
Abstract
Background
Rahman syndrome (RMNS) is a rare genetic disorder inherited in an autosomal dominant manner caused by a de novo mutation in H1-4 gene. Since there are few cases described in the literature, the prevalence of the syndrome is unknown. RMNS should be suspected in individuals presenting mild to severe intellectual disability associated with behavioural problems.
Case presentation
A novel variant in the H1-4 gene: c.139G > C p.(Ala47Pro), classified as likely pathogenic, was identified in a patient with a phenotype compatible with RMNS. Clubfoot and obesity were described in our patient and should be considered in future reviews of the disease.
Conclusions
This case is added to the reduced number of publications previously reported regarding RMNS and contributes to understanding the genetic characteristics, clinical features and diagnosis of this syndrome.
Similar content being viewed by others

Background
Rahman syndrome (RMNS; MIM#617,537), also known as HIST1H1E syndrome, is a rare genetic disorder first described in 2017, when five subjects reported to have de novo heterozygous variants in H1-4 (formerly HIST1H1E) gene [1]. The prevalence of this syndrome is unknown, as only 47 affected individuals worldwide has been reported to date [2, 3].
RMNS is characterized by mild to severe intellectual disability (ID) associated with behavioural problems such as anxiety/phobias, obsessive behaviours, attention-deficit/hyperactivity disorder (ADHD), autistic spectrum disorder/traits (ASD), head banging, auditory hypersensitivity and aggression. It is also associated with variable somatic overgrowth, which manifests as increased birth length, height, weight and/or head circumference. Skeletal involvement is also present, including scoliosis, kyphoscoliosis and decreased bone mineral density. As the phenotype is highly variable, some individuals may also have other minor anomalies, including dysmorphic facial features, strabismus or camptodactyly. Other findings in some patients could be cryptorchidism (in males), hypotonia, seizures, abnormal dentition and hypothyroidism [1,2,3,4,5,6,7]. Abnormal brain magnetic resonance image (BMRI) due to corpus callosum abnormalities is also frequent [3].
The diagnosis of RMNS is established by conducting genetic studies in patients with these suggestive findings. The presence of loss-of-function mutations in H1-4 gene (MIM*142220), located on the short arm of chromosome 6 (6p22), has been defined as the molecular reasons causing the syndrome [1]. This gene encodes Histone H1.4, one of the epigenetic regulator genes’ family, which acts as a linker histone protein and it is responsible for higher-order chromatin structure [2]. H1.4 is involved in epigenetic regulation, especially during embryonic development [1, 7].
RMNS is inherited in an autosomal dominant manner, and it is caused by a de novo mutation. To date, 17 pathogenic variants in H1-4 have been reported in genetic databases associated with RMNS. All of them are germline frameshift mutations involving the C-terminal tail of H1.4 in exon 1 [7].
Differential diagnosis of RMNS comprises other ID syndromes associated with behavioural problems. These disorders present a high overlap of clinical features, making necessary molecular studies to stablish the definitive clinical diagnosis.
We present a case of a child, 12 years and 4 months old, with intelligence quotient (IQ) limit, ASD, ADHD and obesity in which a novel variant in H1-4 gene was identified throughout a whole-exome sequencing (WES) Trio study. This case is added to the reduced number of publications previously reported regarding RMNS and contributes to a better knowledge of genetic characteristics, diagnostic approach and clinical management of this syndrome.
Case presentation and genetic findings
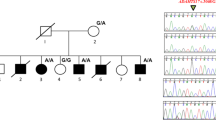
We reported a clinical case of a male 12 years and 4 months old, only child of a non-consanguineous couple. Family history of two first cousins (paternal branch) with diagnosis of ASD. No previous abortions. Well controlled pregnancy with echographic findings of clubfoot. Born at 38 gestational weeks, weight at birth 3,580 kg, size 52 cm., cp 34,5 cm., Apgar 9/10. Normal neonatal screening. No episodes of hospitalization in the neonatal period.
