14i, and j). Therefore, the combined analysis of ATPase and oxidative reactions provides very useful information about the nature of the type 1 small fibres.
Immunohistochemical and molecular findings in this series
All patients had been submitted to muscle biopsy in advance to molecular studies according to the investigation guidelines that were used at the time of their admission. Immunohistochemical studies demonstrated complete telethonin deficiency with the mouse monoclonal antibody that recognizes the most C-terminal 109 amino acids (58–167).
Molecular studies demonstrated in all patients the homozygous c.157 C > T, p.(Gln53*) variant in exon 2 of the titin-cap (TCAP) gene.
Discussion
The results of a review of the literature are presented in Table 2 (Almeida et al. 2012; Almeida et al. 2013; Barresi et al. 2015; Bevilacqua et al. 2020; Blanco-Palmero et al. 2019; Brusa et al. 2018; Chakravorty et al. 2020; Chamova et al. 2017; Chamova et al. 2018; Chen et al. 2020; Chen et al. 2023; Cotta et al. 2014; de Fuenmayor-Fernández de la Hoz et al. 2016; Ferreiro et al. 2011; Fichna et al. 2018; Francis et al. 2014; Huang et al. 2022; Hudson et al. 2017; Ikenberg et al. 2017; Liang et al. 2020; Lima et al. 2005; Lin et al. 2023; Lorenzoni et al. 2023; Lv et al. 2021; Lv et al. 2023; Magri et al. 2017; Moreira et al. 1997; Moreira et al. 2000; Moutinho-Pereira et al. 2022; Nalini et al. 2015; Negrão et al. 2010; Olivé et al. 2008; Paim et al. 2013; Savarese et al. 2016; Seong et al. 2016; Tian et al. 2015; Vainzof et al. 2002; Waddell et al. 2012; Winckler et al. 2019; Yee et al. 2007; Zatz et al. 2000; current paper). As Patient 3 and Patient 4 were from the same small city, they had reported affected cousins, and they shared one common surname, they were counted as one single family.
Patients with telethoninopathy may present proximal, proximal and distal, or fascioscapular-humeral muscle weakness (Chen et al. 2023). In this series, a characteristic pattern of muscle fat replacement could be observed in patients between 22 and 32 years old, and between 7 and 12 years of disease. Patients with less than 7 years duration of disease presented very subtle abnormalities and after 12 years duration of disease they presented end stage muscle fat replacement. The main muscle imaging abnormalities in this series were moderate asymmetric muscle fat replacement of glutei, adductor magnus, quadriceps femoris, biceps femoris long head, semitendinosus, semimembranosus, gastrocnemius medialis, and peroneus muscles, with compensatory hypertrophy of sartorius, and gracilis.
Some particular findings could be noticed by the comparative analysis of the patients from this series to those described in the articles with imaging findings in LGMD R7 (Blanco-Palmero et al. 2019; Brusa et al. 2018; Chamova et al. 2018; Chen et al. 2020; Chen et al. 2023; de Fuenmayor-Fernandez de la Hoz et al. 2016; Ferreiro et al. 2011; Francis et al. 2014; Ikenberg et al. 2017; Lv et al. 2021; Lv et al. 2023; Negrao et al. 2010; Olive et al. 2008; and Paim et al. 2013): (1) muscle fat replacement was usually worse in the posterior compartment of the thighs (biceps femoris, semitendinosus, and semimembranosus) compared to adductor magnus (in the medial compartment), and quadriceps femoris (in the anterior compartment); (2) preservation or hypertrophy of sartorius was a constant finding. One exception was disproportionate preservation of vastus medialis with involvement of the other quadriceps femoris muscles (vastus lateralis, rectus femoris, and vastus intermedius) in two Chinese siblings with the compound heterozygous variants c.110 + 5G > A and c.26_33AGGTGTCG in the TCAP gene (Chen et al. 2020).
