
Abstract
Background
Empirical field studies allow us to view how ecological and environmental processes shape the biodiversity of our planet, but collecting samples in situ creates inherent challenges. The majority of empirical vertebrate gut microbiome research compares multiple host species against abiotic and biotic factors, increasing the potential for confounding environmental variables. To minimize these confounding factors, we focus on a single species of passerine bird found throughout the geologically complex island of Sulawesi, Indonesia. We assessed the effects of two environmental factors, geographic Areas of Endemism (AOEs) and elevation, as well as host sex on the gut microbiota assemblages of the Sulawesi Babbler, Pellorneum celebense, from three different mountains across the island. Using cloacal swabs, high-throughput-amplicon sequencing, and multiple statistical models, we identified the core microbiome and determined the signal of these three factors on microbial composition.
Results
The five most prevalent bacterial phyla within the gut microbiome of P. celebense were Proteobacteria (32.6%), Actinobacteria (25.2%), Firmicutes (22.1%), Bacteroidetes (8.7%), and Plantomycetes (2.6%). These results are similar to those identified in prior studies of passeriform microbiomes. Overall, microbiota diversity decreased as elevation increased, irrespective of sex or AOE. A single ASV of Clostridium was enriched in higher elevation samples, while lower elevation samples were enriched with the genera Perlucidibaca (Family Moraxellaceae), Lachnoclostridium (Family Lachnospiraceae), and an unidentified species in the Family Pseudonocardiaceae.
Conclusions
While the core microbiota families recovered here are consistent with other passerine studies, the decreases in diversity as elevation increases has only been seen in non-avian hosts. Additionally, the increased abundance of Clostridium at high elevations suggests a potential microbial response to lower oxygen levels. This study emphasizes the importance of incorporating multiple statistical models and abiotic factors such as elevation in empirical microbiome research, and is the first to describe an avian gut microbiome from the island of Sulawesi.
Similar content being viewed by others


Background
The complex relationship between microbial symbionts and their hosts is a functionally important, medically relevant, and often understudied component of global biodiversity. In every habitable natural system, there exists communities of microscopic organisms, including those residing in and on other organisms. Humans and other vertebrates harbor communities of microbes in the gastrointestinal (GI) tract known as the gut microbiota that directly contribute to nutrient uptake, immune function, and fitness plasticity in stochastic environments [1,2,3,4,5]. Studies of vertebrate hosts report various ecological and evolutionary factors driving gut microbiota community assemblage, including diet [6,7,8], sex [9], reproductive behavior [10], habitat type [11], movement [12] and host phylogeny [13,14,15,16]; although many of these factors are interrelated in terms of microbe-specific selection pressures. However, as the majority of the vertebrate gut microbiota literature focuses heavily on mammalian systems [17, 18], there is a significant knowledge gap in terms of how these processes affect other major taxa. Additional studies within non-mammalian hosts, particularly in birds, will allow for generalizable insights regarding host-gut micriobiota dynamics across vertebrates. Avian hosts provide an ideal study system as they are known vectors of human diseases (e.g. West Nile Virus, High-pathogenic Bird Flu), demonstrate diverse ecologies and mating systems, and serve as model systems for comparative analyses in ecology and evolutionary biology [19].
Songbirds (Order: Passeriformes) make up more than 50% of global bird diversity [20]. In general, the gut microbiota of Passeriformes are dominated by the bacterial phyla Firmicutes, Actinobacteria, Tenericutes, Bacteroidetes, and Proteobacteria, with a particularly high abundance of Firmicutes in bird species that feed on insects [21,22,23,24,25,26]. Comparative field studies have suggested that host phylogeny is the most significant, albeit weakly associated [19], driver influencing the gut microbiome structure of birds. This relationship is independent of other factors such as diet or habitat [6, 19, 21, 26,27,28]. While placental mammals inoculate their offspring with a portion of their microbiota through live birth, avian young are exposed to microbes in nesting material and receive parental microbiota via incubation and parental saliva [12, 29], suggesting birds are more susceptible to environmental variation in microbial source pools (e.g., due to geographic or ecological gradient effects) than viviparous organisms [15, 19, 26, 28]. While evidence suggests only small differences in avian microbial communities for intraspecific bird populations from both temperate and tropical locations [15, 20, 21, 26, 30], compositional microbiome differences were found between syntopic resident and migratory populations within a species (the barn swallow, Hirundo rustica [12]). Whether these differences were due to migration stress or region-specific microbe uptake is difficult to determine. An alternative approach would be to compare the microbial composition within allopatric populations of a single, widespread avian host species, occurring across a repeated environmental gradient (e.g., elevation). To resolve if region-specificity affects the microbial composition of avian hosts in Southeast Asia, we sampled one passerine species in three areas of endemism across a 1000 m elevational range on the island of Sulawesi, Indonesia.
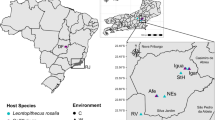
Sulawesi is the eleventh largest island in the world (Fig. 1) and has a complex geological history, with different landmasses accreting and breaking apart over the past 30 million years [31, 32]. Recent studies have revealed Sulawesi remained partially submerged until less than 1 MYA [32]. As a result of these processes, seven different geographically-isolated Areas of Endemism (AOEs) have been identified, supported by clear species boundaries within terrestrial vertebrates [33,34,35]. Sulawesi is also unusual for an island in that it harbors 25 high-elevation mountains (> 2000 m), which have likely contributed to its unusually high percentage of endemic flora and fauna [31, 33, 36, 37]; endemic species make up approximately 48% of all birds and 36% of all mammals described on the island [38]. The mountains on Sulawesi naturally encompass steep gradients in abiotic variables over short distances, making it possible to rapidly sample multiple individuals of a single species spanning a broad environmental cline. The replicate sampling of high-elevation mountains on Sulawesi provides an opportunity to investigate how avian gut microbiome diversity is shaped by both geography (allopatric isolation on different mountains) and steep clines in abiotic variables (elevational gradients).

