
Abstract
Bruton’s tyrosine kinase (BTK), a nonreceptor tyrosine kinase, plays a remarkable role in the transmission and amplification of extracellular signals to intracellular signaling pathways. Various types of cells use the BTK pathway to communicate, including hematopoietic cells particularly B cells and T cells. The BTK pathway plays a role in controlling the proliferation, survival, and functions of B cells as well as other myeloid cells. First, second, and third-generation BTK inhibitors are currently being evaluated for the treatment of immune-mediated diseases in addition to B cell malignancies. In this article, the available evidence on the action mechanisms of BTK inhibitors is reviewed. Then, the most recent data obtained from preclinical studies and ongoing clinical trials for the treatment of autoimmune diseases, such as pemphigus vulgaris, pemphigus foliaceus, bullous pemphigoid, systemic lupus erythematosus, Sjögren’s disease, rheumatoid arthritis, systemic sclerosis, multiple sclerosis, myasthenia gravis, and inflammatory diseases such as psoriasis, chronic spontaneous urticaria, atopic dermatitis, and asthma are discussed. In addition, adverse effects and complications associated with BTK inhibitors as well as factors predisposing patients to BTK inhibitors complications are discussed.
Similar content being viewed by others


Introduction
In 1952, Ogden Bruton first reported X-linked agammaglobulinemia (XLA), which is a primary immunodeficiency disease, in an 8-year-old boy who complained of recurrent bacterial sepsis, otitis, and osteomyelitis, manifested by a notably decreased B-cell number and decreased serum immunoglobulin levels [1]. In 1993, the genetic basis of XLA was discovered as a mutation in a coding sequence of protein-tyrosine kinase and named after Bruton as Bruton’s tyrosine kinase (BTK) [2]. BTK is a 659 amino acid protein forming five signaling domains, which enable it to transmit and amplify signals from various cell surface receptors involved in the transmission of extracellular signals to intracellular signaling pathways [3]. As a non-receptor tyrosine kinase, BTK is expressed in most hematopoietic cells, especially in B cells, leading to the development and activation of B cells through B-cell antigen receptor (BCR) and Toll-like receptor (TLR) signaling [4,5,6]. BTK signaling contribute to the pathogenesis of autoimmune disease in synergy with TLR-mediated pathways [5, 7, 8]. Indeed, antigens bind to BCR and activate BTK, leading to phospholipase-Cγ (PLC-γ) signaling, which in turn activates the NF-κB and MAP kinase pathways, triggering the expression of CD40, CD86, and CD69 on B cells that promote B cell activation and proliferation [9,10,11,12]. BTK remarkably activates the BCR signaling pathway leading to differentiation of B cells into self-reactive B cells as seen in autoimmune diseases [13, 14]. BTK also activates innate immune cells, including macrophages, mast cells, basophils, and neutrophils [15].
While multiple nonsteroidal anti-inflammatory drugs (NSAIDs), corticosteroids, and biologics are available for the treatment of immune-mediated diseases, many patients still do not achieve disease remission with available agents. Rituximab leads to some degrees of disease control by targeting B cell-dependent pathways; that being the case, BTK inhibitors (BTKIs), which also target B-cell-related pathways might be useful as independent therapies or adjuncts to the current treatment options. In the absence of BTK, BCR signaling is insufficient to induce B cell differentiation into mature peripheral B cells, which leads to impaired proliferation of B cells, expression of activation markers, production of antibodies and cytokines, and defective immune responses against infections. BTK inhibition represents a promising therapeutic approach for the treatment of immune-mediated diseases, as it has shown remarkable efficacy in the treatment of B-cell malignancies such as chronic lymphocytic leukemia (CLL), marginal zone lymphoma (MZL), Waldenström macroglobulinemia (WM), mantle cell lymphoma (MCL), various B-cell lymphomas, and chronic graft-versus-host disease (GvHD) [16].
In comparison with chemo-immunotherapy, BTKIs increased the treatment efficacy of B cell malignancies in patients with high-risk features and showed a better tolerability in frail older patients [3]. As the BTKIs target the ATP-binding site, they are classified into three categories, namely covalent irreversible inhibitors, covalent reversible inhibitors, and non-covalent reversible inhibitors. The first covalent irreversible BTKI for the treatment of B-cell tumors, ibrutinib that binds covalently to the cysteine-481 binding site of BTK, was approved by the Food and Drug Administration (FDA) in 2013 and has brought a promising idea for the treatment of immune-mediated diseases. These reversible covalent inhibitors dissociate from common thiols while maintaining sustained inhibition of a protein with a conserved cysteine, providing selectivity [17]. Non-covalent BTKi inhibits BTK by different mechanisms to covalent BTKi such as blocking ATP binding site of BTK, forming the hydrogen bonds, or decreasing surface expression of B-cell activation markers, but not by binding to the C481 site on BTK [18]. Therefore, non-covalent BTKi is considered a potential alternative therapeutic option for patients who developed acquired resistance due to BTK C481 mutations.
Herein, the available evidence on the action mechanisms, efficacy, safety, and side effects of BTKIs is reviewed. Then, the recent data obtained from preclinical studies and clinical trials of BTKIs for the treatment of autoimmune diseases such as pemphigus vulgaris, pemphigus foliaceus, bullous pemphigoid, systemic lupus erythematosus, Sjögren’s disease, rheumatoid arthritis, systemic sclerosis, multiple sclerosis, myasthenia gravis, and inflammatory diseases such as psoriasis, chronic spontaneous urticaria, atopic dermatitis, and asthma are discussed. In addition, BTKIs-related complications and dermatological toxicity are reviewed.
BTK signaling
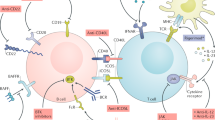
Malignant B cells such as CLL cells, MCL cells, and other stromal cells such as monocyte-derived nurse-like cells (NLC), called lymphoma-associated macrophages (LAM), and T lymphocytes reside in secondary lymphatic organs (i.e., lymph nodes, spleen, and tonsils) constituting the marrow or lymphoid tissue microenvironments [19] (Fig. 1). Chemokine receptors and adhesion molecules establish communication between cells in the microenvironment. Two pathways activate BCR signaling: (1) soluble or surface-bound antigens, (2) homotypic interactions of two BCR molecules. NLC/LAM express B cell-activating factor (BAFF), tumor necrosis factor (TNF) family members, and APRIL (also known as TNFSF13), which activate corresponding receptors on malignant B cells such as BAFF-receptor (BAFF-R), B cell maturation antigen (BCMA), and TACI (also known as TNFRSF13B) to trigger proliferation and survival signals. Activated T helper cells express CD40 ligand (CD154) on its surface to interact with CD40, leading to proliferation and growth of malignant B cells. NLCs and other stromal cells secrete chemokines, such as CXCL12 and CXCL13 and express CD31 on surface to interact with CD38 on the surface of malignant B cells. Activated CD38 engages with ZAP-70, resulting in downstream survival pathways. Cell-to-cell adhesion is established by integrins, particularly VLA-4 integrin (CD49d) on the surface of malignant B cells, and chemokine receptors. Stimulation of the BCR complex (BCR and CD79a, b) also activates SYK and ZAP-70. Stimulated NLC/LAM secret chemotactic factors such as chemokines CXCL12 and CXCL13 to attract malignant B cells such as CLL cells to the microenvironment via CXCR4 and CXCR5 receptors on the CLL cells. BTK is expressed by B cells, NLC, and LAM, contributing to the signaling of other surface receptors, such as CXCR4, CXCR5, and adhesion molecules (integrins).

The figure discusses the interaction and signaling between NLCs, LAMs, and malignant B cells [19]. NLC/LAM expresses BAFF, APRIL, and TNF family members, which activate receptors on malignant B cells such as BAFF-R, BCMA, and TACI. Additionally, activated T helper cells express CD154 to interact with CD40, promoting the proliferation of malignant B cells. NLCs and stromal cells express CD31, which interacts with CD38 on malignant B cells, activating survival pathways. Cell-to-cell adhesion involves integrins and chemokine receptors, particularly VLA-4 integrins and chemokines CXCL12 and CXCL13. BTK is expressed by B cells, NLCs, and LAMs, contributing to signaling from other receptors and molecules
BTK as a therapeutic target
BTKIs target BCR signaling cascade (Fig. 2). As a result, BTKIs disrupt the B cells’ microenvironment, which explains the redistribution of lymphocytosis interactions in well-treated CLL patients [3]. To date, studies have shown that BTKIs limit the expression and upregulation of CD69, and co-stimulatory molecules such as CD80 and CD86 through the phosphatidylinositol 3 kinase (PI3K)/ protein kinase B (PKB, also known as AKT)/ mammalian target of rapamycin (mTOR) pathway, which all are responsible for the induction of B cell activation [20]. In detail, BTKIs decrease the phosphorylation of AKT and suppresses AKT activity. Inhibition of either AKT, PI3K, or mTOR pathways reduces the expression of co-stimulatory molecules such as CD80 and CD86 [20]. BTKIs also affect the B-T cells interactions by decreasing polyclonal proliferation of both CD8 + and CD4 + T cells and cellular expression of pro-inflammatory cytokines of Th17 and Th1, interferon gamma (IFN-γ), and TNF-α. BTKIs also modulate B cell metabolic processes through reducing B cell mitochondrial respiration, selectively for activated B cells. Since mitochondrial respiration is more important for B cell’s co-stimulatory molecule expression than glycolysis, inhibition of mitochondrial respiration leads to reduced B cell activation [20]. B cells stimulate T cells in response to myelin basic protein to secrete pro-inflammatory cytokines such as IFN-γ and TNF-α [21]. Hence, decreased activation of B cells reduces the stimulation of T cells. In conclusion, BTKIs regulate B cell mitochondrial metabolism and limit the expression of pro-inflammatory cytokines in both B and T cells.

The BTK inhibitors limit the expression and upregulation of CD69 and co-stimulatory molecules CD80 and CD86 through the PI3K/AKT/mTOR pathway, which is responsible for B cell activation [20]. BTKi reduces AKT phosphorylation and activity, leading to decreased expression of CD80 and CD86 [20]. Moreover, BTKi decreases the polyclonal proliferation of CD8 + and CD4 + T cells and their production of pro-inflammatory cytokines [21]. It also modulates B cell metabolic processes by reducing mitochondrial respiration, which is important for B cell activation. These effects result in decreased B cell stimulation of T cells and limited expression of pro-inflammatory cytokines from both B cells and T cells
BTK inhibitors
In terms of mechanism of action, BTK inhibitors (BTKis) are classified into two main groups, reversible and irreversible BTKIs, based on the binding mode. Furthermore, based on the chemical interatomic linkage, BTKIs are classified into two groups, covalent and non-covalent BTKIs [22]. Covalent irreversible BTKIs bind cysteine 481 (C481) in the ATP-binding site of BTK by covalent irreversible bonds, resulting in blockage of the phosphorylation of downstream kinases in the BCR signaling pathway, thus blocking B cell activation. Covalent irreversible BTKIs consist of ibrutinib (Imbruvica), acalabrutinib (Calquence (ACP-196)), zanubrutinib (Brukinsa), evobrutinib, remibrutinib (LOU064), elsubrutinib, tolebrutinib (SAR442168), orelabrutinib, branebrutinib (BMS-986195), poseltinib, tirabrutinib hydrochloride (Velexbru) (GS-4059), spebrutinib (CC-292), SHR1459, TAS5315, AC0058TA, and BI-BTK-1. Ibrutinib is the first-generation of BTKIs and is approved to treat several B cell malignancies such as chronic lymphocytic leukemia (CLL), small lymphocytic lymphoma (SLL), Waldenström macroglobulinemia (WM), chronic graft versus host disease (GvHD) (after the failure of ≥ 1 lines of systemic therapy), mantle cell lymphoma (MCL) (after ≥ 1 prior therapy), and marginal zone lymphoma (MZL) (requiring systemic therapy and having received at least 1 prior anti-CD20-based therapy) [23]. Although the efficacy of ibrutinib is satisfying against the mentioned malignancies in clinics, off-target toxicities and drug resistance are reported. Thus, second-generation of BTKIs are developed such as acalabrutinib and zanubrutinib [24,25,26]. Non-covalent reversible inhibitors do not bind to the C481 site of BTK, but share the cysteine 481 binding site and are useful in patients with B cell malignancies resistant to prior therapy with covalent BTKIs. Despite being less advanced compared to irreversible BTKIs, reversible inhibitors have shown to be more effective for the treatment of autoimmune diseases, including rheumatoid arthritis, multiple sclerosis, systemic lupus erythematosus, and graft versus host disease (GvHD). Non-covalent reversible inhibitors consist of fenebrutinib (GDC-0853) and nemtabrutinib. Covalent reversible inhibitors consist of rilzabrutinib (PRN1008), BMS-986142, BIIB091. PRN473 (SAR444727) is both non-covalent and covalent reversible BTKI [6]. BTK inhibitors and their classifications are shown in Table 1.