A selective involvement of adductor magnus, quadriceps femoris, semitendinosus, semimembranosus, and relative preservation of sartorius, and gracilis had been previously reported by Lv et al. (2021) in two Chinese sisters with the homozygous c. 165–166 ins G variant in the TCAP gene. Olive et al. (2008) observed involvement of biceps femoris, semitendinosus, semimembranosus, quadriceps femoris, and tibialis anterior with relative preservation of sartorius in a female patient with the homozygous c.75G > A (p.Trp25*) variant in the TCAP gene in a patient from Moldova. This pattern was similar to that described in a 30 years old male with compound heterozygous mutation comprising c.110 + 5G > A and c. 26_33AGGTGTCG, variants in the TCAP gene described by Chen et al. 2020. The imaging pattern described by Olive et al. (2008) was similar to that described by Negrao et al. (2010) in a 50 years old man with the homozygous c.157C > T (p.Gln53*) variant in the TCAP gene, except that the later did not present the relative preservation of the sartorius. Francis et al. (2014) demonstrated involvement of addutor magnus, biceps femoris long head, and semimembranosus, with hypertrophy of sartorius, and gracilis in a 21 years old male patient (F97-1) with the heterozygous c.26_33dup (p.Glu12Argfs*20) c.100delC/ c.166insG. Therefore, involvement of the medial, anterior, and posterior compartments of the thigh with relative preservation or hypertrophy of the sartorius with or without gracilis involvement was observed with pathogenic variants in the TCAP gene.
The phenomenon of asymmetry was observed in 85.7% (6/7) of the patients in this series, it was moderate in 42.9% (3/7), subtle in 42.9% (3/7), and absent in 14.3% (1/7). The review of the literature (Table 2) revealed asymmetry in only 30.4% (14/46) of the patients. All patients included in this series were analysed for asymmetry both in clinical and imaging exams. Therefore, the hypothesis that the use of ancillary studies might have increased the sensitivity of the detection of asymmetry in cases of subclinical involvement was raised.
Previous imaging studies of telethoninopathy presented either early tibialis anterior muscle fat replacement (Negrão et al. 2010; Olivé et al. 2008) or tibialis anterior preservation (Cotta et al. 2014; de Fuenmayor-Fernandez de la Hoz et al. 2016; Francis et al. 2014; Ikenberg et al. 2017). The phenomenon of divergent leg involvement in different patients with the same disease had been previously described in another Limb Girdle Muscular Dystrophy (LGMDR2, previously known as LGMD2B), related to the dysferlin (DYSF) gene. Most patients with dysferlinopathy usually present posterior leg muscle fat replacement, but some patients present early tibialis anterior involvement (Illa et al. 2001). The muscle imaging differential diagnosis between telethoninopathy and anoctaminopathy is a potential pitfall as anoctaminopathy frequently presents as asymmetric muscle fat replacement (Sarkozy et al. 2012).
Two previous publications described partial deficiency or reduced immunohistochemical reaction for telethonin. The first article (Barresi et al. 2015) described a 49 years old man of Indian descent with the homozygous non-sense mutation, c.244C > T (p.Gln82X), in exon 2 of the TCAP gene, who was tested with two different antibodies, one directed to the full-length protein (clone 53/ Telethonin, BD 612328) that presented partial retention of telethonin reaction and the second was the same antibody used for the patients in this series, directed to the aa 58–167 of telethonin (G-11:sc-25327, Santa Cruz) that showed a complete lack of signal. The second article (Chen et al. 2023) described a 32 years old Chinese man with the heterozygous c.[26 33AGGTGTCG] + [110 + 5 G > A]; p.[Glu12Argfs] + [?] variant in the TCAP gene, that presented partial retention of telethonin immunohistochemical reaction by Chen et al. (2023) with the full length human recombinant protein of human TCAP (NP_003664) produced in E. coli. (LS-C340495; LifeSpan BioSciences, USA). No muscle imaging studies were available for this two patients to correlate the partial deficiency of the expression of telethonin with any particular muscle imaging pattern. The subjective evaluation of the intensity of the myofibrillar sarcoplasmic immunohistochemical reaction of telethonin compared to the control is not easy. Therefore, based on these findings, the routine use of the antibody directed to the C-terminus telethonin should be recommended to avoid false positive results with the antibody directed to the full-length protein.
Morphological examination of the patients in this series demonstrated rimmed vacuoles in 42.9% (3/7) of the patients, and autophagic vacuoles in the same patients 42.9% (3/7). The review in the literature demonstrated rimmed vacuoles in 38.8% (19/49) of the patients, and autophagic vacuoles in 33% (3/9) of the patients. Filamentous inclusions of 17 nm diametre were found in the sarcoplasm of one patient in this series and in one patient described by Chen et al. 2023.