Map of collecting sites on the island of Sulawesi, Indonesia. Grey bars indicate the seven Areas of Endemism (AOEs) described in [32]. The axis values are in degrees
Evidence for a relationship between elevation and gut microbial diversity and abundance in mammals, squamates, and birds has been mixed [21, 30, 39,40,41,42]. However, we predict a decrease in both diversity and relative abundances of microbial taxa in individuals with increasing elevation, corresponding to the lower diversity and abundance of prey, parasites, and environmental microbiota found at higher elevations [30, 40, 43, 44]. Further, although AOEs on Sulawesi represent distinct population barriers for terrestrial vertebrates, this may not extend to volant organisms and their gut microbiota. We therefore predicted that higher elevation would be negatively correlated with microbial diversity and relative abundances of dominant taxa, and that elevation would be a more significant factor in microbial composition than area of endemism within intraspecific populations of endemic Sulawesi passerines.
To determine the effect of AOE and elevation on avian gut microbiota assemblages within these montane systems, we chose the Sulawesi babbler (Pellorneum celebense) as a focal taxon. This species is a common, endemic insectivorous passerine widespread throughout the island, where it occupies dense forest undergrowth from sea-level to 1500 m in elevation, and was the most abundant species found [45,46,47]. We sampled P. celebense individuals along 1000 m elevational transects on three mountains, each within a different AOE, to determine if the diversity and relative abundances of gut microbial assemblages correlate with both elevation and area of endemism on the island of Sulawesi (Fig. 1). Host sex was also included in analyses to account for potential sex-dependent variation. We tested the hypotheses that H1: diversity and abundance of P. celebense gut microbiota is negatively correlated with elevation; and H2: areas of endemism do not have significant effect on host intraspecific microbial variation.
Results
A total of 4339 ASVs from 40 Pellorneum celebense individuals were recovered for downstream analyses. The R package “decontam” did not find any reads from positive or negative controls that were found in all corresponding samples, so no matching reads were removed from the dataset. After transforming raw read counts and removing low abundance ASVs, 3445 were retained in the transformed dataset, with 3028 ASVs recovered after removing individuals with < 3000 reads (n = 2) and rarefying the remaining 38 individuals to the lowest read count (n = 3389 reads, Table 1). A t-test comparing ASV richness by sample between the transformed and rarefied datasets did not reveal a difference in microbial richness (t = 0.81, p = 0.42), so only the results from unrarefied data are reported.
P. celebense microbiome composition
Only one ASV, an unidentified member of the Enterobacteriaceae family (Phylum Proteobacteria), was found in 90% of individuals. An additional ASV in the Enterococcaceae family (Phylum Firmicutes) was shared among 90% of the Torompupu individuals. The five most prevalent bacterial phyla within the gut microbiome of P. celebense were Proteobacteria (32.6%), Actinobacteria (25.2%), Firmicutes (22.1%), Bacteroidetes (8.7%), and Plantomycetes (2.6%). The relative abundances of these phyla by individual are visualized in Fig. 2.

Absolute frequencies in all samples (A) and the relative abundances of each individual (B) of the 5 most abundant phyla of ASVs from the P. celebense cloacal microbiome compositional dataset, identified using the 16S SILVA ribosomal RNA gene database
Trends in P. celebense microbe diversity
By Sex and Mountain To determine if host sex or AOE is directly correlated with changes in alpha diversity, ANOVAs of individual Shannon indices using mountains and sex as explanatory variables were run. Results did not reveal differences in mean group diversity (FMountain = 1.594, pMountain = 0.217; FSex = 1.420, pSex = 0.255, Fig. 3). These results did not change when these ANOVAs were replicated using Chao1 as the diversity index (Fig. 3). However, PERMANOVAs comparing Unifrac beta diversity indices indicated a slight difference in microbial membership (unweighted Unifrac) among mountains (F = 1.216, r2 = 0.062, p = 0.062, Fig. 4a), though this difference was no longer significant when relative abundance of ASVs (weighted Unifrac) was included in the analysis (Fw = 1.283, r2w = 0.065, pw = 0.254, Fig. 4b). A PCoA plot of weighted Unifrac distances revealed a higher clustering of samples from Dako, suggesting that microbial communities of this sample population may have more phylogenetic similarity than populations on the other mountains, as a non-dimensional ordination did not show the same clustering of Dako samples (Fig. 4c).