BTKI in autoimmune diseases
Autoimmune blistering disorders
Pemphigus is an autoimmune disease characterized by painful blisters and erosions. In pemphigus, the immune system mistakenly attacks cells in the epidermis and the mucous membranes by immunoglobulin type G (IgG) autoantibodies against desmogleins, the adhesion proteins that bind keratinocytes to one another. When the bonds are disrupted, fluid collects between the epidermis layers, forming blisters. Pemphigus can be classified into two primary subtypes: pemphigus vulgaris (PV), in which blisters form in the mouth and other mucosal surfaces in addition to the skin and causes agonizing oral erosions, and pemphigus foliaceus (PF), which only affects the skin [27]. In pemphigus, activated T cells initiate an autoimmune cascade, which induces activated B lymphocytes to synthesize anti-desmoglein antibodies [28]. In pemphigus, activated neutrophils, eosinophils, and mast cells of the innate immune system accumulate in the lesions’ infiltrates [27]. Therefore, treatments have to target both adaptive and innate immune pathways. Although systemic corticosteroids are the mainstay of treatment (moderate to high doses of oral prednisone or prednisolone, or intravenous methylprednisolone), long time corticosteroid therapy may result in serious side effects such as gastritis, hypertension, diabetes mellitus, and osteoporosis. In bullous pemphigoid, IgG +/- IgE antibodies and activated T lymphocytes attack the basement membrane of the epidermis. The target is the protein BP180 (also known as type XVII collagen), or less frequently, BP230, a plakin. BP180 and BP230 are associated with the hemidesmosomes, structures that bind the epidermal keratinocytes to the dermis. Binding of the autoantibodies to proteins releases cytokines from T cells, leading to complement activation, recruitment of neutrophils, and release of proteolytic enzymes. Proteolytic enzymes destroy the hemidesmosomes and trigger the formation of subepidermal tense blisters. Most patients with bullous pemphigoid receive steroids, either prednisone or prednisolone. The dose is adjusted until the blisters and inflammatory lesions stop appearing, which usually takes several weeks. As mentioned earlier, systemic steroids have many undesirable side effects. Rilzabrutinib (PRN1008), a covalent reversible BTKI, combined with low doses of corticosteroid or as monotherapy is safe and efficient based on the clinical response in patients with pemphigus vulgaris [29]. A phase II trial of 27 patients with PV and PF showed promising results for using rilzabrutinib. More than half of the patients achieved disease control within 4 weeks without administration of prednisolone [30]. Furthermore, rilzabrutinib is granted Orphan Drug Designation by the United States Food and Drug Administration (FDA) for the treatment of PV (ANZCTR No. ACTRN12614000359639) [31]. Frequent mild gastrointestinal side effects were observed in rilzabrutinib therapy [31]. A phase III trial evaluated the efficacy and safety of oral rilzabrutinib in moderate to severe PV or pemphigus foliaceus (NCT03762265). It was reported that the proportion of patients meeting the primary endpoint on rilzabrutinib was not significantly different from placebo (NCT03762265) [32]. Rilzabrutinib in combination with corticosteroid was evaluated in another phase III clinical trial as a promising for its self-limited immunomodulatory effects for the treatment of newly diagnosed or relapsing PV; disease control was observed early and improved with continued treatment, and a favorable benefit-risk profile was achieved (NCT02704429) [33]. Regarding human and animal studies, rilzabrutinib has shown promising therapeutic results in humans with pemphigus, while PRN437 is more effective than rilzabrutinib in animal models with pemphigus [34]. Further case reports showed that ibrutinib, a covalent irreversible BTKI, could be administrated for the treatment of chronic lymphocytic leukemia (CLL) and acquired paraneoplastic pemphigus (PNP) [35, 36]. Tirabrutinib hydrochloride (Velexbru) (GS-4059) is a covalent irreversible BTKI, which reduces IgG production and impairs IgG autoantibody-mediated signaling pathway involved in the pathogenesis of pemphigus, thus could be an alternative therapy for resistant pemphigus [37]. Tirabrutinib is approved in Japan for the treatment of plasma cell lymphoma, Waldenström macroglobulinemia (WM), and primary lymphoma of the central nervous system. To evaluate the safety and efficacy of tirabrutinib, sixteen patients with refractory pemphigus were included in a phase II trial (JapicCTI-184231) [38]. Treatment with tirabrutinib caused remission in patients with refractory pemphigus and led to reduced oral corticosteroid exposure [38]. In in vivo studies, oral PRN473 that is both non-covalent and covalent reversible BTKi, is efficacious and well-tolerated in the treatment of canine pemphigus foliaceus (PF) [34, 39, 40]. Results of in vitro studies demonstrate that PRN473 is highly selective and has prolonged effect on BTK with minimal systemic effects [41]. The administered BTKIs in pemphigus are shown in Table 2.
Systemic lupus erythematosus
Systemic lupus erythematosus (SLE) is a multi-organ multi-factorial disease characterized by the autoreactive T and B cells and production of autoantibodies against self-antigens such as nucleic acids, DNA in both the double-stranded (Anti-dsDNA) and the single-stranded (Anti-ssDNA) conformations, RNA nuclear antigens such as the Ro/SAA, ribonucleoprotein, and non-nuclear components, and phospholipids. Indeed, autoantibodies progressively accumulate in tissues years before the clinical onset of SLE and form antigen-antibody complex deposits, causing inflammation and tissue injury [42, 43]. SLE flare is accompanied by an increase in autoantibodies (primarily anti-dsDNA) [44]. Therefore, B cell–targeting therapies can lead to B cell depletion, which is accompanied by a reduction in autoantibodies. Although corticosteroids and B cell–targeting therapies (monoclonal antibodies against CD20, CD19, and CD22) are essential components in SLE treatment, therapeutic outcomes are associated with severe side effects [45, 46]. Medications that inhibit more than one pathway in SLE pathogenesis would help to reach higher therapeutic efficacy. As mentioned earlier, BTK in B cells plays a key role in B cell activation and its differentiation; thus, targeting and depleting B cells via BTKI can be a viable alternative therapeutic modality. A study in the lupus nephritic mouse model showed that BTK inhibition dampened humoral autoimmunity [47]. Study on ibrutinib in lupus-prone B6.Sle1 or B6.Sle1.Sle3 mice revealed that humoral and cellular autoimmunity reduced; some autoantibodies, including antinucleosome antibodies and antihistone antibodies, but not antidsDNA antibodies, reduced and led to improvement of lupus nephritis [48].
BI-BTK-1, a highly selective irreversible BTKI, is used to target both myeloid cell (particularly macrophage) and B cell activation and function in the MRL-lpr/lpr murine model of SLE. It is reported that lupus-associated cutaneous and neuropsychiatric disease decreased and cognitive function improved following reduced accumulation of macrophages, T cells, and B cells within the central nervous system, particularly the choroid plexus. Finally, skin lesions improved macroscopically and histologically in the mice model [49]. In a phase II trial of SLE, the efficacy and safety of fenebrutinib (GDC-0853), a non-covalent reversible BTKI, was assessed in 260 patients with moderate to severe SLE. Although levels of phosphorylated BTK, CD19 + B cells, autoantibodies (mainly antidsDNA antibodies) decreased and the BTK pathway was inhibited, fenebrutinib did not achieve a treatment benefit over the placebo group (NCT02908100) [50]. In another phase Ib/IIa trial, safety, tolerability, and preliminary efficacy of orelabrutinib (ICP-022) were evaluated. Orelabrutinib, a covalent irreversible BTKI, has shown to reduce levels of anti-dsDNA and IgG, total B cells, and increase C4 in patients with SLE. Orelabrutinib was generally safe and well tolerated in patients with mild to moderate SLE [51]. Zanubrutinib, a covalent irreversible BTKI, is in an ongoing phase II study to evaluate its efficacy in patients with active proliferative lupus nephritis (NCT04643470). Evobrutinib, a covalent irreversible BTKI, was evaluated in a phase II study for the efficacy and safety in patients with active autoantibody-positive SLE. It was reported that evobrutinib was not an effective therapeutic intervention for patients with SLE, but it was well tolerated at all doses, with no dose effect observed for treatment-emergent adverse event (NCT02975336) [52].
A phase II trial evaluated the safety and efficacy of elsubrutinib, a covalent irreversible BTKI, alone or in combination with upadacitinib (ABT-494), a JAK1 selective inhibitor, in patients with moderately to severely active SLE (NCT03978520). ABBV-599HD (Elsubrutinib 60 mg + upadacitinib 30 mg) resulted in significant improvements in SLE disease activity and reduced overall flares and time to first flares with acceptable safety through 48 weeks (NCT03978520) [53]. In another phase II trial, the safety of ABBV-599HD is being evaluated for adult patients with moderately to severely active SLE to assess change in disease state (NCT04451772).
Branebrutinib (BMS-986195), a covalent irreversible BTKI, was evaluated in a phase II trial for its safety and effectiveness in patients with active SLE; however, the data have not been published (NCT04186871). AC0058TA, a covalent irreversible BTKI, was evaluated for the safety, tolerability, pharmacokinetics, and pharmacodynamics in adult SLE patients with positive ANA levels in a phase Ib trial; however, no study results were posted or published about the clinical trial (NCT03878303). The administered BTKIs in SLE are shown in Table 3.
Sjögren’s syndrome
Sjögren’s syndrome (SS) is a chronic inflammatory disease manifesting with dryness of the eyes (xerophthalmia), mouth (xerostomia), skin, mucosal surfaces, and extra-glandular involvement including arthritis, renal complications, vasculitis (mainly cryoglobulinemic vasculitis), and extranodal lymphoproliferation (causing lymphocytic interstitial pneumonitis) [54, 55]. The extraglandular manifestations of Sjögren’s syndrome are mainly associated with increase in auto-reactive B-cell stimulation, B-cell hyperactivity, increased levels of circulating immunoglobulins (autoantibodies), and alterations in B-cell subpopulations [56]. B cell–targeting therapies such as monoclonal antibodies against CD20 (such as rituximab) did not reach remarkable results in patients with Sjögren’s syndrome in two clinical trials [57, 58]. Remibrutinib (LOU064), a covalent irreversible BTKI, provides an alternative therapy for diseases driven by B cells, mast cells, and basophils such as Sjögren’s syndrome and has been assessed for its safety and tolerability in a phase I trial [59]. To assess basophil suppression by remibrutinib, CD203c inhibition was applied twice daily showing positive outcomes [60]. Remibrutinib (LOU064) has fewer side effects, higher specificity and potency of blocking activity than its ancestor molecules [61]. Remibrutinib is well-tolerated at all doses without any dose-limiting toxicity and demonstrates a safe profile and strong BTK inhibition in blood and skin pharmacodynamics in healthy human subjects and in healthy subjects with asymptomatic atopic diathesis [59]. Remibrutinib was evaluated in a phase II trial for its efficacy in patients with Sjögren’s syndrome, the trial has been terminated, but the data have not been published yet (NCT04035668). Tirabrutinib has been evaluated for its efficacy and safety in patients with moderate to severe active Sjögren’s syndrome, either primary or associated with a concomitant systemic autoimmune disease, through a phase II study (NCT03100942) [62]. Tirabrutinib demonstrated no significant differences versus placebo in primary and secondary endpoints [62]. Branebrutinib (BMS-986195) is well-tolerated and safe enough to be administrated in healthy humans in a phase I study [63]. Branebrutinib is rapidly absorbed with 100% occupancy of BTK after a single dose and inactivates BTK rapidly (NCT02705989) [63]. Branebrutinib was evaluated in a phase II trial to assess its safety and effectiveness in patients with primary active Sjögren’s syndrome; however, the data have not been published yet (NCT04186871). The administered BTKIs in Sjögren’s syndrome are shown in Table 4.