Rimmed vacuoles are abnormal empty spaces or cavities within the sarcoplasm, irregular or round, localized in any part of the fibre, in which the rim of the vacuole contains a basophilic granular material (HE) or red granular material (modified Gomori trichrome) (Engel and Franzini-Armstrong 2004; Weihl 2019). Rimmed vacuoles were described in several acquired myopathies including inflammatory myopathies such as inclusion body myositis, toxic myopathies, and hereditary myopathies as autophagic vacuolar myopathies, ion channel muscle diseases, and related to several other genes, such as ANO5, CAV3, COL6A1, CRYAB, DES, DMD, DNAJB6, DUX4/SMCHD1, DYSF, EMD, FHL1, FKRP, GAA, GNE, GYG1, LAMA2, LDB3, LMNA, MATR3, MYHC-IIA, MYH2, MYH7, MYOT, PABPN1, PNPLA2, TCAP, TIA1, TNPO3, TTN, and TRIM32 (Cotta et al. 2021; Engel and Franzini-Armstrong 2004; Mair et al. 2020).
Rimmed vacuoles may contain products of cytoplasmic degradation that reflect a dysfunction in membrane trafficking with autophagic vacuoles that contain endosomal, nuclear, and endoplasmic reticulum proteins (Weihl 2019). Filamentous inclusions may be intrasarcoplasmic or intranuclear. In inclusion body myositis, tubulofilamentous inclusions of 15–21 nm are pair helical filaments containing p-tau protein that accumulate as a consequence of impaired autophagy and dysfunction of both ubiquitin-proteosome system, and molecular chaperones (Guglielmi et al. 2024).
In hereditary diseases, it is believed that rimmed vacuoles accumulate structural proteins such as intermediate filaments, RNA-binding proteins, and proteins associated with protein folding, degradation, and RNA-binding (Weihl 2019). Therefore, rimmed vacuoles may be a consequence of cellular dysfunction due to several acquired or hereditary causes. Specifically in telethoninopathy, Lv et al. 2021 raised the hypothesis that autophagolysosomes may be of mitochondrial origin, related to impaired mitophagy, as they found vacuoles surrounded by double membranes.
Lobulated fibres were observed in 57.1% (4/7) of the patients in this series and in 43.1% (22/51) of the patients in the review of the literature. Type 1 atrophic fibres were observed in 42.9% (3/7) of the patients in this series and in 58.3% (7/12) of the patients from the review of the literature. In this series, morphometric studies demonstrated smallness of Type 1 fibres compared to Type 2 fibres with fibre disproportion that varied from 34 to 57%. No similar results were found in the review of the literature.
Telethoninopathy morphologic presentation is different from Congenital fibre type disproportion (CFTD). CFTD is a morphologic hallmark of several types of structural congenital myopathies, a group of non progressive myopathies that usually start in the first two years of life, and are clinically different from the progressive muscular dystrophies. In CFTD there is type 1 fibre type disproportion, i.e., type 1 fibres are at least 25% (Dubowitz et al. 2020) or 35% to 40% (Clarke 2011; Cotta et al. 2021) smaller than type 2 fibres, combined with type 1 fibre predominance. In CFTD, lobulated or trabeculated fibres are usually not observed.
Nemaline bodies were observed in 14% (1/7) in this series and had been previously described by our group (Paim et al. 2013), in the patient with the longest duration of disease (46 years). The review of the literature revealed nemaline bodies in the muscle biopsy of a 22 years old female Bulgarian Muslim (Chamova et al. 2018). Therefore, nemaline bodies may occur earlier in the course of the disease than previously thought (Paim et al. 2013).
Patient 2 in this series presented ectopic dystrophin sarcoplasmic deposits in immunohistochemistry that may indicate myofibrillar abnormalities. Myofibrillar deposits had been previously described in telethoninopathy by Chen et al. 2023 in a 32 years old Chinese man. Those authors suggested that myofibrillar myopathy in telethoninopathy may be under-reported due to the small number of electron microscopy studies.
Multifocal loss of the sarcolemmal membrane was observed in 14.3% (1/7) of the patients in this series. In this review of the literature, no description of multifocal sarcolemmal loss was described in the LGMD R7 articles with electron microscopy results (Barresi et al. 2015; Brusa et al. 2018; Chen et al. 2023; Cotta et al. 2014; Lv et al. 2021; Olive et al. 2008; Paim et al. 2013). On the other hand, multifocal sarcolemmal loss had been previously described in Anoctaminopathy, Limb Girdle Muscular Dystrophy anoctamin 5-related (LGMD-R12, previously known as LGMD2L) (Bolduc et al. 2010).