Microbial alpha diversity plots and associated p values using number of ASVs and Shannon Index compared by the variables: A sex (adults) and juveniles, B mountain, C elevational as a factor (low = < 700 m, mid = 700–1200 m, high = > 1200 m), and D continuous elevation

Beta-diversity PCoA plots using unweighted Unifrac (A), weighted Unifrac (B), and NDMS (C) ordinations. Stress value for the NDMS was 0.190. Individuals are represented by a point shaped by sex and colored by elevation. Variations by mountain are represented by ordination ellipses
By Elevation While an ANOVA of individual Shannon diversity by elevational category did not show a significant decrease in mean diversity as elevation increases (F = 5.174, p = 0.072, Fig. 3c), a linear regression using numerical elevation data revealed a significant decrease as elevation increases (t = 4.967, p = 1.56e−05, Fig. 3d). PERMANOVAs of beta diversity indices revealed a significant difference by elevational category only in microbial composition (weighted Unifrac), not membership alone (unweighted Unifrac) (Fw = 2.521, r2w = 0.120, pw = 0.044; F = 1.180, r2 = 0.060, p = 0.071). A PERMANOVA of CCA ordination residuals testing the effect of elevation as an environmental gradient also revealed a clear negative trend between elevation and microbial relative abundance (F = 1.230, p = 0.004, Fig. 5a). When this analysis was repeated adding mountain as a conditional variable, these changes in abundance became less significant, though plotting regression lines suggests a continued negative correlation with elevation (Fm = 1.202, pm = 0.077, Fig. 5b).

Canonical Correspondence Analyses (CCAs) using only elevation as an environmental gradient (A) and with both elevation as a gradient and mountain as a conditional variable (B). Mountains are represented as separate regression lines, with shaded areas represent 95% confidence intervals. P values of each model indicates that the correlation between microbial diversity and elevation is less significant when sampling location is considered
Modeling interacting factors
To determine if mountain or host sex is producing a random effect potentially confounding the influence of elevation on microbial assemblages, three robust mixed-effect models were run with elevation as the predictor effect and mountain and sex as separate and interacting random effects. Because robust models do not generate p values, the significance of models are confirmed if the slope and 95% CI intervals do not intercept 0 [48]. All three models were significant under these conditions and resulting predictor plots revealed identical negative correlations between microbial diversity and elevation, regardless of which categorical variable was used as the random effect (Additional file 1: Fig. S2 and Table S2).
Elevational effects on microbe taxonomic abundance
To determine which microbial families changed in abundance at each end of the elevational gradient, logfold values were only compared between the “low” and “high” elevation categories. The families Pseudonocardiaceae, Lachnospiraceae, and Moraxellaceae were significantly more abundant at lower elevations, whereas Clostridiaceae was enriched at higher elevations (Fig. 6).

Differential abundances between low and high elevational categories. ASV’s enriched in higher elevations fall to the left side of the x-axis, while those enriched in lower elevations fall to the right
Discussion
In this study, we determined the effects of elevation, mountain (representing independent AOEs), and sex on microbial assemblages in a Sulawesi songbird, Pellorneum celebense. In general, cloacal microbiota appeared to decrease in abundance and diversity as elevation increases, regardless of mountain or host sex. Surprisingly, there seems to be no influence of host sex on microbiome composition in this system. While a PERMANOVA comparing microbial membership (unweighted Unifrac distances) did reveal a significant influence of mountain, the most likely explanation is that this species was only found above 900 m on Mt. Dako. Therefore, the differences are most likely attributed to a lack of sampling at lower elevations at this locality, as this significance was lost when abundance-weighted microbial composition (weighted Unifrac distances) was compared (Additional file 1: Fig. S1).
While previous avian gut microbiota studies did not reveal correlations with elevation, decreases in microbial alpha and beta diversities at higher elevations are also seen across populations of the toad-headed lizard, Phrynocephalus vlangalii [39]. This decrease is thought to be influenced by hypoxic conditions resulting from decreased oxygen partial pressure, though it is worth noting the elevational range of P. vlangalii (2900–4250 m) is much higher than that of P. celebense. Conversely, a study of high-altitude pikas (Ochotona curzoniae) found higher diversity and functional enrichment as elevation increased [40], while studies of Tibetan ruminants (Bos spp. and Ovis spp.) and mesquite lizards (Sceloporus grammicus) did not find any relationship between diversity and elevation that was not consistent with subsequent dietary shifts [49, 50]. While we can assume the decrease in diversity at higher elevations is associated with lower diversity of invertebrate prey species, a lack of dietary analyses prevents a definitive conclusion. Additionally, selection pressures, especially those related to oxygen levels and air pressure, are likely increasing as elevation increases. Expanding elevational analyses to include avian host species with a wide elevational range, especially those differing in feeding guilds, would further validate this negative correlation with microbial diversity at the community scale.
Observed patterns from specific microbial taxa found in P. celebense cloacal samples also offer insights into the underlying factors influencing the microbiome community. Clostridium sensu stricto 1 is an obligate anaerobic fermenter metabolizing a range of compounds such as carbohydrates, amino acids, alcohols, and purines [51]. The increased presence of Clostridium in higher elevation hosts may suggest an increased dependency on microbial symbionts for metabolism due to lower oxygen levels, as microbes related to host metabolic pathways were also seen in higher proportions in high-elevation S. grammicus lizard populations [50]. The genus Perlucidibaca has only been isolated from aquatic samples, which does not provide a clear biological explanation for its increased abundance in hosts at low elevations [52]. A possible explanation is that these microbes are present in sampling sites near bodies of standing water (which are typically seen only in lower elevations), though no environmental samples were collected in this study so this could not be confirmed. Members of the family Pseudonocardiaceae are known to produce antibacterial metabolites, especially in high nitrogen environments [53, 54]. The genus Lachnoclostridium is associated with metabolism of similar metabolites as Clostridium, but has been identified as an indicator of early stages of colorectal cancer and therefore may indicate localized immune activity in the cloaca [55, 56]. Both families were also significantly more abundant in hosts at lower elevation, which may suggest increased immune activity. Because the majority of functional analyses on gut microbes are human-based, however, these potential associations are merely speculative. Future gut microbiota studies incorporating measured immune activity could discern if these microbes are associated with avian immune function.