Rheumatoid arthritis
Rheumatoid Arthritis (RA) is an autoimmune and inflammatory disease, in which the immune system attacks healthy cells causing inflammation (painful swelling) in the affected tissues [64]. In detail, RA involves dysregulated T and B lymphocyte proliferation, particularly B cells, via BCR signaling leading to the production of autoantibodies and inflammatory cytokines [65]. As mentioned earlier, BTK is expressed in myeloid cells, including neutrophils, mast cells, monocytes, and macrophages infiltrating into synovium in RA [66, 67]. Immune complexes containing IgG are present in the joints, affecting synovial macrophages to produce cytokines and matrix metalloproteinases (MMPs) that contribute to RA pathophysiology [65]. In addition, BTK mediates bone resorption by stimulating osteoclast proliferation and differentiation as a contributor to RA [65, 68]. Due to the significant role of BTK in the pathogenic pathways of RA, BTKIs could be promising options. Ibrutinib demonstrated good efficacy with antiarthritic activity in arthritic DBA/1 mice models [69]. By longitudinal integrative whole-exome, whole-transcriptome sequencing and targeted sequencing, it has been reported that the Long-term need for ibrutinib to treat chronic RA leads to development of acquired resistance in patients, particularly developing a C481S mutation, which promotes BTK activation [70]. Thus, ibrutinib can cause a potential problem for all BTKIs, reversible or irreversible, which target Cys-481 in patients with mantle cell lymphoma with acquired resistance to ibrutinib [70]. Spebrutinib (CC-292), a covalent irreversible BTKI, has demonstrated sustained BTK occupancy, with low, even undetectable plasma levels of the drug in healthy humans in a preliminary phase I trial, and occupied all circulating BTK protein [11]. However, spebrutinib did not achieve significant clinical efficacy in a phase IIa trial in active RA patients on background methotrexate therapy (NCT01975610) [71]. Despite the lack of clinical efficacy in the trial, patients treated with spebrutinib showed a statistical reduction in chemokines CXCL13 and MIP-1β (implicated in B cell trafficking) and serum CTX-I (a measure of osteoclastic activity) compared to placebo [71]. Acalabrutinib (ACP-196), second-generation covalent irreversible BTKI, was also assessed in a phase IIa trial in 31 active RA patients on background methotrexate but did not show a meaningful clinical response after 4 weeks of treatment (NCT02387762). Fenebrutinib (GDC-0853) has demonstrated proper efficacy in a phase II trial in older/unfit patients and those with high-risk and/or relapsed CLL [25]. The primary outcome of higher doses of fenebrutinib (150–200 mg twice daily) was more than 50% clinical improvement (ACR50) compared to placebo, at 12 weeks of treatment according to the American College of Rheumatology criteria [25]. Poseltinib (LY3337641/HM71224), a novel BTKI acting on B cell activation and osteoclast formation, is evaluated through in vitro studies. Poseltinib blocks phosphorylation of BTK, ERK, and PLCγ2 resulting in suppression of osteoclast formation and inhibition of the upregulation of activation markers such as CD40, CD86, and CD69 on stimulated B cells [72]. To evaluate the efficacy and safety of poseltinib, a phase II trial was designed which was terminated after interim data did not demonstrate significant efficacy with no difference between doses of poseltinib and placebo at week 12 in moderate-to-severe RA patients (NCT02628028) [73]. BMS-986142, a covalent reversible BTKI, was evaluated for safety and efficacy in patients with moderate to severe RA with an inadequate response to methotrexate alone or methotrexate with up to 2 TNF Inhibitors, but the data have not been published (NCT02638948). Evobrutinib, a covalent irreversible BTKI, was assessed to determine efficacy, dose-response, and safety in active moderate to severe RA with previous methotrexate treatment, and it was well tolerated across indications at all doses (NCT03233230) [74]. Tirabrutinib (GS-4059) is evaluated for its safety profile, tolerability, and effect on disease-specific clinical markers and outcomes in patients with RA in a phase I trial [75]. Patients who received tirabrutinib 20 mg daily for four weeks achieved ACR20 in 38% of patient group compared to 20% for the placebo group, up to one month after treatment [75]. The safety and efficacy of elsubrutinib were evaluated on a background of upadacitinib, conventional synthetic disease-modifying anti-rheumatic drugs (csDMARDs), in a phase II trial to define the optimal dose for further development in patients with rheumatoid arthritis and inadequate response or intolerance to biological disease-modifying antirheumatic drugs; and it was reported that significant improvements in disease activity metrics of RA was achieved (NCT03682705) [76]. Branebrutinib (BMS-986195) was investigated in a phase I trial to evaluate the effects in healthy male subjects and in patients with moderate to severe RA; however, the trial was terminated without reporting the results (NCT03245515). Another phase I trial was conducted to evaluate the effects in healthy male subjects and in patients with moderate to severe RA and the trial has been completed, but the data have not been published (NCT02638948). Another phase I trial was conducted to assess the effect of branebrutinib on the pharmacokinetics of methotrexate, caffeine, montelukast, flurbiprofen, omeprazole, midazolam, digoxin, and pravastatin; the trial has been completed, but the data have not been published (NCT03131973). TAS5315, an irreversible covalent BTKI, was evaluated in a phase II trial to assess the efficacy and safety of TAS5315 in combination with methotrexate in 12 weeks or 36 weeks in patients with moderate to severe RA with inadequate response to maximally tolerated methotrexate dose, and it was reported that some bleeding risks occured, and nevertheless demonstrated numerical differences, compared with placebo, in the improvement rates of all measures of RA disease activity (NCT03605251) [77]. The administered BTKIs in RA are shown in Table 5.
Systemic sclerosis
Systemic sclerosis (SSc) is an autoimmune disorder, in which the immune system attacks the connective tissue of the skin, internal organs, and blood vessels resulting in fibrosis formation [78]. Pulmonary and cardiac fibrosis and particularly pulmonary hypertension are severe fatal complications [79, 80]. Accumulating evidence suggests that impaired function of regulatory and effector B cells leads to immune dysregulation, hyperreactivity, and chronic activation of effector B cells, which in turn, increases the production of autoantibodies, vasculopathy, and chronic activation of fibroblasts [81, 82]. Moreover, B cell-derived profibrotic IL-6 and TNF-α in response to TLR9 stimulation contribute to the pathogenesis of SSc [83]. Tocilizumab (Actemra), an IL-6-receptor-α inhibitor, failed to reduce skin thickening but caused modification of Rodnan Skin Score and improved pulmonary function in a phase III study [84]. Immunomodulators addressing B cells such as rituximab and tocilizumab in patients with SSc showed mixed efficacy with complete B cell depletion in a case-control study [84, 85]. Ibrutinib was assessed in 24 patients with SSc and showed suppressed production of the profibrotic cytokines IL-6 and TNF-α of effector B cells and also less activated phosphorylated NF-κB in an in vitro model of SSc sample [81]. In addition, autologous stem cell transplantation is an available treatment but only for selective patients with severe disease and high risk of major organ failure. The administered BTKIs in SS are shown in Table 6.
Multiple sclerosis
Multiple sclerosis (MS) is an autoimmune disease, in which the immune system attacks the myelin sheath of neurons, resulting in slowed and disrupted nerves’ conduction. In the cerebrospinal fluid of MS patients, a significant increase in the expression of B cell co-stimulatory molecules such as CD80 and CD86 is observed [21]. B cells stimulate T cells in response to myelin basic protein to secrete pro-inflammatory cytokines such as IFN-γ and TNF-α [20, 21]. BTK is expressed in microglia (myeloid cells) and B cells of the central nervous system. As mentioned earlier, BTKI reduces B cells’ mitochondrial respiration; thus treatment with BTKI can be considered as therapeutic agents in patients with MS [20]. Tolebrutinib (SAR442168) that crosses the blood-brain barrier is a covalent irreversible BTKI, which was assessed in a phase IIb trial for its efficacy and safety in relapsing-remitting MS or relapsing secondary progressive MS [86]. Reduction in the number of gadolinium-enhancing lesions was reported after 12 weeks of treatment (NCT03889639) [86]. In another phase II trial, tolebrutinib at a 60 mg daily dose for 48 weeks (nearly one year) was evaluated to see if it could help clear chronically inflamed brain’s white matter lesions in MS (NCT04742400) [87]. None of the paramagnetic rim lesions (PRLs) had disappeared despite nearly a year of treatment, suggesting that tolebrutinib had no effect on smoldering inflammation [87]. Since the patients needed to transition to the higher dose of 120 mg daily for the next 48 weeks and the higher dose of tolebrutinib might lead to liver damage, the study had been hold [87]. In a phase III trials, tolebrutinib is currently being evaluated in delaying disability progression in nonrelapsing in primary progressive MS (PPMS) to determine the efficacy, safety, tolerability, pharmacokinetics, pharmacodynamics, and the efficacy on clinical endpoints, magnetic resonance imaging (MRI) lesions, cognitive performance, physical function, and patient’s quality of life (NCT04458051). Moreover, in another phase III trials, tolebrutinib was evaluated in delaying disability progression in nonrelapsing in secondary progressive MS (NRSPMS) (NCT04411641). However, the trial was placed on hold because of reported cases of drug-induced liver injury in patients, potentially caused by a preexisting factors related to hepatic dysfunction (NCT04411641). In another phase III trial in patients with relapsing MS, efficacy, safety, tolerability, and pharmacodynamics of daily tolebrutinib is being assessed compared to teriflunomide (Aubagio) on disability progression, MRI lesions, cognitive performance, and quality of life (NCT04410991). Evobrutinib that affects B cell activation both in vitro and in vivo, was assessed in a phase II trial in patients with relapsing MS. Patients who received 75 mg of evobrutinib once daily had fewer lesions than those receiving placebo after 12 weeks of treatment. However, patients who received 25 mg once daily or 75 mg twice daily did not show any significant difference versus placebo. Longer and larger trials are necessary to assess the efficacy of evobrutinib [88]. A phase III trial evaluated the efficacy and safety of evobrutinib administered orally twice daily versus teriflunomide (Aubagio) administered orally once daily in patients with relapsing MS (NCT04338022). However, results of the trial revealed that evobrutinib did not lead to a more superior reduction in annualized relapse rates than teriflunomide (NCT04338022). Orelabrutinib in a phase II trial was evaluated to detect the number of new brain lesions with active inflammation after 12 weeks and also its efficacy, safety, and relapse rates after 120 weeks (NCT04711148) and led to significant reductions in new active brain lesions among patients with relapsing-remitting MS (RRMS) (NCT04711148). BIIB091, a novel selective covalent reversible small-molecule BTKI, has been evaluated in vivo and in a phase I trial so far. In in vivo studies, BIIB091 inhibited B cell activation and autoantibodies production [89]. In a phase I trial, BIIB091 inhibited naïve and memory B cell activation with a minor impact on myeloid or lymphoid cell survival after 14 days of dosing in healthy volunteers [89]. Fenebrutinib is currently in an ongoing phase III clinical trial for evaluation of its efficacy and safety on disability progression and relapse rate in adult participants with PRMS (NCT04586023). The administered BTKIs in MS are shown in Table 6.
Myasthenia gravis
Myasthenia gravis (MG) is a chronic autoimmune neuromuscular disease that causes weakness in the skeletal muscles such as arms, legs, and breathing muscles worsening after periods of activity and improving after periods of rest. In MG, antibodies against acetylcholine receptors (AChR), muscle-specific kinase (MuSK), and lipoprotein receptor related protein 4 (LRP4) block and destroy the receptors at the neuromuscular junction [90, 91]. Circulating antibodies against AChR are detected in blood samples of most MG patients [92]. Interaction between activated T and B cells leads to the production of IgG-type antibodies [93]. BTKI may be a promising therapeutic option for the treatment of MG; however, there has been only one clinical trial to assess the effect of BTKIs on MG patients. Tolebrutinib was assessed in phase III trial to evaluate its efficacy and safety in adult patients with moderate-to-severe generalized MG (NCT05132569). The trial was terminated due to the drug-induced liver injury in patients, potentially caused by a preexisting factors related to hepatic dysfunction (NCT05132569). The administered BTKIs in MG are shown in Table 6.