Conclusion
Telethoninopathy is a rare subtype of Limb Girdle Muscular Dystrophy with heterogeneous morphologic presentation. Patients with the c.157C > T variant in the TCAP gene seemed to present similar morphological characteristics to patients with other variants, but this information need to be confirmed with larger samples. Intrasarcoplasmic 17 nm filamentous inclusions, multifocal loss of the sarcolemmal membrane, pyknosis, nuclear membrane invaginations, and intranuclear sarcoplasmic pseudo-inclusions may be found. A morphological aspect reminiscent of fibre type disproportion with small type 1 lobulated/ trabeculated fibres may be observed without type 1 fibre predominance. Anti-telethonin immunohistochemistry may be helpful in unsolved cases with myopathic abnormalities without dystrophic features. Detailed clinical and imaging correlation may be necessary in cases of nonspecific abnormalities to confirm the dystrophic nature of the disease. Knowledge of the morphologic phenotypic variability may help establish early and precise diagnosis to guide appropriate genetic counselling, clinical and rehabilitation care.
Availability of data and materials
Not applicable.
Abbreviations
- AdL:
-
Adductor longus Muscle
- AdM:
-
Adductor magnus Muscle
- ANO5 gene:
-
Anoctamin 5 gene
- Bf:
-
Biceps femoris Muscle
- CFTD:
-
Congenital fibre type disproportion
- CT:
-
Computed tomography
- DYSF gene:
-
Dysferlin gene
- E:
-
Extensor group of muscles of the leg (extensor digitorum longus and extensor hallucis longus)
- G:
-
Gracilis Muscle
- Gl:
-
Gastrocnemius lateralis Muscle
- Gm:
-
Gastrocnemius medialis Muscle
- Gmax:
-
Gluteus maximus Muscle
- Gmed:
-
Gluteus medius Muscle
- Gmin:
-
Gluteus minimus Muscle
- LGMD:
-
Limb Girdle Muscular Dystrophy
- LGMDR2:
-
Limb Girdle Muscular Dystrophy R2, dysferlin-related
- LGMD2B:
-
Limb Girdle Muscular Dystrophy 2B, dysferlin-related (previous nomenclature)
- LGMDR7:
-
Limb Girdle Muscular Dystrophy R7, telethonin-related
- LGMD2G:
-
Limb Girdle Muscular Dystrophy 2G, telethonin-related (previous nomenclature)
- LGMDR12:
-
Limb Girdle Muscular Dystrophy R12, anoctamin 5-related
- LGMD2L:
-
Limb Girdle Muscular Dystrophy 2L, anoctamin 5-related (previous nomenclature)
- MRI:
-
Magnetic resonance imaging
- P:
-
Peroneus Group of muscles (peroneus longus and peroneus brevis)
- Rf:
-
Rectus femoris Muscle
- S:
-
Sartorius Muscle
- Sm:
-
Semimembranosus Muscle
- So:
-
Soleus Muscle
- St:
-
Semitendinosus Muscle
- Ta:
-
Tibialis anterior Muscle
- TCAP gene:
-
Titin-cap gene or telethonin gene
- Tp:
-
Tibialis posterior Muscle
- Vi:
-
Vastus intermedius Muscle
- VL:
-
Vastus lateralis Muscle
- Vmed:
-
Vastus medialis Muscle
References
Almeida CF, Lima BL, Onofre-Oliveira PCG, Pavanello RCM, Zatz M, Vainzof M. LGMD2G with clinical presentation of congenital muscular: a rare phenotype. Neuromuscul Disord. 2012;22:804. https://doi.org/10.1016/j.nmd.2012.06.099.
Almeida CF, Onofre-Oliveira PCG, Zatz M, Negrao L, Vainzof M. Why is LGMD2G rare? Neuromuscul Disord. 2013;23:S738. https://doi.org/10.1016/j.nmd.2013.06.460.
Barresi R, Morris C, Hudson J, Curtis E, Pickthall C, Bushby K, et al. Conserved expression of truncated telethonin in a patient with limb-girdle muscular dystrophy 2G. Neuromuscul Disord. 2015;25:349. https://doi.org/10.1016/j.nmd.2014.12.006.
Benarroch L, Bonne G, Rivier F, Hamroun D. The 2024 version of the gene table of neuromuscular disorders (nuclear genome). Neuromuscul Disord. 2024;34:126. https://doi.org/10.1016/j.nmd.2023.12.007.
Bevilacqua JA, Guecaimburu Ehuletche MDR, Perna A, Dubrovsky A, Franca MC Jr, Vargas S, et al. The Latin American experience with a next generation sequencing genetic panel for recessive limb-girdle muscular weakness and Pompe disease. Orphanet J Rare Dis. 2020;15:11. https://doi.org/10.1186/s13023-019-1291-2.