Conclusions
The results of this study illuminate the importance of assessing abiotic and biotic factors in empirical gut microbiota research by documenting intraspecific variation seen in wild host populations. Studies focusing on host phylogeny and diet in wild systems may miss the potential confounding effects of environmental factors if they are not included in these analyses. In the absence of elevational data, this dataset would reveal limited spatial or sex-dependent variation in microbial communities. Sulawesi montane ecosystems provide an ideal study system of an isolated, endemic avian community. By incorporating an elevational gradient and testing for interacting factors, we show that the cloacal microbiota membership, structure, and overall abundance in P. celebense populations significantly decreases in higher elevations in all three different areas of endemism that were sampled. Additionally, the higher abundance of metabolic microbe ASVs at high elevations suggests altitude-related shifts in community structure. Future community studies can confirm if these elevational shifts are consistent across host feeding guilds and phylogeny. This study lays the foundation for future work on montane host communities on Sulawesi, and contributes to a growing global microbiome dataset by providing the first report of avian gut microbiota from this remarkably unique island.
Methods
Study region
Each of the three mountains surveyed shared general habitat gradients relative to elevation (Fig. 1). We categorized elevations under 1000 m as lowland forest, dominated by large canopy trees with thick undergrowth. Above 1000 m we observed mossy transitional forests, with understory dominated by rattan vines (subfamily Calamoideae). Summit ecosystems (> 1600 m) are categorized as mossy forests with a sparse understory of Rhododendron spp., though no P. celebense individuals were found in ecosystems above 1450 m.
-
Gunung Torompupu NW central Core AOE. Summit is 2495 m. Located west of the Palu-Koro fault, which has been shown to be a species boundary for terrestrial vertebrates [34]. The understory at all elevations was dominated by dense rattan.
-
Gunung Katopasa Eastern peninsula AOE. Summit is 2835 m. Sampling sites between 700 and 1300 m included fire damage and clearings for small plantations.
-
Gunung Dako Northern Peninsula AOE. Summit is 2260 m. Because the mountain is more accessible than most, lower elevation habitat was fragmented by small plantations with cleared understories. Given that this species specializes in dense forest undergrowth, the level of cultivation likely explains why we did not observe P. celebense below 900 m on this mountain.
Sample collection
Birds were sampled during three, month-long expeditions during the dry seasons between 2017 and 2018 (Fig. 1, Table 2) using mist-net transects spanning elevational gradients at each mountain. Once captured in mist-nets, individuals were promptly removed and placed in cloth holding bags while transported to camp for processing. To profile the avian gut microbiota, cloacal swabs (Copan Diagnostics, Murrieta, CA) were collected from live animals once at camp. First, 3% H2O2 and a fresh KimWipe® was used to sterilize the skin surrounding the cloaca. A sterile flocked nylon swab was gently inserted, turned ½ rotation clockwise and ½ rotation counterclockwise, and placed into a tube of 96% ethanol. This reagent was chosen over RNAlater® as it is less likely to degrade DNA when stored at warmer temperatures, which is unavoidable during remote tropical fieldwork [57, 58]. A tube of ethanol was used as a negative for every new site to account for potential reagent contamination. Individuals were sexed by dissection and visual inspection of gonads. If there were no developed ova or testes and the skull was not fully ossified, birds were defined as juvenile. Upon returning from the field, cloacal swabs were stored in a – 80° freezer until processed. All sampling protocols were approved by institutional animal care and use committees at the University of California, Berkeley (AUP-2016-04-8665-1) and the American Museum of Natural History (AMNHIACUC-20171020). Research permits (Surat Izin Penelitian) for three expeditions undertaken in 2017 and 2018 were obtained from Ministry of Research, Technology and Higher Education (KEMENRISTEKDIKTI) (no. 213/SIP/FRP/E5/Dit.KI/VIII/2017), with sample export documents for each expedition provided by the Research Center for Biology, Indonesian Institute of Sciences (LIPI).
Microbiome sequencing
DNA extractions were conducted using the MoBio® Power Soil kit (MoBio Laboratories Inc., Carlsbad, CA) with an extra wash step to maximize DNA recovery. DNA was PCR-amplified in triplicate using the Earth Microbiome Project’s 16S rRNA PCR protocol primers 515RB (5′-GTGYCAGCMGCCGCGGTAA-3′) and 806RB (5′-GGACTACNVGGGTWTCTAAT-3′) and sequenced on the Illumina® MiSeq platform, generating 250 base pair paired-end amplicon reads [59, 60]. PCRs and sequencing, including of negative controls, were performed at the Argonne National Laboratory Sample Processing Facility (Lemont, IL, USA).
Data preprocessing
Preprocessing of raw reads was conducted using packages implemented in QIIME2 v.2018.11 [60]. With the DADA2 plugin, single-end reads were trimmed and chimeric sequences were removed before being paired and classified as Amplicon Sequence Variants (ASVs) [61]. A multiple sequence alignment of paired-end reads was generated using the ‘mafft’ program [62], and FastTree was used to create a midpoint-rooted phylogenetic tree [63]. Taxonomic identification of each ASV was generated using the Naïve Bayesian q-2 feature classifier trained on the 16S SILVA 132 ribosomal RNA gene database [64]. Downstream analyses were conducted in RStudio v.1.2.1335. The R package ‘decontam’ was used to assess potential contamination using field and laboratory controls [65]. Reads that matched chloroplast and host mitochondrial sequences were then removed. Due to the compositional nature of Illumina MiSeq data, [66] reads were transformed to relative abundances and ASVs with a relative abundance of < 0.00001 were removed for certain downstream analyses using the R package ‘phyloseq’ [67]. However, a rarefied dataset using the ‘phyloseq’ command rarefy_even_depth was analyzed in parallel to rule out potential effects of these preprocessing steps.