BTKI in inflammatory diseases
Psoriasis
Psoriasis is characterized by raised plaques and scales on the skin caused by dysfunction of the immune system [94]. Indeed, an overactive immune system speeds up skin cell growth, thus skin epithelial keratinocytes pile up forming pink or red patches, and white or silvery scales [95]. Oxidative stress is known as an important contributor to the pathogenesis of psoriasis. Neutrophils secrete oxidants through the BTK pathway that maintains inflammation in psoriasis [96, 97]. In addition, BTK executes signaling functions in dendritic cells and γδ + T cells. In detail, activation of the BTK pathway upregulates inflammatory cytokines such as IL-23/TNF-α in dermal CD11c dendritic cells and IL-17 A in γδ + T cells. Ibrutinib was evaluated in dermal psoriasis-like inflammation of the imiquimod-induced (IMQ) mouse model [98]. Preventive treatment with ibrutinib in the IMQ mouse model led to the reduction in IL-23/TNF-α levels of CD11c dendritic cells and IL-17 A levels of γδ + T cells; thus, ibrutinib reduces oxidative stress in these innate immune cells, which makes a promising therapeutic option for psoriasis [37]. The administered BTKIs in psoriasis are shown in Table 7.
Chronic spontaneous urticaria
Chronic spontaneous urticaria (CSU), also known as chronic idiopathic urticaria, is the presence of urticaria (hives) on most days of the week, for a duration of six weeks or longer. Mechanistically, CSU happens because of infiltration of mainly T helper 2 cells (Th2) around small venules of the skin [99]. BTK is required in the activation of mast cells via FcεRI and producing autoantibodies by B cells. Thus, BTKI might be effective in CSU. Fenebrutinib (GDC-0853) was assessed in a phase II trial in adult patients with CSU for more than six months and symptomatic despite treatment with H1 antihistamines (up to fourfold the approved dose); IgG-anti-FcεRI autoantibodies significantly decreased at week 8 at all dose levels compared to placebo, which demonstrated good efficacy in patients with CSU, but the long-term extension of the trial was also terminated due to the transient transaminase elevations in a limited number of patients and safety issues (NCT03137069) [100]. Notably, fenebrutinib did not result in remarkable reductions in IgG subtypes such as IgG1 and IgG3 [101]. Remibrutinib (LOU064) provides an alternative therapy for diseases driven by B cells, mast cells, and basophils such as CSU. Remibrutinib was evaluated for its clinical safety and pharmacodynamics in CSU with asymptomatic atopic diathesis in a phase I clinical trial (NCT03918980) [59]. Remibrutinib was well-tolerated at all doses without any dose‐limiting toxicity with a favorable safety profile and near complete basophil or skin prick test (SPT) inhibition was achieved at greater than or equal to 50 mg q.d. for CD63 and at greater than or equal to 100 mg q.d [59]. An ongoing phase III trial is designed to evaluate the efficacy and safety of remibrutinib in the treatment of CSU in adults which was inadequately controlled by H1 antihistamines (NCT05030311). Rilzabrutinib (SAR444671) is currently being assessed in an ongoing phase II trial for the safety and effectiveness of 3 oral doses, compared with placebo for decreasing the frequency and severity of pruritus and urticaria in patients with CSU (NCT05107115). The administered BTKIs in CSU are shown in Table 7.
Atopic dermatitis
Atopic dermatitis (AD), the most common form of eczema, is a chronic inflammatory disorder causing dry, itchy, inflamed, and cracked skin [102]. AD is usually a chronic condition and common in young children but also occurs at any age [103, 104]. PRN437 (SAR444727), both non-covalent and covalent reversible topically administered BTKI, inhibits three pathways including the activation of monocyte and neutrophil migration mediated by IgG (FcgR), the activation of mast cell and basophil mediated by IgE (FceR), and the activation of the β2-integrin c-1 and subsequently neutrophil recruitment into inflamed tissue [105, 106]. PRN473 (SAR444727) was evaluated for the safety, tolerability, and efficacy in phase IIa in 40 patients with mild to moderate AD; the trial has been completed, but the data have not been published (NCT04992546). It is reported in a study that ibrutinib (PCI-32765) therapy suppresses IgE-mediated basophil activation and reduces mast cell and basophil reactivity to the allergens in adults suffering from allergy to peanut or tree nut (NCT03149315) [107]; therefore, ibrutinib eliminates aeroallergen skin test [107, 108]. Branebrutinib (BMS-986166) was evaluated in phase II trial to evaluate the efficacy, safety, and tolerability for the treatment of patients with moderate to severe AD; the trial has been completed, but the data have not been published (NCT05014438). The administered BTKIs in AD are shown in Table 7.
Asthma
Asthma is associated with chronic inflammation, airway hyper-responsiveness, and reversible airflow obstruction. Accumulation of inflammatory mediators, cytokines, chemokines, infiltrating immune cells in airways leading to remodeling of the airways, including subepithelial fibrosis, myofibroblast hyperplasia, goblet cell hyperplasia, wall thickening, smooth muscle cell hyperplasia and hypertrophy, epithelial hypertrophy, and airway wall thickening [109,110,111]. B cells produce antibodies such as IgE [112]. IgE binds to its receptor (FcqRII or CD23) and induces CD23-mediated eosinophilic infiltration causing airway hyper-responsiveness of asthma [113]. Stimulation of tyrosine kinases such as SYK, ZAP-70, BTK, and ITK on B cells is the earliest signaling response in inflammatory cells; thus, BTKI can be a therapeutic option for asthma. Rilzabrutinib is being assessed in an ongoing phase II trial to evaluate its efficacy, safety, and tolerability in patients with moderate-to-severe asthma (NCT05104892). As mentioned earlier, BTK deficiency is characterized by decreased B cell level and serum immunoglobulin level. It is expected that patients with BTK deficiency be protected from atopy such as allergic rhinitis, asthma, and atopic dermatitis (eczema). Surprisingly, in a case report, a 7-year-old boy with agammaglobulinemia presented with allergic rhinitis, severe papular urticaria, asthma symptoms, and a positive skin prick test to aeroallergens and food allergens. He had a mutation in the BTK gene revealed by genetic analysis [114]. The administered BTKIs in asthma are shown in Table 7.
BTKIs adverse events
Both on-target and variable off-target activities of BTKI on the cellular process are suggested to be linked with adverse events (AEs), since some AEs cannot be explained by BTK inhibition alone [115]. Clinically, AEs develop during long-time therapy with BTKIs because of unlimited inhibition of BTK, and consequently lead to significant rates of dosage reduction or treatment cessation. So, toxicity and AEs profile of BTKIs are related to their pattern of kinase binding [116]. The most observed AEs of BTKIs include bleeding, rash, diarrhea, and atrial fibrillation (AF).
Bleeding is assumed to be related to the effect of BTKI on BTK and TEC family proteins and their role in collagen-induced platelet aggregation, GPIb-IX, and integrin αIIbβ3 [117,118,119]. Rash and diarrhea are epidermal growth factor receptor (EGFR)-related AEs in BTKI-treated patients. AF is attributed to the BTKIs effect on C-terminal Src kinase (CSK) [120]. Another suggested mechanism of BTKIs-related AF is the inhibition of PI3K signaling which is responsible for cardiac protection under stress and is regulated by BTK and TEC family proteins [121]. Clinicians are advised to monitor cardiac symptoms, such as light-headedness, syncope, and palpitations, in patients on all BTKIs [116]. All BTKi AEs are shown in Table 8.
Ibrutinib
While evaluating the efficacy and safety of ibrutinib in phase III RESONATE (PCYC-1112) in patients with relapsed/refractory (R/R) CLL with a median age of 67 years, the most common AEs were diarrhea, fatigue, nausea, pyrexia, anemia, neutropenia, thrombocytopenia, pneumonia, and AF. A subdural hematoma was reported in 1 patient in this trial [122]. In phase III studies RESONATE-2 (PCYC-1115-CA) and ILLUMINATE in patients with CLL/SLL and with a median age of 73 years, the most common AEs were cough, hypertension, and AF, in addition to the AEs mentioned earlier [123, 124]. In a case report, a 68-year-old man with CLL received ibrutinib. His initial response was lymphocytosis. After 6 months, he reported migratory arthralgias and fatigue [116]. Myalgia and arthralgia, mostly migratory arthralgias, are observed in a retrospective analysis of CLL patients treated with ibrutinib [125]. With longer-term follow-up, some cases are reported with ventricular arrhythmias and cardiac death, as ibrutinib is associated with reduced QT duration [126, 127]. Another AE is minor bleeding (low-grade ecchymoses and petechiae) emerging in up to two-thirds of patients associated with impaired platelet function and decreased platelet count rather than thrombocytopenia [128]. Major bleeding is reported less frequently in 2–9% of patients [122, 123, 129]. Bleeding among ibrutinib-treated patients can occur in the presence or absence of thrombocytopenia [130,131,132,133,134,135]. Some opportunistic infections, especially invasive fungal infections, such as Pneumocystis jirovecii and Aspergillus fumigatus have emerged in patients with CLL on BTKIs, particularly on ibrutinib [136,137,138,139]. Aspergillus fumigatus induces BTK phosphorylation in macrophages, and impairs nuclear factor of activated T-cells (NFAT) and nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) responses [140], ITK kinase [141], and M1 polarization in macrophages [142]. These mechanisms increase the susceptibility of ibrutinib-treated patients to fungal infections. There are a few reports of Pneumocystis jirovecii in XLA patients, as the ibrutinib-sensitive TEC kinase is a substitute for BTK in non-B-cells [143]. Thus, both inhibition of BTK and TEC may predispose XLA patients to fungal infection. Ibrutinib inhibits the platelet adhesion to lymphatic endothelial cells through phospho-SRC/spleen tyrosine kinase (SYK) and C-type lectin-like receptor 2 (CLEC-2) that is another proposed mechanism for increased rates of invasive fungal infections [144]. In phase Ib/II PCYC-1102 and extension study PCYC-1103 with up to 8 years of follow-up, the most sustained AE was hypertension in 28% of patients [145]. The proposed mechanism is the inhibition and downregulation of PI3K-p110α and nitrous oxide production [121, 146]. From 2009 to 2016, hypertension rates were studied in 562 patients treated with ibrutinib for malignancies, amongst which 440 (78.3%) patients developed new or worsened high blood pressure over a median follow-up of 30 months. The effect of new-onset or worsened hypertension on major cardiovascular events was assessed; hypertension was accompanied by arrhythmia, myocardial infarction, heart failure, stroke, and cardiovascular death [146]. Non-palpable asymptomatic petechial rash (which is associated with ibrutinib-induced platelet dysfunction), pruritic palpable rash (which is associated with EGFR inhibition and infiltration of the inflammatory cells) [147, 148], erythema nodosum, brittle fingernails or toenails, and formation of vertical nail ridges are observed in two-thirds of patients on ibrutinib [149]. Conversely, another mechanism underlying rash is the ibrutinib-induced increase of EGFR expression in dermal fibroblasts in the HDF3CGF system [150,151,152]. Unfortunately, 60% of patients on ibrutinib in the long-term had acquired resistance to covalent inhibitors, caused by cysteine C481 to serine substitution in BTK [153,154,155]. Two possible explanation for BTKI’s contribution to the occurrence of AF is discussed earlier; another possible explanation for ibrutinib-induced AF is the simultaneous binding to HER2 and HER4, whereas acalabrutinib inhibits HER4 and TEC, but not HER2; zanubrutinib inhibits TEC and HER4, but not HER2; tirabrutinib inhibits TEC but neither HER4 nor HER2. Ibrutinib’s simultaneous targeting of HER2 and HER4 is suggested to be responsible for AF [156].