Blanco-Palmero VA, Hernández-Laín A, Uriarte-Pérez de Urabayen D, Cantero-Montenegro D, Olivé M, Domínguez-González C. Late onset distal myopathy: a new telethoninopathy. Neuromuscul Disord. 2019;29:80. https://doi.org/10.1016/j.nmd.2018.11.001.
Bolduc V, Marlow G, Boycott KM, Saleki K, Inoue H, Kroon J, et al. Recessive mutations in the putative calcium-activated chloride channel Anoctamin 5 cause proximal LGMD2L and distal MMD3 muscular dystrophies. Am J Hum Genet. 2010;86:213. https://doi.org/10.1016/j.ajhg.2009.12.013.
Brusa R, Magri F, Papadimitriou D, Govoni A, Del Bo R, Ciscato P, et al. A new case of limb girdle muscular dystrophy 2G in a Greek patient, founder effect and review of the literature. Neuromuscul Disord. 2018;28:532. https://doi.org/10.1016/j.nmd.2018.04.006.
Chakravorty S, Nallamilli BRR, Khadilkar SV, Singla MB, Bhutada A, Dastur R, et al. Clinical and genomic evaluation of 207 genetic myopathies in the Indian subcontinent. Front Neurol. 2020;11:559327. https://doi.org/10.3389/fneur.2020.559327. eCollection 2020.
Chamova T, Bichev S, Gospodinova M, Zlatareva D, Taneva A, Kastreva K, et al. Limb girdle muscular dystrophy 2G in a religious minority of Bulgarian muslims homozy-gous for the c.75G>A, p.Trp25* TCAP mutation. Neuromuscul Disord. 2017;27:S96. https://doi.org/10.1016/j.nmd.2017.06.181.
Chamova T, Bichev S, Todorov T, Gospodinova M, Taneva A, Kastreva K, et al. Limb girdle muscular dystrophy 2G in a religious minority of Bulgarian Muslims homozygous for the c.75G>A, p.Trp25X mutation. Neuromuscul Disord. 2018;28:625. https://doi.org/10.1016/j.nmd.2018.05.00.
Chen H, Xu G, Lin F, Jin M, Cai N, Qiu L, et al. Clinical and genetic characterization of limb girdle muscular dystrophy R7 telethonin-related patients from three unrelated Chinese families. Neuromuscul Disord. 2020;30:137. https://doi.org/10.1016/j.nmd.2019.12.004.
Chen Z, Saini M, Koh JS, Prasad K, Koh SH, Tay KSS, et al. Unique clinical, radiological and histopathological characteristics of a Southeast Asian cohort of patients with limb-girdle muscular dystrophy 2G/LGMD-R7-telethonin-related. J Neuromuscul Dis. 2023;10:91. https://doi.org/10.3233/JND-221517.
Clarke NF. Congenital fiber-type disproportion. Semin Pediatr Neurol. 2011;18:264. https://doi.org/10.1016/j.spen.2011.10.008.
Cotta A, Carvalho E, da-Cunha-Júnior AL, Valicek J, Navarro MM, Baptista Junior S, et al. Muscle biopsy essential diagnostic advice for pathologists. Surg Exp Pathol. 2021;4:3. https://doi.org/10.1186/s42047-020-00085-w.
Cotta A, Paim JF. Muscle Biopsy Reporting. Arch Pathol Lab Med. 2016;140:879. https://doi.org/10.5858/arpa.2016-0070-LE.
Cotta A, Paim JF, da-Cunha-Junior AL, Neto RX, Nunes SV, Navarro MM, et al. Limb girdle muscular dystrophy type 2G with myopathic-neurogenic motor unit potentials and a novel muscle image pattern. BMC Clin Pathol. 2014;14:41. https://doi.org/10.1186/1472-6890-14-41.
Dastgir J, Rutkowski A, Alvarez R, Cossette SA, Yan K, Hoffmann RG, et al. Common Data Elements for Muscle Biopsy Reporting. Arch Pathol Lab Med. 2016;140:51. https://doi.org/10.5858/arpa.2014-0453-OA.
de Fuenmayor-Fernández de la Hoz CP, Hernández-Laín A, Olivé M, Fernández-Marmiesse A, Domínguez-González C. Novel mutation in TCAP manifesting with asymmetric calves and early-onset joint retractions. Neuromuscul Disord. 2016;26:749. https://doi.org/10.1016/j.nmd.2016.07.003.