Statistical analyses
All statistical analyses and visualizations were performed in R 4.1.2 (R Core Team 2020). The top 20 microbial phyla were identified, and the shared microbial taxa of all samples were determined using the core_members function of ‘phyloseq’. Mountain and host sex were used as categorical variables, while elevation was analyzed as either a continuous or categorical variable: “low” for samples from elevations under 700 m (n = 16), “mid” for 700–1200 m (n = 9), and “high” for elevations over 1200 m (n = 15). Alpha diversity means were calculated using Shannon diversity and Chao1 diversity indices using the estimate_richness function of ‘phyloseq’ and compared using ANOVAs (for categorical factors) with post-hoc Tukey tests and a general linear model (for elevation). Beta diversity indices were calculated as the sum of phylogenetic branch lengths (Unifrac distances) to compare variances in microbial membership (unweighted Unifrac) and composition (abundance-weighted Unifrac) among categorical factors using PERMANOVAS with the adonis function [68]. We also included an NDMS ordination based on Bray–Curtis distances. To evaluate elevation as a continuous environmental gradient, Canonical Correspondence Analyses (CCAs) were run [69], using categorical factors as conditional variables. To determine directional correlations between microbial diversity and elevation, robust mixed-effect models were created and evaluated using the R package “robustlmm,” as the robust model is more appropriate than other mixed models for datasets with small sample sizes and high degrees of freedom [48]. We used Shannon diversity index as the response variable in this model to detect shifts in ASV abundance and richness, using elevation as the continuous fixed effect and combinations of sex and mountain as random effects. DESeq2, a logistical regression of dispersions weighted by normalized mean counts of unfiltered taxonomic data [70], was used to assess which microbial taxa changed in abundance among mountain, host sex, and elevational category.
Availability of data and materials
All data files and reproduceable code can be found at https://github.com/rjoakim/babbler_microbiome
References
Spor A, Koren O, Ley R. Unravelling the effects of the environment and host genotype on the gut microbiome. Nat Rev Microbiol. 2011;9:279–90. https://doi.org/10.1038/nrmicro2540.
Russell JA, Dubilier N, Rudgers JA. Nature’s microbiome: introduction. Mol Ecol. 2014;23:1225–37. https://doi.org/10.1111/mec.12676.
Carding S, Verbeke K, Vipond DT, Corfe BM, Owen LJ. Dysbiosis of the gut microbiota in disease. Microb Ecol Health Dis. 2015. https://doi.org/10.3402/mehd.v26.26191.
Koskella B, Hall LJ, Metcalf CJE. The microbiome beyond the horizon of ecological and evolutionary theory. Nat Ecol Evol. 2017;1:1606–15. https://doi.org/10.1038/s41559-017-0340-2.
Suzuki TA, Ley RE. The role of the microbiota in human genetic adaptation. Science. 2020. https://doi.org/10.1126/science.aaz6827.
Phillips CD, Phelan G, Dowd SE, McDonough MM, Ferguson AW, Delton Hanson J, et al. Microbiome analysis among bats describes influences of host phylogeny, life history, physiology and geography. Mol Ecol. 2012;21:2617–27.
Delsuc F, Metcalf JL, Wegener Parfrey L, Song SJ, González A, Knight R. Convergence of gut microbiomes in myrmecophagous mammals. Mol Ecol. 2014;23:1301–17.
Hale VL, Tan CL, Niu K, Yang Y, Knight R, Zhang Q, et al. Diet versus phylogeny: a comparison of gut microbiota in captive colobine monkey species. 2018;75:515–27. https://doi.org/10.1007/s00248-017-1041-8
Roggenbuck M, Bærholm Schnell I, Blom N, Bælum J, Bertelsen MF, Pontén TS, et al. The microbiome of new world vultures. Nat Commun. 2014. https://doi.org/10.1038/ncomms6498.
Jacob S, Parthuisot N, Vallat A, Ramon-Portugal F, Helfenstein F, Heeb P. Microbiome affects egg carotenoid investment, nestling development and adult oxidative costs of reproduction in Great tits. Funct Ecol. 2015;29:1048–58. https://doi.org/10.1111/1365-2435.12404.
Sullam KE, Rubin BER, Dalton CM, Kilham SS, Flecker AS, Russell JA. Divergence across diet, time and populations rules out parallel evolution in the gut microbiomes of Trinidadian guppies. ISME J. 2015;9:1508–22. https://doi.org/10.1038/ismej.2014.231.
Turjeman S, Corl A, Wolfenden A, Tsalyuk M, Lublin A, Choi O, et al. Migration, pathogens and the avian microbiome: a comparative study in sympatric migrants and residents. Mol Ecol. 2020;29:4706–20. https://doi.org/10.1111/mec.15660.
Sanders JG, Powell S, Kronauer DJC, Vasconcelos HL, Frederickson ME, Pierce NE. Stability and phylogenetic correlation in gut microbiota: lessons from ants and apes. Mol Ecol. 2014;23:1268–83.