Acalabrutinib
Efficacy and safety of acalabrutinib was evaluated in the phase III study ELEVATE-TN in patients with CLL with a median age of 70 years. The most common AEs included headache, diarrhea, fatigue, cough, upper respiratory tract infection, arthralgia, bleeding events such as contusion and petechiae, neutropenia, anemia, thrombocytopenia, urinary tract infection, pneumonia, dyspnea, back pain, AF, acute myocardial infarction, brain injury, and cardiac failure [157]. Headache is uniquely observed with acalabrutinib; nearly 70% of patients experienced headaches during weeks 1 to 3 of treatment [158]. In phase III ASCEND in patients with relapsed or refractory CLL with a median age of 67 years, increased levels of alanine aminotransferase, hepatotoxicity, and major bleeding were also reported in addition to the earlier mentioned AEs [159]. The same AEs in addition to sepsis were reported in the phase II ACE-CL-001 [158, 160]. In clinical studies, rates of discontinuation due to AEs are lower with acalabrutinib rather than ibrutinib (9–11% at 28.3-month follow-up) [157]. In in vitro study of human platelets, acalabrutinib does not inhibit TEC, suggesting a reduced number of bleeding cases [161, 162]. Acalabrutinib has a lower rate of AF in comparison with ibrutinib [158]. As mentioned earlier, diarrhea is the most common AE in patients on BTKIs, which occurs before month 6 of treatment [122, 157, 163], and the rates of diarrhea reported in patients on acalabrutinib are similar to those on ibrutinib [158, 164].
Zanubrutinib
In a phase II study, zanubrutinib was evaluated in Chinese patients with R/R CLL/SLL with a median age of 61 years; the most common AEs were neutropenia, thrombocytopenia, lung infection/pneumonia, upper respiratory infection, and anemia [165]. In the phase III SEQUOIA trial on patients with del(17p) CLL/SLL with a median age of 70 years, the most common AEs were contusion, diarrhea, nausea, constipation, rash, back pain, cough, arthralgia, fatigue, minor bleeding, bruising, dermatological malignancies, non-skin second malignancies, AF, sepsis secondary to pseudomonas, melanoma, and acute renal failure, and 4 of the patients died in this trial; two due to disease progression, one due to an adverse event after disease progression (acute kidney injury), and one after disease progression due to septic shock [166]. In another study, 0.3–2.2% of major bleedings were seen in zanubrutinib-treated patients [167]. Based on the results from clinical trials, fewer AF cases were reported in patients on zanubrutinib or acalabrutinib (mentioned earlier) than ibrutinib. Moreover, in the phase III trial ASPEN study, zanubrutinib versus ibrutinib in patients with WM was compared. Both AF and hypertension were reported in lower rates for zanubrutinib than ibrutinib with a median follow-up of 19.4 months [168]. Thus, treatment with zanubrutinib or acalabrutinib leads to fewer AF cases [156]. Among ibrutinib-treated patients, the frequency of diarrhea is reported in 32% of patients, while it is reported in 21% of patients treated with zanubrutinib, which is associated with a less potent inhibition of EGFR [166].
Tirabrutinib
In a low-patient-enrolled phase II study in treatment-naïve patients or patients with relapsed/refractory WM (27 patients in total), the most common AEs were rash, neutropenia, lymphopenia, and leukopenia. The trial was a short-term follow-up and the available dataset on AEs was limited, but the trial met the primary endpoint [169]. In clinical trials, diarrhea was reported in 7–44% of patients receiving tirabrutinib [127, 170, 171].
Fenebrutinib
In the phase II ANDES study in patients with active RA who were on fenebrutinib, the most common AEs were upper respiratory tract infections, nausea, headache, and anemia [172].
Conclusion and future directions
BTKI target BCR signaling cascades that are responsible for both normal and malignant B cells’ survival and proliferation. BTKI binds to the ATP-binding site of BTK and blocks the phosphorylation of kinases in the BCR signaling cascade and also reduces B cell mitochondrial respiration resulting in less B cell activation, less secretion of b cell-derived pro-inflammatory cytokines, and less co-activation of T cell. First, second, and third-generation, reversible, and irreversible BTKIs, based on binding mode, are all developed and evaluated in clinical trials. Clearly, ibrutinib as a first-generation BTKI and the best-studied BTKI so far has already shown remarkable efficacy in the treatment of various B cell malignancies such as high-risk CLL, MZL, WM, relapsed/refractory MCL, and chronic GvHD. Ongoing clinical trials have clarified that BTKIs, particularly highly selective second and third-generation BTKIs, can provide therapeutic options in immune-mediated diseases where B cells and T cells are responsible for the disease etiopathogenesis. Application of BTKIs is still challenging due to the diverse AEs and as it cannot guarantee adequate safety and efficacy in immune-mediated diseases. Therefore, further research on the unexplored aspects of BTKI are strongly recommended.
Data availability
Not applicable.
References
Bruton OC, Agammaglobulinemia. Pediatrics. 1952;9(6):722–8.
Vetrie D, Vorechovský I, Sideras P, Holland J, Davies A, Flinter F, et al. The gene involved in X-linked agammaglobulinaemia is a member of the src family of protein-tyrosine kinases. Nature. 1993;361(6409):226–33.
Burger JA. BTK inhibitors: present and future. Cancer J (Sudbury Mass). 2019;25(6):386.
Szilveszter KP, Németh T, Mócsai A. Tyrosine kinases in autoimmune and inflammatory skin diseases. Front Immunol. 2019;10:1862.
Rip J, De Bruijn MJ, Appelman MK, Pal Singh S, Hendriks RW, Corneth OB. Toll-like receptor signaling drives btk-mediated autoimmune disease. Front Immunol. 2019;10:95.
Robak E, Robak T. Bruton’s kinase inhibitors for the treatment of Immunological diseases: current status and perspectives. J Clin Med. 2022;11(10):2807.
Hendriks RW, Yuvaraj S, Kil LP. Targeting Bruton’s tyrosine kinase in B cell malignancies. Nat Rev Cancer. 2014;14(4):219–32.
Kil LP, De Bruijn MJ, Van Nimwegen M, Corneth OB, Van Hamburg JP, Dingjan GM, et al. Btk levels set the threshold for B-cell activation and negative selection of autoreactive B cells in mice. Blood J Am Soc Hematol. 2012;119(16):3744–56.
Park JK, Byun J-Y, Park JA, Kim Y-Y, Lee YJ, Oh JI, et al. HM71224, a novel Bruton’s tyrosine kinase inhibitor, suppresses B cell and monocyte activation and ameliorates arthritis in a mouse model: a potential drug for rheumatoid arthritis. Arthritis Res Therapy. 2016;18(1):1–9.
Liu L, Di Paolo J, Barbosa J, Rong H, Reif K, Wong H. Antiarthritis effect of a novel Bruton’s tyrosine kinase (BTK) inhibitor in rat collagen-induced arthritis and mechanism-based pharmacokinetic/pharmacodynamic modeling: relationships between inhibition of BTK phosphorylation and efficacy. J Pharmacol Exp Ther. 2011;338(1):154–63.
Evans EK, Tester R, Aslanian S, Karp R, Sheets M, Labenski MT, et al. Inhibition of Btk with CC-292 provides early pharmacodynamic assessment of activity in mice and humans. J Pharmacol Exp Ther. 2013;346(2):219–28.
Niiro H, Clark EA. Regulation of B-cell fate by antigen-receptor signals. Nat Rev Immunol. 2002;2(12):945–56.
Zarrin AA, Bao K, Lupardus P, Vucic D. Kinase inhibition in autoimmunity and inflammation. Nat Rev Drug Discovery. 2021;20(1):39–63.
McDonald C, Xanthopoulos C, Kostareli E. The role of Bruton’s tyrosine kinase in the immune system and disease. Immunology. 2021;164(4):722–36.
Zhang D, Gong H, Meng F. Recent advances in BTK inhibitors for the treatment of inflammatory and autoimmune diseases. Molecules. 2021;26(16):4907.
Buggy JJ, Elias L. Bruton tyrosine kinase (BTK) and its role in B-cell malignancy. Int Rev Immunol. 2012;31(2):119–32.
Owens TD, Brameld KA, Verner EJ, Ton T, Li X, Zhu J, et al. Discovery of Reversible Covalent Bruton’s tyrosine kinase inhibitors PRN473 and PRN1008 (Rilzabrutinib). J Med Chem. 2022;65(7):5300–16.
Lewis KL, Cheah CY, Non-Covalent BTK. Inhibitors-the New BTKids on the Block for B-Cell malignancies. J Pers Med. 2021;11(8).
Burger JA, Ghia P, Rosenwald A, Caligaris-Cappio F. The microenvironment in mature B-cell malignancies: a target for new treatment strategies. Blood J Am Soc Hematol. 2009;114(16):3367–75.
Li R, Tang H, Burns JC, Hopkins BT, Le Coz C, Zhang B, et al. BTK inhibition limits B-cell–T-cell interaction through modulation of B-cell metabolism: implications for multiple sclerosis therapy. Acta Neuropathol. 2022;143(4):505–21.
Fraussen J, Claes N, Van Wijmeersch B, van Horssen J, Stinissen P, Hupperts R, et al. B cells of multiple sclerosis patients induce autoreactive proinflammatory T cell responses. Clin Immunol. 2016;173:124–32.
Zain R, Vihinen M. Structure-function relationships of covalent and non-covalent BTK inhibitors. Front Immunol. 2021:2675.
LLC P. IMBRUVICA®(ibrutinib) Prescribing Information. Pharmacyclics, LLC Sunnyvale, CA; 2017.
Ran F, Liu Y, Wang C, Xu Z, Zhang Y, Liu Y et al. Review of the development of BTK inhibitors in overcoming the clinical limitations of ibrutinib. Eur J Med Chem. 2021:114009.
Thompson PA, Burger JA. Bruton’s tyrosine kinase inhibitors: first and second generation agents for patients with chronic lymphocytic leukemia (CLL). Expert opinion on investigational drugs. 2018;27(1):31–42.
Yosifov DY, Wolf C, Stilgenbauer S, Mertens D. From biology to therapy: the CLL success story. HemaSphere. 2019;3(2).
Naik PP. Translational autoimmunity in pemphigus and the role of novel Bruton tyrosine kinase inhibitors. J Translational Autoimmun. 2022:100156.
Schmidt E, Kasperkiewicz M, Joly. P1: CAS: 528: DC% 2BC1MXhsleltrjO. vol. 394, issue 10201. Pemphigus Lancet. 2019:882– 94.
Murrell D, Patsatsi A, Stavropoulos P, Baum S, Zeeli T, Kern J, et al. Proof of concept for the clinical effects of oral rilzabrutinib, the first Bruton tyrosine kinase inhibitor for pemphigus vulgaris: the phase II BELIEVE study. Br J Dermatol. 2021;185(4):745–55.
Altman EM. Novel therapies for Pemphigus Vulgaris. Am J Clin Dermatol. 2020;21(6):765–82.
Smith PF, Krishnarajah J, Nunn PA, Hill RJ, Karr D, Tam D, et al. A phase I trial of PRN1008, a novel reversible covalent inhibitor of Bruton’s tyrosine kinase, in healthy volunteers. Br J Clin Pharmacol. 2017;83(11):2367–76.
Sanofi provides update on Phase. 3 study evaluating rilzabrutinib for the treatment of pemphigus: Sanofi. https://www.sanofi.com/en/media-room/press-releases/2021/2021-09-09-05-00-00-2293920
Murrell DF, Patsatsi A, Stavropoulos P, Baum S, Zeeli T, Kern JS et al. Phase 2 BELIEVE study part B: efficacy and safety of rilzabrutinib for patients with pemphigus vulgaris. J Eur Acad Dermatol Venereol. 2022.
Goodale EC, Varjonen KE, Outerbridge CA, Bizikova P, Borjesson D, Murrell DF, et al. Efficacy of a Bruton’s tyrosine kinase inhibitor (PRN-473) in the treatment of canine pemphigus foliaceus. Vet Dermatol. 2020;31(4):291–e71.