As a clinical history, he went twice (first time around 2 months of birth and the second one around 20 months of age) through surgical interventions in order to correct clubfoot. Since then, he was from time to time controlled by orthopaedists. At 3 years old, he was evaluated by neuropediatricians because of a language delay, motor clumsiness and ASD. Biochemical analyses and initially performed genetic studies (high-resolution karyotype and array comparative genomic hybridization (aCGH)) showed no significant results.
At the age of 10, he was again referred to the neuropediatricians due to poor school performance. After evaluation, he was diagnosed with ASD, ADHD and ID with low IQ (80). On physical examination, weight (60.6 kg., > P99, 2.4 SD), height (149 cm, P92, 1.43 SD) and head circumference (56 cm., P91, 1.34 SD) indicated obesity. The morphological examination revealed thick middle and distal third eyebrows sparsely populated, large eyes and a downward ophthalmic inclination, astigmatism, hypermetropia, ogival palate, anteverted nostrils, thin upper lip and a coffee-and-milk spot in the right lumbar area measuring 2 × 3. The body examination revealed genu valgum, clubfoot and flat feet. The cranial nerves, force, tone and osteotendinous reflexes were normal, but he presented motor clumsiness. Biochemical and hormonal analyses were normal, so hypothyroidism and hormonal overgrowth were discarded.
Motivated by these nonspecific dysmorphic features and the phenotype being indistinguishable from others ID syndromes, he was requested to be genetically studied, so a WES Trio assay was carried out on the patient and her biological parents. In the study, an heterozygous variant (c.139G > C; p.(Ala47Pro)) was identified de novo in the first exon of H1-4 gene (NM_005321.2). This variant had not been previously reported in genetic databases (gnomAD, OMIM, PubMed, ClinVar) associated with a specific phenotype. However, the presence of other mutations in this gene was associated with patients suffering from RMNS or autism [1, 2, 5, 7,8,9].
According to the American College of Medical Genetics and Genomics (ACMG) variant classification guidelines [10], this variant could have been classified as a variant of uncertain significance (VUS), since it meets the following criteria: it is a missense variant that exists at extremely low frequency in general population databases (one heterozygote in gnomAD, and absent in 1000G and ESP) (PM2), it is predicted to be deleterious in 6 of 8 in silico prediction systems (SIFT, Polyphen2_HDIV, Polyphen2_HVAR, MutationTaster, MutationAssessor, LRT, FATHMM and MetaSVM), and it is found in a highly conserved residue (PP3). However, the variant was absent in parents and there was not family history. Thus, applying de novo criteria in a proband within the phenotypic spectrum of H1-4-related disorders (PS2), the variant should be classified as likely pathogenic.
There are no similar mutations in H1-4 described associated with RMNS. Of the 17 pathogenic variants in H1-4 that been reported to date associated with RMNS in genetic databases, no one is similar to that described in this report. All of them are frameshift mutations in exon 1 (Table 1). Our patient’s clinical features were compatible with RMNS. Therefore, we can consider this novel variant c.139G > C; p.(Ala47Pro) in the H1-4 gene, as a cause of RMNS.
The patient was treated with methylphenidate hydrochloride with great response. In school, he attended a classroom adapted for ASD children. Nowadays, at age of 12, he presents the following physical features: weight: 81 kg (> P99, 2.62 SD); height: 156 cm (P94, 1.54 SD); head circumference: 57.5 cm (P98, 2 SD); and full pubertal development (Tanner scale-male = 5).
Discussion
RMNS is a rare genetic disorder caused by a de novo mutation in H1-4 gene, described to date in only 47 individuals [1,2,3]. Mutations in H1-4 are supposed to disrupt epigenetic regulation, especially during embryonic development [1, 7]. In this work, we presented the first missense variant in exon 1 of H1-4 gene associated with RMNS.