Carrillo-Araujo M, Tas N, Alcántara-Hernández RJ, Gaona O, Schondube JE, Medellín RA, et al. Phyllostomid bat microbiome composition is associated to host phylogeny and feeding strategies. Front Microbiol. 2015;6:1–9.
Kropáčková L, Těšický M, Albrecht T, Kubovčiak J, Čížková D, Tomášek O, et al. Codiversification of gastrointestinal microbiota and phylogeny in passerines is not explained by ecological divergence. Mol Ecol. 2017;26:5292–304. https://doi.org/10.1111/mec.14144.
Ingala MR, Becker DJ, Bak Holm J, Kristiansen K, Simmons NB. Habitat fragmentation is associated with dietary shifts and microbiota variability in common vampire bats. Ecol Evol. 2019;9:6508–23.
Amato KR. Co-evolution in context: the importance of studying gut microbiomes in wild animals. Microbiome Sci Med. 2013;1:10–29.
Zmora N, Suez J, Elinav E. You are what you eat: diet, health and the gut microbiota. Nat Rev Gastroenterol Hepatol. 2019;16:35–56. https://doi.org/10.1038/s41575-018-0061-2.
Bodawatta KH, Hird SM, Grond K, Poulsen M, Jønsson KA. Avian gut microbiomes taking flight. Trends Microbiol. 2021. https://doi.org/10.1016/j.tim.2021.07.003.
Bodawatta KH, Sam K, Jønsson KA, Poulsen M. Comparative analyses of the digestive tract microbiota of New Guinean Passerine birds. Front Microbiol. 2018;9:1–13.
Hird SM, Sánchez C, Carstens BC, Brumfield RT. Comparative gut microbiota of 59 neotropical bird species. Front Microbiol. 2015. https://doi.org/10.3389/fmicb.2015.01403.
Kreisinger J, Čížková D, Kropáčková L, Albrecht T. Cloacal microbiome structure in a long-distance migratory bird assessed using deep 16sRNA pyrosequencing. 2015. https://doi.org/10.1371/journal.pone.0137401.
Lewis WB, Moore FR, Wang S. Characterization of the gut microbiota of migratory passerines during stopover along the northern coast of the Gulf of Mexico. J Avian Biol. 2016;47:659–68.
Kropáčková L, Pechmanová H, Vinkler M, Svobodová J, Velová H, Těšičký M, et al. Variation between the oral and faecal microbiota in a free-living passerine bird, the great tit (Parus major). PLoS ONE. 2017. https://doi.org/10.1371/journal.pone.0179945.
Grond K, Sandercock BK, Jumpponen A, Zeglin LH. The avian gut microbiota: community, physiology and function in wild birds. J Avian Biol. 2018. https://doi.org/10.1111/jav.01788.
Capunitan DC, Johnson O, Terrill RS, Hird SM. Evolutionary signal in the gut microbiomes of 74 bird species from Equatorial Guinea. Mol Ecol. 2020;29:829–47. https://doi.org/10.1111/mec.15354.
García-Amado MA, Shin H, Sanz V, Lentino M, Martínez LM, Contreras M, et al. Comparison of gizzard and intestinal microbiota of wild neotropical birds. PLoS ONE. 2018;13:1–16.
Videvall E, Strandh M, Engelbrecht A, Cloete S, Cornwallis CK. Measuring the gut microbiome in birds: comparison of faecal and cloacal sampling. Mol Ecol Resour. 2018;18:424–34. https://doi.org/10.1111/1755-0998.12744.
van Veelen HPJ, Falcao Salles J, Tieleman BI. Multi-level comparisons of cloacal, skin, feather and nest-associated microbiota suggest considerable influence of horizontal acquisition on the microbiota assembly of sympatric woodlarks and skylarks. Microbiome. 2017;5:156.
Woodhams DC, Bletz MC, Becker CG, Bender HA, Buitrago-Rosas D, Diebboll H, et al. Host-associated microbiomes are predicted by immune system complexity and climate. Genome Biol. 2020;21:1–20.
Hall R. Southeast Asia’s changing palaeogeography. Blumea - Biodiversity, Evol Biogeogr Plants. 2009;54:148–61. https://doi.org/10.3767/000651909X475941.
Nugraha AMS, Hall R. Late Cenozoic palaeogeography of Sulawesi, Indonesia. Palaeogeogr Palaeoclimatol Palaeoecol. 2018;490:191–209.
Evans BJ, Andayani N, Melnick DJ, Supriatna J, Setiadi MI, Cannatella DC. Monkeys and toads define areas of endemism on Sulawesi. Evolution. 2006;57:1436.
Shekelle M, Meier R, Wahyu I, Ting N, Meier R, Wahyu I, et al. Molecular phylogenetics and chronometrics of tarsiidae based on 12S mtDNA haplotypes: evidence for miocene origins of crown tarsiers and numerous species within the Sulawesian Clade. Int J Primatol. 2010;31:1083–106.
Allen R, Damayanti CS, Frantz LAF, Leus K, Hulme-Beaman A, Gillemot S, et al. Synchronous diversification of Sulawesi’s iconic artiodactyls driven by recent geological events. Proc R Soc B Biol Sci. 2018;285:20172566.
Moss SJ, Wilson MEJ. Biogeographic implications of the Tertiary palaeogeographic evolution of Sulawesi and Borneo. Biogeogr Geol Evol SE Asia. 1987;133:63.