Lee A, Sandhu S, Imlay-Gillespie L, Mulligan S, Shumack S. Successful use of Bruton’s kinase inhibitor, ibrutinib, to control paraneoplastic pemphigus in a patient with paraneoplastic autoimmune multiorgan syndrome and chronic lymphocytic leukaemia. Australas J Dermatol. 2017;58(4):e240–2.
Ito Y, Makita S, Maeshima AM, Hatta S, Suzuki T, Yuda S et al. Paraneoplastic pemphigus associated with B-cell chronic lymphocytic leukemia treated with ibrutinib and rituximab. Intern Med. 2018:0578–17.
Ariza Y, Murata M, Ueda Y, Yoshizawa T. Bruton’s tyrosine kinase (btk) inhibitor tirabrutinib suppresses osteoclastic bone resorption. Bone Rep. 2019;10:100201.
Yamagami J, Ujiie H, Aoyama Y, Ishii N, Tateishi C, Ishiko A, et al. A multicenter, open-label, uncontrolled, single-arm phase 2 study of tirabrutinib, an oral Bruton’s tyrosine kinase inhibitor, in pemphigus. J Dermatol Sci. 2021;103(3):135–42.
Hertl M, Jedlickova H, Karpati S, Marinovic B, Uzun S, Yayli S, et al. Pemphigus. S2 Guideline for diagnosis and treatment–guided by the European Dermatology Forum (EDF) in cooperation with the European Academy of Dermatology and Venereology (EADV). J Eur Acad Dermatol Venereol. 2015;29(3):405–14.
Yuan H, Pan M, Chen H, Mao X. Immunotherapy for Pemphigus: Present and Future. Front Med. 2022:1551.
Xing Y, Chu KA, Wadhwa J, Chen W, Zhu J, Bradshaw JM, et al. Preclinical mechanisms of topical PRN473, a Bruton Tyrosine Kinase inhibitor, in immune-mediated skin disease models. ImmunoHorizons. 2021;5(7):581–9.
Shlomchik MJ, Craft JE, Mamula MJ. From T to B and back again: positive feedback in systemic autoimmune disease. Nat Rev Immunol. 2001;1(2):147–53.
Arbuckle MR, McClain MT, Rubertone MV, Scofield RH, Dennis GJ, James JA, et al. Development of autoantibodies before the clinical onset of systemic lupus erythematosus. N Engl J Med. 2003;349(16):1526–33.
Swaak A, Groenwold J, Aarden L, Van Eps LS, Feltkamp E. Prognostic value of anti-dsDNA in SLE. Ann Rheum Dis. 1982;41(4):388–95.
Lorenzo-Vizcaya A, Fasano S, Isenberg DA. Bruton’s tyrosine kinase inhibitors: a new therapeutic target for the treatment of SLE? ImmunoTargets Therapy. 2020;9:105.
Ringheim GE, Wampole M, Oberoi K. Bruton’s tyrosine kinase (BTK) inhibitors and autoimmune diseases: making sense of BTK inhibitor specificity profiles and recent clinical trial successes and failures. Front Immunol. 2021:4494.
Chan VS-F, Tsang HH-L, Tam RC-Y, Lu L, Lau C-S. B-cell-targeted therapies in systemic lupus erythematosus. Cell Mol Immunol. 2013;10(2):133–42.
Hutcheson J, Vanarsa K, Bashmakov A, Grewal S, Sajitharan D, Chang BY, et al. Modulating proximal cell signaling by targeting Btk ameliorates humoral autoimmunity and end-organ disease in murine lupus. Arthritis Res Therapy. 2012;14(6):1–13.
Chalmers SA, Wen J, Doerner J, Stock A, Cuda CM, Makinde HM, et al. Highly selective inhibition of Bruton’s tyrosine kinase attenuates skin and brain disease in murine lupus. Arthritis Res Therapy. 2018;20(1):1–11.
Isenberg D, Furie R, Jones NS, Guibord P, Galanter J, Lee C, et al. Efficacy, Safety, and Pharmacodynamic effects of the Bruton’s tyrosine kinase inhibitor fenebrutinib (GDC-0853) in systemic Lupus Erythematosus: results of a phase II, Randomized, Double‐Blind, Placebo‐Controlled Trial. Arthritis Rheumatol. 2021;73(10):1835–46.
Li R, Zhu X, Liu S, Zhang X, Xie C, Fu Z, LB0005 ORELABRUTINIB, AN IRREVERSIBLE INHIBITOR OF BRUTON’S TYROSINE KINASE (BTK), FOR THE TREATMENT OF SYSTEMIC LUPUS ERYTHEMATOSUS (SLE).: RESULTS OF A RANDOMIZED, DOUBLE-BLIND, PLACEBO-CONTROLLED, PHASE IB/IIA DOSE-FINDING STUDY. BMJ Publishing Group Ltd; 2022.
Wallace DJ, Dörner T, Pisetsky DS, Sanchez-Guerrero J, Patel AC, Parsons-Rich D, et al. Efficacy and safety of the Bruton’s tyrosine kinase inhibitor evobrutinib in systemic Lupus Erythematosus: results of a phase II, Randomized, Double-Blind, placebo-controlled dose-ranging trial. ACR Open Rheumatol. 2023;5(1):38–48.
Efficacy and Safety of ABBV-599 High Dose. (Elsubrutinib 60 mg and Upadacitinib 30 mg) and Upadacitinib Monotherapy for the Treatment of Systemic Lupus Erythematosus: A Phase 2, Double-blind, Placebo-controlled Trial. https://acrabstracts.org/abstract/efficacy-and-safety-of-abbv-599-high-dose-elsubrutinib-60-mg-and-upadacitinib-30-mg-and-upadacitinib-monotherapy-for-the-treatment-of-systemic-lupus-erythematosus-a-phase-2-double-blind-placebo-c/
Kvarnström M, Ottosson V, Nordmark B, Wahren-Herlenius M. Incident cases of primary Sjögren’s syndrome during a 5-year period in Stockholm County: a descriptive study of the patients and their characteristics. Scand J Rheumatol. 2015;44(2):135–42.
Nocturne G, Mariette X. Advances in understanding the pathogenesis of primary Sjögren’s syndrome. Nat Rev Rheumatol. 2013;9(9):544–56.
Mavragani CP. Mechanisms and new strategies for primary Sjögren’s syndrome. Annu Rev Med. 2017;68:331–43.
Bowman SJ, Everett CC, O’Dwyer JL, Emery P, Pitzalis C, Ng WF, et al. Randomized controlled trial of rituximab and cost-effectiveness analysis in treating fatigue and oral dryness in primary Sjögren’s syndrome. Arthritis Rheumatol. 2017;69(7):1440–50.
Devauchelle-Pensec V, Mariette X, Jousse-Joulin S, Berthelot J-M, Perdriger A, Puéchal X, et al. Treatment of primary Sjögren syndrome with rituximab: a randomized trial. Ann Intern Med. 2014;160(4):233–42.
Kaul M, End P, Cabanski M, Schuhler C, Jakab A, Kistowska M, et al. Remibrutinib (LOU064): a selective potent oral BTK inhibitor with promising clinical safety and pharmacodynamics in a randomized phase I trial. Clin Transl Sci. 2021;14(5):1756–68.
MacGlashan D Jr. Expression of CD203c and CD63 in human basophils: relationship to differential regulation of piecemeal and anaphylactic degranulation processes. Clin Experimental Allergy. 2010;40(9):1365–77.
Gabizon R, London N. A fast and clean BTK inhibitor. J Med Chem. 2020;63(10):5100–1.
Price E, Bombardieri M, Kivitz A, Matzkies F, Gurtovaya O, Pechonkina A et al. Safety and efficacy of filgotinib, lanraplenib and tirabrutinib in Sjögren’s syndrome: a randomized, phase 2, double-blind, placebo-controlled study. Rheumatology. 2022.
Catlett IM, Nowak M, Kundu S, Zheng N, Liu A, He B, et al. Safety, pharmacokinetics and pharmacodynamics of branebrutinib (BMS-986195), a covalent, irreversible inhibitor of Bruton’s tyrosine kinase: randomised phase I, placebo‐controlled trial in healthy participants. Br J Clin Pharmacol. 2020;86(9):1849–59.
McInnes IB, Schett G. The pathogenesis of rheumatoid arthritis. N Engl J Med. 2011;365(23):2205–19.
Gillooly KM, Pulicicchio C, Pattoli MA, Cheng L, Skala S, Heimrich EM, et al. Bruton’s tyrosine kinase inhibitor BMS-986142 in experimental models of rheumatoid arthritis enhances efficacy of agents representing clinical standard-of-care. PLoS ONE. 2017;12(7):e0181782.
Firestein GS, Corr M. Common mechanisms in immune-mediated inflammatory disease. J Rheumatol Supplement. 2005;73:8–13.
de Weers M, Verschuren MC, Kraakman ME, Mensink RG, Schuurman RK, van Dongen JJ, et al. The Bruton’s tyrosine kinase gene is expressed throughout B cell differentiation, from early precursor B cell stages preceding immunoglobulin gene rearrangement up to mature B cell stages. Eur J Immunol. 1993;23(12):3109–14.
Arneson LC, Carroll KJ, Ruderman EM. Bruton’s tyrosine kinase inhibition for the treatment of rheumatoid arthritis. ImmunoTargets Therapy. 2021;10:333.
Honigberg LA, Smith AM, Sirisawad M, Verner E, Loury D, Chang B et al. The Bruton tyrosine kinase inhibitor PCI-32765 blocks B-cell activation and is efficacious in models of autoimmune disease and B-cell malignancy. Proceedings of the National Academy of Sciences. 2010;107(29):13075-80.
Chiron D, Di Liberto M, Martin P, Huang X, Sharman J, Blecua P, et al. Cell-cycle reprogramming for PI3K inhibition overrides a relapse-specific C481S BTK mutation revealed by Longitudinal Functional Genomics in Mantle Cell LymphomaOverriding Relapse-Specific BTK mutation in MCL. Cancer Discov. 2014;4(9):1022–35.
Schafer PH, Kivitz AJ, Ma J, Korish S, Sutherland D, Li L, et al. Spebrutinib (CC-292) affects markers of B cell activation, chemotaxis, and osteoclasts in patients with rheumatoid arthritis: results from a mechanistic study. Rheumatol Therapy. 2020;7(1):101–19.
Park J, Park J, Lee Y, Song J, Oh J, Lee Y-M, et al. THU0499 HM71224, a novel oral BTK inhibitor, inhibits human immune cell activation: new drug candidate to treat B-cell associated autoimmune diseases. Ann Rheum Dis. 2014;73(Suppl 2):355–6.
Genovese MC, Spindler A, Sagawa A, Park W, Dudek A, Kivitz A, et al. Safety and Efficacy of Poseltinib, Bruton’s tyrosine kinase inhibitor, in patients with rheumatoid arthritis: a Randomized, Double-blind, Placebo-controlled, 2-part phase II study. J Rhuematol. 2021;48(7):969.
Montalban X, Wallace D, Genovese MC, Tomic D, Parsons-Rich D, Le Bolay C, et al. Characterisation of the safety profile of evobrutinib in over 1000 patients from phase II clinical trials in multiple sclerosis, rheumatoid arthritis and systemic lupus erythematosus: an integrated safety analysis. J Neurol Neurosurg Psychiatry. 2023;94(1):1–9.
Study to evaluate safety and pharmacokinetics of GS-. 4059 (tirabrutinib) in healthy volunteers and participants with Rheumatoid Arthritis (RA). https://classic.clinicaltrials.gov/ct2/show/NCT02626026
Fleischmann R, Friedman A, Drescher E, Singhal A, Cortes-Maisonet G, Doan T, et al. Safety and efficacy of elsubrutinib or upadacitinib alone or in combination (ABBV-599) in patients with rheumatoid arthritis and inadequate response or intolerance to biological therapies: a multicentre, double-blind, randomised, controlled, phase 2 trial. Lancet Rheumatol. 2022;4(6):e395–406.
Takeuchi T, Tanaka S, Murata M, Tanaka Y. Irreversible covalent Bruton’s tyrosine kinase inhibitor, TAS5315 versus placebo in rheumatoid arthritis patients with inadequate response to methotrexate: a randomised, double-blind, phase IIa trial. Ann Rheum Dis. 2023;82(8):1025–34.