Dubowitz V, Sewry CA, Oldfors A. Muscle biopsy: a practical approach. 5th ed. Saunders Elsevier, Printed in China; 2020. p. 1–600 https://www.elsevier.com/books/muscle-biopsy/dubowitz/978-0-7020-7471-4.
Engel AG, Franzini-Armstrong C. Myology. 3rd ed. New York: McGraw-Hill, Medical Publishing Division; 2004. p. 1–1960.
Ferreiro A, Mezmezian M, Olivé M, Herlicoviez D, Fardeau M, Richard P, et al. Telethonin-deficiency initially presenting as a congenital muscular dystrophy. Neuromuscul Disord. 2011;21:433. https://doi.org/10.1016/j.nmd.2011.03.005.
Fichna JP, Macias A, Piechota M, Korostyński M, Potulska-Chromik A, Redowicz MJ, et al. Whole-exome sequencing identifies novel pathogenic mutations and putative phenotype-influencing variants in Polish limb-girdle muscular dystrophy patients. Hum Genomics. 2018;12:34. https://doi.org/10.1186/s40246-018-0167-1.
Francis A, Sunitha B, Vinodh K, Polavarapu K, Katkam SK, Modi S, et al. Novel TCAP mutation c.32C>A causing limb girdle muscular dystrophy 2G. PLoS One. 2014;9:e102763. https://doi.org/10.1371/journal.pone.0102763.
Guglielmi V, Cheli M, Tonin P, Vattemi G. Sporadic Inclusion body myositis at the crossroads between muscle degeneration, inflammation, and aging. Int J Mol Sci. 2024;25:2742. https://doi.org/10.3390/ijms25052742.
Hayashi T, Arimura T, Itoh-Satoh M, Ueda K, Hohda S, Inagaki N, et al. Tcap gene mutations in hypertrophic cardiomyopathy and dilated cardiomyopathy. J Am Coll Cardiol. 2004;44(11):2192–201. https://doi.org/10.1016/j.jacc.2004.08.058.
Huang K, Li QX, Duan HQ, Luo YB, Bi FF, Yang H. Findings of limb-girdle muscular dystrophy R7 telethonin-related patients from a Chinese neuromuscular center. Neurogenetics. 2022;23:37. https://doi.org/10.1007/s10048-021-00681-2.
Hudson J, Cameron S, Graham E, Evangelista T, Straub V, Guglieri M, et al. Telethonin gene mutations detected by next generation sequencing. Neuromuscul Disord. 2017;27:S5. https://www.clinicalkey.com/#!/content/journal/1-s2.0-S0960896617302602.
Ikenberg E, Karin I, Ertl-Wagner B, Abicht A, Bulst S, Krause S, et al. Rare diagnosis of telethoninopathy (LGMD2G) in a Turkish patient. Neuromuscul Disord. 2017;27:856. https://doi.org/10.1016/j.nmd.2017.05.0174.
Illa I, Serrano-Munuera C, Gallardo E, Lasa A, Rojas-García R, Palmer J, et al. Distal anterior compartment myopathy: a dysferlin mutation causing a new muscular dystrophy phenotype. Ann Neurol. 2001;49:130. https://doi.org/10.1002/1531-8249(200101)49:1%3c130::AID-ANA22%3e3.0.CO;2-0.
Liang WC, Jong YJ, Wang CH, Wang CH, Tian X, Chen WZ, et al. Clinical, pathological, imaging, and genetic characterization in a Taiwanese cohort with limb-girdle muscular dystrophy. Orphanet J Rare Dis. 2020;15:160. https://doi.org/10.1186/s13023-020-01445-1.
Lima BL, Gouveia TL, Pavanello RC, Faulkner G, Valle G, Zatz M, et al. LGMD2G - screening for mutations in a large sample of Brazilian patients allows the identification of new cases. Neuromuscul Disord. 2005;15:676. https://doi.org/10.1016/j.nmd.2005.06.006.
Lin F, Yang K, Lin X, Jin M, Chen L, Zheng FZ, et al. Clinical features, imaging findings and molecular data of limb-girdle muscular dystrophies in a cohort of Chinese patients. Orphanet J Rare Dis. 2023;18:356. https://doi.org/10.1186/s13023-023-02897-x.
Liu W, Pajusalu S, Lake NJ, Zhou G, Ioannidis N, Mittal P, et al. Estimating prevalence for limb-girdle muscular dystrophy based on public sequencing databases. Genet Med. 2019;21:2512. https://doi.org/10.1038/s41436-019-0544-8.