Esselstyn JA, Achmadi AS, Handika H, Giarla TC, Rowe KC. A new climbing shrew from Sulawesi highlights the tangled taxonomy of an endemic radiation. J Mammal. 2019;100:1713–25. https://doi.org/10.1093/jmammal/gyz077.
Wikramanayake E, Dinerstein E, Loucks CJ, Olson DM, Morrison J, Lamoreaux J, et al. Terrestrial ecoregions of the Indo-Pacific: a conservation assessment. Electr Green J. 2002. https://doi.org/10.5860/choice.40-0287.
Zhang W, Li N, Tang X, Liu N, Zhao W. Changes in intestinal microbiota across an altitudinal gradient in the lizard Phrynocephalus vlangalii. Ecol Evol. 2018;8:4695–703. https://doi.org/10.1002/ece3.4029.
Li H, Zhou R, Zhu J, Huang X, Qu J. Environmental filtering increases with elevation for the assembly of gut microbiota in wild pikas. Microb Biotechnol. 2019;12:976–92.
Bodawatta KH, Koane B, Maiah G, Sam K, Poulsen M, Jønsson KA. Species-specific but not phylosymbiotic gut microbiomes of New Guinean passerine birds are shaped by diet and flight-associated gut modifications. Proc R Soc B Biol Sci. 2021. https://doi.org/10.1098/rspb.2021.0446.
Herder EA, Spence AR, Tingley MW, Hird SM. Elevation correlates with significant changes in relative abundance in hummingbird fecal microbiota, but composition changes little. Front Ecol Evol. 2021;8:534.
Foster JT, Woodworth BL, Eggert LE, Hart PJ, Palmer D, Duffy DC, et al. Genetic structure and evolved malaria resistance in Hawaiian honeycreepers. Mol Ecol. 2007;16:4738–46. https://doi.org/10.1111/j.1365-294X.2007.03550.x.
Galen SC, Witt CC. Diverse avian malaria and other haemosporidian parasites in Andean house wrens: evidence for regional co-diversification by host-switching. J Avian Biol. 2014;45:374–86.
Moyle RG, Oliveros CH, Andersen MJ, Hosner PA, Benz BW, Manthey JD, et al. ARTICLE tectonic collision and uplift of Wallacea triggered the global songbird radiation. 2016. https://doi.org/10.1038/ncomms12709
Gelang M, Cibois A, Pasquet E, Olsson U, Alström P, Ericson PGP. Phylogeny of babblers (Aves, Passeriformes): major lineages, family limits and classification. Zool Scr. 2009;38:225–36. https://doi.org/10.1111/j.1463-6409.2008.00374.x.
Cai T, Cibois A, Alström P, Moyle RG, Kennedy JD, Shao S, et al. Near-complete phylogeny and taxonomic revision of the world’s babblers (Aves: Passeriformes). Mol Phylogenet Evol. 2019;2019(130):346–56. https://doi.org/10.1016/j.ympev.2018.10.010.
Koller M. Robustlmm: an R package for Robust estimation of linear Mixed-Effects models. J Stat Softw. 2016. https://doi.org/10.18637/jss.v075.i06.
Zhang Z, Xu D, Wang L, Hao J, Wang J, Zhou X, et al. Convergent evolution of rumen microbiomes in high-altitude mammals. Curr Biol. 2016;26:1873–9.
Montoya-Ciriaco N, Gómez-Acata S, Muñoz-Arenas LC, Dendooven L, Estrada-Torres A, Aníbal H, de la Vega-Pérez D, Navarro-Noya YE. Dietary effects on gut microbiota of the mesquite lizard Sceloporus grammicus (Wiegmann, 1828) across different altitudes. Microbiome. 2020. https://doi.org/10.1186/s40168-020-0783-6.
Alou M, Ndongo S, Frégère L, Labas N, Andrieu C, Richez M, et al. Taxonogenomic description of four new clostridium species isolated from human gut: `Clostridium amazonitimonense’, `Clostridium merdae’, `Clostridium massilidielmoense’ and `Clostridium nigeriense’. N Microbes N Infect. 2018;21:128–39. https://doi.org/10.1016/j.nmni.2017.11.003.
Song J, Choo YJ, Cho JC. Perlucidibaca piscinae gen. nov., sp. Nov., a freshwater bacterium belonging to the family Moraxellaceae. Int J Syst Evol Microbiol. 2008;58:97–102. https://doi.org/10.1099/ijs.0.65039-0.
Platas G, Morón R, González I, Collado J, Genilloud O, Peláez F, et al. Production of antibacterial activities by members of the family pseudonocardiaceae: influence of nutrients. World J Microbiol Biotechnol. 1998;14:521–7. https://doi.org/10.1023/A:1008874203344.
Thoemmes MS, Cove MV. Comparing the microbial communities of natural and supplemental nests of an endangered ecosystem engineer. bioRxiv. 2019. https://doi.org/10.1101/727966.
Uchimura Y, Wyss M, Brugiroux S, Limenitakis JP, Stecher B, McCoy KD, et al. Complete genome sequences of 12 species of stable defined moderately diverse mouse microbiota 2. Genome Announc. 2016;4:951–67. https://doi.org/10.1128/genomeA.00951-16.
Liang JQ, Li T, Nakatsu G, Chen YX, Yau TO, Chu E, et al. A novel faecal Lachnoclostridium marker for the non-invasive diagnosis of colorectal adenoma and cancer. Gut. 2019;69:1248–57. https://doi.org/10.1136/gutjnl-2019-318532.