Steen VD, editor. Clinical manifestations of systemic sclerosis. Seminars in cutaneous medicine and surgery; 1998.
Rubio-Rivas M, Royo C, Simeón CP, Corbella X, Fonollosa V, editors. Mortality and survival in systemic sclerosis: systematic review and meta-analysis. Seminars in arthritis and rheumatism. Elsevier; 2014.
Tyndall AJ, Bannert B, Vonk M, Airò P, Cozzi F, Carreira PE, et al. Causes and risk factors for death in systemic sclerosis: a study from the EULAR Scleroderma trials and Research (EUSTAR) database. Ann Rheum Dis. 2010;69(10):1809–15.
Einhaus J, Pecher A-C, Asteriti E, Schmid H, Secker K-A, Duerr-Stoerzer S, et al. Inhibition of effector B cells by ibrutinib in systemic sclerosis. Arthritis Res Therapy. 2020;22(1):1–8.
Matsushita T, Hamaguchi Y, Hasegawa M, Takehara K, Fujimoto M. Decreased levels of regulatory B cells in patients with systemic sclerosis: association with autoantibody production and disease activity. Rheumatology. 2016;55(2):263–7.
Matsushita T, Kobayashi T, Mizumaki K, Kano M, Sawada T, Tennichi M, et al. BAFF inhibition attenuates fibrosis in scleroderma by modulating the regulatory and effector B cell balance. Sci Adv. 2018;4(7):eaas9944.
Khanna D, editor. Efficacy and safety of tocilizumab for the treatment of systemic sclerosis: results from a phase 3 randomized controlled trial. 2018 ACR/ARHP Annual Meeting; 2018: ACR.
Jordan S, Distler JH, Maurer B, Huscher D, Van Laar JM, Allanore Y, et al. Effects and safety of rituximab in systemic sclerosis: an analysis from the European Scleroderma Trial and Research (EUSTAR) group. Ann Rheum Dis. 2015;74(6):1188–94.
Reich DS, Arnold DL, Vermersch P, Bar-Or A, Fox RJ, Matta A, et al. Safety and efficacy of tolebrutinib, an oral brain-penetrant BTK inhibitor, in relapsing multiple sclerosis: a phase 2b, randomised, double-blind, placebo-controlled trial. Lancet Neurol. 2021;20(9):729–38.
ACTRIMS. 2024: Tolebrutinib fails to eliminate iron rim lesions in Phase 2 trial. https://multiplesclerosisnewstoday.com/news-posts/2024/03/06/actrims-2024-tolebrutinib-fails-eliminate-prls-small-trial/
Montalban X, Arnold DL, Weber MS, Staikov I, Piasecka-Stryczynska K, Willmer J, et al. Placebo-controlled trial of an oral BTK inhibitor in multiple sclerosis. N Engl J Med. 2019;380(25):2406–17.
Bame E, Tang H, Burns JC, Arefayene M, Michelsen K, Ma B, et al. Next-generation Bruton’s tyrosine kinase inhibitor BIIB091 selectively and potently inhibits B cell and fc receptor signaling and downstream functions in B cells and myeloid cells. Clin Translational Immunol. 2021;10(6):e1295.
Nogales-Gadea G, Ramos-Fransi A, Suarez-Calvet X, Navas M, Rojas-Garcia R, Mosquera JL, et al. Analysis of serum miRNA profiles of myasthenia gravis patients. PLoS ONE. 2014;9(3):e91927.
Querol L, Illa I. Myasthenia gravis and the neuromuscular junction. Curr Opin Neurol. 2013;26(5):459–65.
Patrick J, Lindstrom J. Autoimmune response to acetylcholine receptor. Science. 1973;180(4088):871–2.
Conti-Fine BM, Milani M, Kaminski HJ. Myasthenia gravis: past, present, and future. J Clin Investig. 2006;116(11):2843–54.
Krueger JG, Bowcock A. Psoriasis pathophysiology: current concepts of pathogenesis. Ann Rheum Dis. 2005;64(suppl 2):ii30.
Boehncke W-H, Schön MP, Psoriasis. Lancet. 2015;386(9997):983–94.
Kusuba N, Kitoh A, Miyachi Y, Kabashima K. Role of neutrophils in the pathogenesis of imiquimod-induced psoriasis-like skin lesions. J Dermatol Sci. 2016;84(1):e73.
Papagrigoraki A, Maurelli M, Del Giglio M, Gisondi P, Girolomoni G. Advanced glycation end products in the pathogenesis of psoriasis. Int J Mol Sci. 2017;18(11):2471.
Al-Harbi NO, Nadeem A, Ahmad SF, Bakheet SA, El-Sherbeeny AM, Ibrahim KE, et al. Therapeutic treatment with Ibrutinib attenuates imiquimod-induced psoriasis-like inflammation in mice through downregulation of oxidative and inflammatory mediators in neutrophils and dendritic cells. Eur J Pharmacol. 2020;877:173088.
Giménez-Arnau AM, DeMontojoye L, Asero R, Cugno M, Kulthanan K, Yanase Y, et al. The pathogenesis of chronic spontaneous urticaria: the role of infiltrating cells. J Allergy Clin Immunology: Pract. 2021;9(6):2195–208.
Metz M, Sussman G, Gagnon R, Staubach P, Tanus T, Yang WH, et al. Fenebrutinib in H1 antihistamine-refractory chronic spontaneous urticaria: a randomized phase 2 trial. Nat Med. 2021;27(11):1961–9.
Soundararajan S, Kikuchi Y, Joseph K, Kaplan AP. Functional assessment of pathogenic IgG subclasses in chronic autoimmune urticaria. J Allergy Clin Immunol. 2005;115(4):815–21.
Lawton S. Practical issues for emollient therapy in dry and itchy skin. Br J Nurs. 2009;18(16):978–84.
Kay J, Gawkrodger DJ, Mortimer MJ, Jaron AG. The prevalence of childhood atopic eczema in a general population. J Am Acad Dermatol. 1994;30(1):35–9.
Spergel JM, Paller AS. Atopic dermatitis and the atopic march. J Allergy Clin Immunol. 2003;112(6):S118–27.
Jolly PS, Berkowitz P, Bektas M, Lee H-E, Chua M, Diaz LA, et al. p38MAPK signaling and desmoglein-3 internalization are linked events in pemphigus acantholysis. J Biol Chem. 2010;285(12):8936–41.
Berkowitz P, Diaz LA, Hall RP, Rubenstein DS. Induction of p38MAPK and HSP27 phosphorylation in pemphigus patient skin. J Invest Dermatol. 2007;128(3):738–40.
Dispenza MC, Pongracic JA, Singh AM, Bochner BS. Short-term ibrutinib therapy suppresses skin test responses and eliminates IgE-mediated basophil activation in adults with peanut or tree nut allergy. J Allergy Clin Immunol. 2018;141(5):1914–6. e7.
Regan JA, Cao Y, Dispenza MC, Ma S, Gordon LI, Petrich AM, et al. Ibrutinib, a Bruton’s tyrosine kinase inhibitor used for treatment of lymphoproliferative disorders, eliminates both aeroallergen skin test and basophil activation test reactivity. J Allergy Clin Immunol. 2017;140(3):875–9. e1.
Elias JA, Lee CG, Zheng T, Ma B, Homer RJ, Zhu Z. New insights into the pathogenesis of asthma. J Clin Invest. 2003;111(3):291–7.
Elias JA, Zhu Z, Chupp G, Homer RJ. Airway remodeling in asthma. J Clin Invest. 1999;104(8):1001–6.
Wong WS, Leong KP. Tyrosine kinase inhibitors: a new approach for asthma. Biochim Biophys Acta. 2004;1697(1–2):53–69.
Oettgen HC, Geha RS. IgE regulation and roles in asthma pathogenesis. J Allergy Clin Immunol. 2001;107(3):429–40.
Heusser CH, Wagner K, Bews JP, Coyle A, Bertrand C, Einsle K, et al. Demonstration of the therapeutic potential of non-anaphylactogenic anti-IgE antibodies in murine models of skin reaction, lung function and inflammation. Int Arch Allergy Immunol. 1997;113(1–3):231–5.
Shabestari MS, Rezaei N. Asthma and allergic rhinitis in a patient with BTK deficiency. J Investig Allergol Clin Immunol. 2008;18(4):300–4.
Awan F, Schuh A, Brown JR, et al. Acalabrutinib monotherapy in patients with chronic lymphocytic leukemia who are intolerant to ibrutinib. Blood Adv. 2019;3(9):1553–62.
Lipsky A, Lamanna N. Managing toxicities of Bruton tyrosine kinase inhibitors. Hematology 2014, the American Society of Hematology Education Program Book. 2020;2020(1):336– 45.
Lipsky AH, Farooqui MZ, Tian X, Martyr S, Cullinane AM, Nghiem K, et al. Incidence and risk factors of bleeding-related adverse events in patients with chronic lymphocytic leukemia treated with ibrutinib. Haematologica. 2015;100(12):1571.
Dobie G, Kuriri FA, Omar MM, Alanazi F, Gazwani AM, Tang CP, et al. Ibrutinib, but not zanubrutinib, induces platelet receptor shedding of GPIb-IX-V complex and integrin αIIbβ3 in mice and humans. Blood Adv. 2019;3(24):4298–311.
Kamel S, Horton L, Ysebaert L, Levade M, Burbury K, Tan S, et al. Ibrutinib inhibits collagen-mediated but not ADP-mediated platelet aggregation. Leukemia. 2015;29(4):783–7.
Xiao L, Salem J-E, Clauss S, Hanley A, Bapat A, Hulsmans M, et al. Ibrutinib-mediated atrial fibrillation attributable to inhibition of C-terminal src kinase. Circulation. 2020;142(25):2443–55.
McMullen JR, Boey EJ, Ooi JY, Seymour JF, Keating MJ, Tam CS. Ibrutinib increases the risk of atrial fibrillation, potentially through inhibition of cardiac PI3K-Akt signaling. Blood J Am Soc Hematol. 2014;124(25):3829–30.
Byrd JC, Brown JR, O’Brien S, Barrientos JC, Kay NE, Reddy NM, et al. Ibrutinib versus Ofatumumab in previously treated chronic lymphoid leukemia. N Engl J Med. 2014;371(3):213–23.
Burger JA, Tedeschi A, Barr PM, Robak T, Owen C, Ghia P, et al. Ibrutinib as initial therapy for patients with chronic lymphocytic leukemia. N Engl J Med. 2015;373(25):2425–37.
Moreno C, Greil R, Demirkan F, Tedeschi A, Anz B, Larratt L, et al. Ibrutinib plus Obinutuzumab versus chlorambucil plus obinutuzumab in first-line treatment of chronic lymphocytic leukaemia (iLLUMINATE): a multicentre, randomised, open-label, phase 3 trial. Lancet Oncol. 2019;20(1):43–56.
Mato AR, Nabhan C, Thompson MC, Lamanna N, Brander DM, Hill B, et al. Toxicities and outcomes of 616 ibrutinib-treated patients in the United States: a real-world analysis. Haematologica. 2018;103(5):874.
Guha A, Derbala MH, Zhao Q, Wiczer TE, Woyach JA, Byrd JC, et al. Ventricular arrhythmias following ibrutinib initiation for lymphoid malignancies. J Am Coll Cardiol. 2018;72(6):697–8.
Salem J-E, Manouchehri A, Bretagne M, Lebrun-Vignes B, Groarke JD, Johnson DB, et al. Cardiovascular toxicities associated with ibrutinib. J Am Coll Cardiol. 2019;74(13):1667–78.
Berglöf A, Hamasy A, Meinke S, Palma M, Krstic A, Månsson R, et al. Targets for ibrutinib beyond B cell malignancies. Scand J Immunol. 2015;82(3):208–17.
O’Brien S, Jones JA, Coutre SE, Mato AR, Hillmen P, Tam C, et al. Ibrutinib for patients with relapsed or refractory chronic lymphocytic leukaemia with 17p deletion (RESONATE-17): a phase 2, open-label, multicentre study. Lancet Oncol. 2016;17(10):1409–18.