Lorenzoni PJ, Kay CSK, Ducci RD, Fustes OJH, Rodrigues PRDVP, Hrysay NMC, et al. Single-centre experience with autosomal recessive limb-girdle muscular dystrophy: case series and literature review. Arq Neuropsiquiatr. 2023;81:922. https://doi.org/10.1055/s-0043-1772833.
Loughlin M. Muscle biopsy: a laboratory investigation. Oxford: Butterworth Heinemann Ltd.; 1993. p. 1–242.
Lv X, Gao F, Dai T, Zhao D, Jiang W, Geng H, et al. Distal myopathy due to TCAP variants in four unrelated Chinese patients. Neurogenetics. 2021;22:1. https://doi.org/10.1007/s10048-020-00623-4.
Lv X, Lin F, Wu W, Wang H, Luo Y, Wang Z, et al. The clinical features and TCAP mutation spectrum in a Chinese cohort of patients with limb-girdle muscular dystrophy R7. Hum Mol Genet. 2023;32:2502. https://doi.org/10.1093/hmg/ddad090.
Magri F, Nigro V, Angelini C, Mongini T, Mora M, Moroni I, et al. The italian limb girdle muscular dystrophy registry: relative frequency, clinical features, and differential diagnosis. Muscle Nerve. 2017;55:55. https://doi.org/10.1002/mus.25192.
Mair D, Biskup S, Kress W, Abicht A, Brück W, Zechel S, et al. Differential diagnosis of vacuolar myopathies in the NGS era. Brain Pathol. 2020;30:877. https://doi.org/10.1111/bpa.12864.
Moreira ES, Vainzof M, Marie SK, Sertié AL, Zatz M, Passos-Bueno MR. The seventh form of autosomal recessive limb-girdle muscular dystrophy is mapped to 17q11–12. Am J Hum Genet. 1997;61:151. https://doi.org/10.1086/513889.
Moreira ES, Wiltshire TJ, Faulkner G, Nilforoushan A, Vainzof M, Suzuki OT, et al. Limb-girdle muscular dystrophy type 2G is caused by mutations in the gene encoding the sarcomeric protein telethonin. Nat Genet. 2000;24:163. https://doi.org/10.1038/72822.
Moutinho-Pereira S, Morais-de-Sá E, Greenfield H, Pereira PR. Systemic sclerosis in a patient with muscle dystrophy. BMJ Case Rep. 2022;15:e250389. https://doi.org/10.1136/bcr-2022-250389.
Murphy AP, Straub V. The classification, natural history and treatment of the limb girdle muscular dystrophies. J Neuromuscul Dis. 2015;2:S7. https://doi.org/10.3233/JND-150105.
Nalini A, Polavarapu K, Sunitha B, Kulkarni S, Gayathri N, Srinivas Bharath MM, et al. A prospective study on the immunophenotypic characterization of limb girdle muscular dystrophies 2 in India. Neurol India. 2015;63:548. https://doi.org/10.4103/0028-3886.162048.
Negrão L, Matos A, Geraldo A, Rebelo O. Limb-girdle muscular dystrophy in a Portuguese patient caused by a mutation in the telethonin gene. Acta Myol. 2010;29:21. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2954583/.
Olivé M, Shatunov A, Gonzalez L, Carmona O, Moreno D, Quereda LG, et al. Transcription-terminating mutation in telethonin causing autosomal recessive muscular dystrophy type 2G in a European patient. Neuromuscul Disord. 2008;18:929. https://doi.org/10.1016/j.nmd.2008.07.009.
Paim JF, Cotta A, Vargas AP, Navarro MM, Valicek J, Carvalho E, et al. Muscle phenotypic variability in limb girdle muscular dystrophy 2G. J Mol Neurosci. 2013;50:339. https://doi.org/10.1007/s12031-013-9987-6.
Sarkozy A, Deschauer M, Carlier RY, Schrank B, Seeger J, Walter MC, et al. Muscle MRI findings in limb girdle muscular dystrophy type 2L. Neuromuscul Disord. 2012;22:S122. https://doi.org/10.1016/j.nmd.2012.05.012.
Savarese M, Di Fruscio G, Torella A, Fiorillo C, Magri F, Fanin M, et al. The genetic basis of undiagnosed muscular dystrophies and myopathies: Results from 504 patients. Neurology. 2016;87:71. https://doi.org/10.1212/WNL.0000000000002800.