Dominianni C, Wu J, Hayes RB, Ahn J. Comparison of methods for fecal microbiome biospecimen collection. 2014.https://doi.org/10.1186/1471-2180-14-103
Bodawatta KH, Puzejova K, Sam K, Poulsen M, Jønsson KA. Cloacal swabs and alcohol bird specimens are good proxies for compositional analyses of gut microbial communities of Great tits (Parus major). Anim Microbiome. 2020. https://doi.org/10.1186/s42523-020-00026-8.
Caporaso JG, Lauber CL, Walters WA, Berg-Lyons D, Huntley J, Fierer N, et al. Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. ISME J. 2012;6:1621–4. https://doi.org/10.1038/ismej.2012.8.
Bolyen E, Rideout JR, Dillon MR, Bokulich NA, Abnet CC, Al-Ghalith GA, et al. Author correction: reproducible, interactive, scalable and extensible microbiome data science using QIIME 2 (Nat Biotechnol 2019;37(8):(852–857). https://doi.org/10.1038/s41587-019-0209-9). Nat Biotechnol. 2019;37:1091. https://doi.org/10.1038/s41587-019-0252-6.
Callahan BJ, McMurdie PJ, Rosen MJ, Han AW, Johnson AJA, Holmes SP. DADA2: High-resolution sample inference from Illumina amplicon data. Nat Methods. 2016;13:581–3.
Katoh K. MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002;30:3059–66.
Price MN, Dehal PS, Arkin AP. FastTree 2 – approximately maximum-likelihood trees for large alignments. PLoS ONE. 2010;5:e9490. https://doi.org/10.1371/journal.pone.0009490.
Quast C, Pruesse E, Yilmaz P, Gerken J, Schweer T, Yarza P, et al. The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 2013. https://doi.org/10.1093/nar/gks1219.
Davis NM, Proctor DM, Holmes SP, Relman DA, Callahan BJ. Simple statistical identification and removal of contaminant sequences in marker-gene and metagenomics data. Microbiome. 2018. https://doi.org/10.1186/s40168-018-0605-2.
McMurdie PJ, Holmes S. Waste Not, Want not: why rarefying microbiome data is inadmissible. PLoS Comput Biol. 2014;10.
McMurdie PJ, Holmes S. Phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. PLoS ONE. 2013. https://doi.org/10.1371/journal.pone.0061217.
Lozupone C, Knight R. UniFrac: a new phylogenetic method for comparing microbial communities. Appl Environ Microbiol. 2005;71:8228–35. https://doi.org/10.1128/AEM.71.12.8228-8235.2005.
Paliy O, Shankar V. Application of multivariate statistical techniques in microbial ecology. Mol Ecol. 2016;25:1032–57.
Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. https://doi.org/10.1186/s13059-014-0550-8.
Acknowledgements
The authors would like to thank L. Bloch, Daniel, J. Childers, D. Wait, Pungki L, A. Riyanto, Apandi, Suparno, the Sulawesi Mountaineering club, and Mardin Sarkam, Safir, Mursalim for contributions to fieldwork, and Safir for keeping us alive and fed on the summits; L. Smith and C. Claypool-Wang for lab assistance; S. Holmes and the instructors of the WHOI STAMPS workshop for assistance with statistical analyses; Kementerian Riset, Teknologi dan Pendidikan Tinggi (KEMENRISTEKDIKTI RI), Balai Konservasi Sumber Daya Alam (BKSDA) Sulawesi Tengah, and Dinas Kehutanan Provinsi Sulawesi Tengah for providing research permits.
Funding
This research was funded by the National Science Foundation (DEB 1457845 awarded to JAM and RCKB and DEB 1457654/2039257 awarded to SP).
Author information
Authors and Affiliations
Contributions
RLJ collected samples, performed laboratory assays, analyzed the data, and wrote the manuscript. MI, TH, YD, and SA collected samples and edited the manuscript. ASA and JAM led the field expeditions and edited the manuscript. SLP conceived the study, edited the manuscript, and contributed resources. RCKB conceived the study, collected samples, edited the manuscript, and contributed resources. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Capture, handling, and sampling protocols were approved under permit numbers AMNHIACUC-20171020 and AUP-2016–04-8665–1, in accordance with Institutional Animal Care and Use Committees at the University of California, Berkeley and the American Museum of Natural History, respectively.
Consent for publication
All authors listed on the manuscript have consented to the submission of this manuscript for publication in Animal Microbiome, and each participated in its development as stated in Author Contributions.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Table S1.
Specimen data for samples used in this study. Fig. S1. Histogram of total elevation values for each specimen, and total values by mountain. The inconsistency in elevational sampling gradient at each mountain explains variation seen in mountain-based comparisons. Fig. S2. PCoAs of Unweighted and Weighted unifrac distances by mountain. Fig. S3. Robust linear model residual effect plot with elevation as the predictor effect. Tic marks along the x-axis represent elevations of individual data points. All plots were identical, regardless of whether mountain or host sex were set as conditional variables. Table S2. Slope and confidence intervals for robust mixed-effect models using elevation as an environmental gradient and mountain and sex as random effects. All three models were not significant as the confidence intervals do not intersect zero [48].
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Joakim, R.L., Irham, M., Haryoko, T. et al. Geography and elevation as drivers of cloacal microbiome assemblages of a passerine bird distributed across Sulawesi, Indonesia. anim microbiome 5, 4 (2023). https://doi.org/10.1186/s42523-022-00219-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s42523-022-00219-3