Byrd JC, Furman RR, Coutre SE, Flinn IW, Burger JA, Blum KA, et al. Targeting BTK with ibrutinib in relapsed chronic lymphocytic leukemia. N Engl J Med. 2013;369(1):32–42.
de Weerdt I, Koopmans SM, Kater AP, van Gelder M. Incidence and management of toxicity associated with ibrutinib and idelalisib: a practical approach. Haematologica. 2017;102(10):1629.
Shatzel JJ, Olson SR, Tao DL, McCarty OJ, Danilov AV, DeLoughery TG. Ibrutinib-associated bleeding: pathogenesis, management and risk reduction strategies. J Thromb Haemost. 2017;15(5):835–47.
Brown JR. How I treat CLL patients with ibrutinib. Blood. 2018;131(4):379–86.
Gribben JG, Bosch F, Cymbalista F, Geisler CH, Ghia P, Hillmen P, et al. Optimising outcomes for patients with chronic lymphocytic leukaemia on ibrutinib therapy: European recommendations for clinical practice. Br J Haematol. 2018;180(5):666–79.
Stephens DM, Byrd JC. How I manage ibrutinib intolerance and complications in patients with chronic lymphocytic leukemia. Blood J Am Soc Hematol. 2019;133(12):1298–307.
Rogers KA, Mousa L, Zhao Q, Bhat SA, Byrd JC, El Boghdadly Z, et al. Incidence of opportunistic infections during ibrutinib treatment for B-cell malignancies. Leukemia. 2019;33(10):2527–30.
Frei M, Aitken SL, Jain N, Thompson P, Wierda W, Kontoyiannis DP, et al. Incidence and characterization of fungal infections in chronic lymphocytic leukemia patients receiving ibrutinib. Leuk Lymphoma. 2020;61(10):2488–91.
Ryan CE, Cheng MP, Issa NC, Brown JR, Davids MS. Pneumocystis Jirovecii pneumonia and institutional prophylaxis practices in CLL patients treated with BTK inhibitors. Blood Adv. 2020;4(7):1458–63.
Varughese T, Taur Y, Cohen N, Palomba ML, Seo SK, Hohl TM, et al. Serious infections in patients receiving ibrutinib for treatment of lymphoid cancer. Clin Infect Dis. 2018;67(5):687–92.
Bercusson A, Colley T, Shah A, Warris A, Armstrong-James D. Ibrutinib blocks Btk-dependent NF-ĸB and NFAT responses in human macrophages during aspergillus fumigatus phagocytosis. Blood J Am Soc Hematol. 2018;132(18):1985–8.
Dubovsky JA, Beckwith KA, Natarajan G, Woyach JA, Jaglowski S, Zhong Y, et al. Ibrutinib is an irreversible molecular inhibitor of ITK driving a Th1-selective pressure in T lymphocytes. Blood. J Am Soc Hematol. 2013;122(15):2539–49.
Colado A, Franco JLM, Elías EE, Amondarain M, Rubio MV, Martínez VS, et al. Second generation BTK inhibitors impair the anti-fungal response of macrophages and neutrophils. Am J Hematol. 2020;95(7):E174–8.
Jongco AM, Gough JD, Sarnataro K, Rosenthal DW, Moreau J, Ponda P, et al. X-linked agammaglobulinemia presenting as polymicrobial pneumonia, including Pneumocystis jirovecii. Ann Allergy Asthma Immunol. 2014;112(1):74–5. e2.
Perkhofer S, Kehrel BE, Dierich MP, Donnelly JP, Nussbaumer W, Hofmann J, et al. Human platelets attenuate aspergillus species via granule-dependent mechanisms. J Infect Dis. 2008;198(8):1243–6.
Byrd JC, Furman RR, Coutre SE, Flinn IW, Burger JA, Blum K, et al. Ibrutinib Treatment for First-Line and Relapsed/Refractory chronic lymphocytic leukemia: final analysis of the pivotal phase Ib/II PCYC-1102 StudyLong-Term single-Agent Ibrutinib in CLL. Clin Cancer Res. 2020;26(15):3918–27.
Dickerson T, Wiczer T, Waller A, Philippon J, Porter K, Haddad D, et al. Hypertension and incident cardiovascular events following ibrutinib initiation. Blood. 2019;134(22):1919–28.
Iberri DJ, Kwong BY, Stevens LA, Coutre SE, Kim J, Sabile JM, et al. Ibrutinib-associated rash: a single-centre experience of clinicopathological features and management. Br J Haematol. 2016;180(1):164–6.
Sibaud V, Beylot-Barry M, Protin C, Vigarios E, Recher C, Ysebaert L. Dermatological toxicities of Bruton’s tyrosine kinase inhibitors. Am J Clin Dermatol. 2020;21(6):799–812.
Bitar C, Farooqui MZ, Valdez J, Saba NS, Soto S, Bray A, et al. Hair and nail changes during long-term therapy with ibrutinib for chronic lymphocytic leukemia. JAMA Dermatology. 2016;152(6):698–701.
Liu H-b, Wu Y, Lv T-f, Yao Y-w, Xiao Y-y, Yuan D-m, et al. Skin rash could predict the response to EGFR tyrosine kinase inhibitor and the prognosis for patients with non-small cell lung cancer: a systematic review and meta-analysis. PLoS ONE. 2013;8(1):e55128.
Haselmayer P, Camps M, Liu-Bujalski L, Nguyen N, Morandi F, Head J, et al. Efficacy and pharmacodynamic modeling of the BTK inhibitor evobrutinib in autoimmune disease models. J Immunol. 2019;202(10):2888–906.
Angst D, Gessier F, Janser P, Vulpetti A, Wälchli R, Beerli C, et al. Discovery of LOU064 (Remibrutinib), a potent and highly selective covalent inhibitor of Bruton’s tyrosine kinase. J Med Chem. 2020;63(10):5102–18.
Woyach JA, Furman RR, Liu T-M, Ozer HG, Zapatka M, Ruppert AS, et al. Resistance mechanisms for the Bruton’s tyrosine kinase inhibitor ibrutinib. N Engl J Med. 2014;370(24):2286–94.
Hamasy A, Wang Q, Blomberg K, Mohammad D, Yu L, Vihinen M, et al. Substitution scanning identifies a novel, catalytically active ibrutinib-resistant BTK cysteine 481 to threonine (C481T) variant. Leukemia. 2017;31(1):177–85.
Quinquenel A, Fornecker L-M, Letestu R, Ysebaert L, Fleury C, Lazarian G, et al. Prevalence of BTK and PLCG2 mutations in a real-life CLL cohort still on ibrutinib after 3 years: a FILO group study. Blood J Am Soc Hematol. 2019;134(7):641–4.
Estupiñán HY, Berglöf A, Zain R, Smith CE. Comparative analysis of BTK inhibitors and mechanisms underlying adverse effects. Front cell Dev Biology. 2021;9:630942.
Sharman JP, Egyed M, Jurczak W, Skarbnik A, Pagel JM, Flinn IW, et al. Acalabrutinib with or without obinutuzumab versus chlorambucil and obinutuzumab for treatment-naive chronic lymphocytic leukaemia (ELEVATE-TN): a randomised, controlled, phase 3 trial. Lancet. 2020;395(10232):1278–91.
Byrd JC, Wierda WG, Schuh A, Devereux S, Chaves JM, Brown JR, et al. Acalabrutinib monotherapy in patients with relapsed/refractory chronic lymphocytic leukemia: updated phase 2 results. Blood. 2020;135(15):1204–13.
Ghia P, Pluta A, Wach M, Lysak D, Kozak T, Simkovic M, et al. ASCEND: Phase III, randomized trial of acalabrutinib versus idelalisib plus rituximab or bendamustine plus rituximab in relapsed or refractory chronic lymphocytic leukemia. J Clin Oncol. 2020;38(25):2849–61.
Byrd JC, Woyach JA, Furman RR, Martin P, O’Brien S, Brown JR, et al. Acalabrutinib in treatment-naive chronic lymphocytic leukemia. Blood. 2021;137(24):3327–38.
Byrd JC, Harrington B, O’Brien S, Jones JA, Schuh A, Devereux S, et al. Acalabrutinib (ACP-196) in relapsed chronic lymphocytic leukemia. N Engl J Med. 2016;374(4):323–32.
Nicolson PL, Hughes CE, Watson S, Nock SH, Hardy AT, Watson CN, et al. Inhibition of Btk by Btk-specific concentrations of ibrutinib and acalabrutinib delays but does not block platelet aggregation mediated by glycoprotein VI. Haematologica. 2018;103(12):2097.
Burger JA, Barr PM, Robak T, Owen C, Ghia P, Tedeschi A, et al. Long-term efficacy and safety of first-line ibrutinib treatment for patients with CLL/SLL: 5 years of follow-up from the phase 3 RESONATE-2 study. Leukemia. 2020;34(3):787–98.
Sun C, Nierman P, Kendall EK, Cheung J, Gulrajani M, Herman SE, et al. Clinical and biological implications of target occupancy in CLL treated with the BTK inhibitor acalabrutinib. Blood. 2020;136(1):93–105.
Xu W, Yang S, Zhou K, Pan L, Li Z, Zhou J, et al. Treatment of relapsed/refractory chronic lymphocytic leukemia/small lymphocytic lymphoma with the BTK inhibitor zanubrutinib: phase 2, single-arm, multicenter study. J Hematol Oncol. 2020;13(1):1–12.
Tam CS, Robak T, Ghia P, Kahl BS, Walker P, Janowski W, et al. Zanubrutinib monotherapy for patients with treatment-naïve chronic lymphocytic leukemia and 17p deletion. Haematologica. 2021;106(9):2354.
Guo Y, Liu Y, Hu N, Yu D, Zhou C, Shi G, et al. Discovery of Zanubrutinib (BGB-3111), a Novel, Potent, and selective covalent inhibitor of Bruton’s tyrosine kinase. J Med Chem. 2019;62(17):7923–40.
Tam CS, Opat S, D’Sa S, Jurczak W, Lee H-P, Cull G, et al. A randomized phase 3 trial of zanubrutinib vs ibrutinib in symptomatic Waldenström macroglobulinemia: the ASPEN study. Blood. 2020;136(18):2038–50.
Sekiguchi N, Rai S, Munakata W, Suzuki K, Handa H, Shibayama H, et al. A multicenter, open-label, phase II study of tirabrutinib (ONO/GS‐4059) in patients with Waldenström’s macroglobulinemia. Cancer Sci. 2020;111(9):3327–37.
Walter HS, Rule SA, Dyer MJ, Karlin L, Jones C, Cazin B, et al. A phase 1 clinical trial of the selective BTK inhibitor ONO/GS-4059 in relapsed and refractory mature B-cell malignancies. Blood J Am Soc Hematol. 2016;127(4):411–9.
Sharman JP, Black-Shinn JL, Clark J, Bitman B. Understanding ibrutinib treatment discontinuation patterns for chronic lymphocytic leukemia. Blood. 2017;130:4060.
Cohen S, Tuckwell K, Katsumoto TR, Zhao R, Galanter J, Lee C, et al. Fenebrutinib Versus Placebo or Adalimumab in Rheumatoid Arthritis: a Randomized, Double-Blind, phase II trial. Arthritis Rheumatol. 2020;72(9):1435–46.
Acknowledgements
Not applicable.
Funding
This study was supported by a grant from Tehran University of Medical Sciences, Tehran, Iran, under grant number 64014.
Author information
Authors and Affiliations
Contributions
G.MT., N.Y., and N.R. conceptualized the study. G.MT. and N.Y. conducted database search and drafting the initial draft. G.MT., N.Y., and N.R. prepared the final draft. N.R. supervised the project and critically appraised the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Tavakoli, G.M., Yazdanpanah, N. & Rezaei, N. Targeting Bruton’s tyrosine kinase (BTK) as a signaling pathway in immune-mediated diseases: from molecular mechanisms to leading treatments. Adv Rheumatol 64, 61 (2024). https://doi.org/10.1186/s42358-024-00401-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s42358-024-00401-y