Seong MW, Cho A, Park HW, Seo SH, Lim BC, Seol D, et al. Clinical applications of next-generation sequencing-based gene panel in patients with muscular dystrophy: Korean experience. Clin Genet. 2016;89:484. https://doi.org/10.1111/cge.12621.
Straub V, Murphy A, Udd B, LGMD workshop study group. 229th ENMC international workshop: Limb girdle muscular dystrophies - nomenclature and reformed classification Naarden, the Netherlands, 17–19 March 2017. Neuromuscul Disord. 2018;28:702. https://doi.org/10.1016/j.nmd.2018.05.007.
Tian X, Liang WC, Feng Y, Wang J, Zhang VW, Chou CH, et al. Expanding genotype/phenotype of neuromuscular diseases by comprehensive target capture/NGS. Neurol Genet. 2015;1:e14. https://doi.org/10.1212/NXG.0000000000000015.
Udd B, Stenzel W, Oldfors A, Olivé M, Romero N, Lammens M, et al. 1st ENMC European meeting: The EURO-NMD pathology working group Recommended Standards for Muscle Pathology Amsterdam, The Netherlands, 7 December 2018. Neuromuscul Disord. 2019;29:483. https://doi.org/10.1016/j.nmd.2019.03.002.
Vainzof M, Moreira ES, Suzuki OT, Faulkner G, Valle G, Beggs AH, et al. Telethonin protein expression in neuromuscular disorders. Biochim Biophys Acta. 2002;1588:33. https://doi.org/10.1016/s0925-4439(02)00113-8.
Valle G, Faulkner G, De Antoni A, Pacchioni B, Pallavicini A, Pandolfo D, et al. Telethonin, a novel sarcomeric protein of heart and skeletal muscle. FEBS Lett. 1997;415:163. https://doi.org/10.1016/s0014-5793(97)01108-3.
Waddell LB, Lek M, Bahlo M, Bromhead C, Jonse K, North KN, et al. The identification of LGMD2G (TCAP) in Australia [abstract]. Neuromuscul Disord. 2012;22:804. https://doi.org/10.1016/j.nmd.2012.06.100.
Weihl CC. Sporadic inclusion body myositis and other rimmed vacuolar myopathies. Continuum (minneap Minn). 2019;25:1586. https://doi.org/10.1212/CON.0000000000000790.
Winckler PB, da Silva AMS, Coimbra-Neto AR, Carvalho E, Cavalcanti EBU, Sobreira CFR, et al. Clinicogenetic lessons from 370 patients with autosomal recessive limb-girdle muscular dystrophy. Clin Genet. 2019;96:341. https://doi.org/10.1111/cge.13597.
Yee W, Pramono Z, Tan C, Kathiravelu P, Lai P. Limb girdle muscular dystrophy 2G and a novel TCAP mutation in ethnic Chinese. Neuromuscul Disord. 2007;17:764. https://doi.org/10.1016/j.nmd.2007.06.180.
Zatz M, Vainzof M, Passos-Bueno MR. Limb-girdle muscular dystrophy: one gene with different phenotypes, one phenotype with different genes. Curr Opin Neurol. 2000;13:511. https://doi.org/10.1097/00019052-200010000-00002.
Acknowledgements
We would like to thank the patients for participation in this study, and Mr. Cleides Campos de Oliveira for technical assistance.
Funding
Not applicable.
Author information
Authors and Affiliations
Contributions
AC was responsible for the conception, design, organization, photographic documentation, draft, and revision of the final version of the manuscript. EC, and JV contributed with neurophysiological data. ALCJr contributed with muscle imaging data. EBS, MIL, and BAC contributed with electron microscopy information. MMN, FG, RXdSN, APV, MMM, SVNN, and AFC contributed with clinical data. CCS, RIT, and MV contributed with molecular studies. AC, and JFP were responsible for muscle biopsy analyses.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This manuscript has been approved by the Ethics and Research Comittee of The SARAH Network of Rehabilitation Hospitals and “Plataforma Brasil”, number 751.634 CAAE: 34094014.4.0000.0022.
Consent for publication
A written informed consent for publication was obtained from patients.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Cotta, A., Carvalho, E., da-Cunha-Júnior, A.L. et al. Muscle ultrastructure and histopathological findings in a Brazilian single-centre series of genetically classified telethoninopathy patients. Surg Exp Pathol 7, 12 (2024). https://doi.org/10.1186/s42047-024-00155-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s42047-024-00155-3