RMNS should be suspected in individuals presenting mild to severe ID, accompanied by behavioural issues as the most common clinical features. However, the phenotype is highly variable [1, 4]. These clinical features will presumably change as new cases are described.
The patient described above presented ID with low IQ, ASD with ADHD, and obesity as mainly clinical features. In the morphological examination, thick middle and distal third eyebrows sparsely populated, large eyes and a downward ophthalmic inclination, astigmatism, hypermetropia, ogival palate, anteverted nostrils, thin upper lip and a coffee-and-milk spot, were the most characteristic features.
While it has been previously suggested that RMNS belongs to the family of overgrowth-intellectual disability (OGID) syndromes [11], and affected patients could have variable somatic overgrowth manifested as increased birth length, height, weight, and/or head circumference, recent data suggest that most children with RMNS do not have tall stature or macrocephaly [2]. Our case allows the confirmation of obesity, and not somatic overgrowth, as a factor associated with the development of this syndrome.
Although the patient presented motor clumsiness, this condition could be caused by clubfoot, genu valgum and flat feet. Even if these clinical characteristics present in our patient had not been previously described, they should be considered in future reviews of the disease. Brain magnetic resonance imaging (MRI) is frequently abnormal in patients suffering from RMNS [2], unfortunately it could not be performed in our case.
Molecular genetic testing approaches depend on hospital protocols and laboratory possibilities. We discarded single-gene testing or multigene panel because the phenotype was indistinguishable from other ID syndromes. To date, more than 180 of such disorders have been identified [3] and the identification of a pathogenic variant in an altered gene is the only difference between all these clinical entities, so the genetic studies the only way to establish the ultimate diagnosis. aCGH and WES have been defined as the election molecular studies in autism and/or intellectual disability, since the genetic aetiology is recognized in ~ 25–35% of patients with ASD [9]. Therefore, we conducted CGH array as first option, amplifying the genetic study with WES.
Until now, there have been described 17 pathogenic variants in H1-4 associated with RMNS. All of them reported as germline frameshift mutations involving the C-terminal tail of H1-4 [7]. These findings highlight the importance of further H1-4 functional studies. It is hypothesized that protein-truncating variants escape nonsense-mediated RNA decay, resulting in abnormal proteins (which have a reduced net positive charge) [1, 7].
We identified a de novo heterozygous variant c.139G > C; p.(Ala47Pro) in the first exon of H1-4 gene. It had not been previously reported in genetic databases associated with RMNS, and there are no similar mutations in H1-4 described associated with RMNS (Table 1). Only another missense variant in the first exon of the gene, at two amino acids distance of which we reported, has been described. However, it is a somatic mutation associated with multiple myeloma (Table 1). According to the ACMG variant classification guidelines, the variant should be classified as likely pathogenic, with a CADD (Phred) value (15.85) predicting the deleterious effect of the variant [12].
Conclusion
Rahman syndrome is a rare genetic disorder that should be suspected in patients with the coexistence of intellectual disability, behavioural problems and variable somatic overgrowth. In addition, genetic studies are the only option for a differential diagnosis and confirmation of the syndrome.
The presence of a de novo variant, and clinical features being compatible with the OMIM phenotype of Rahman syndrome, allowed us to consider this variant as a cause of RMNS. This case is added to previous works and confirms that de novo variants are the main way of inheritance in RMNS.
Availability of data and materials
The datasets used and/or analysed during the current study are available from the corresponding author on reasonable request.
Abbreviations
- RMNS:
-
Rahman syndrome
- ID:
-
Intellectual disability
- ADHD:
-
Attention deficit hyperactivity disorder
- ASD:
-
Autism spectrum disorder
- BMRI:
-
Brain magnetic resonance image
- IQ:
-
Intelligence quotient
- WES:
-
Whole-exome sequencing
- cp:
-
Craniofacial perimeter
- aCGH:
-
array comparative genomic hybridization
- ACMG:
-
American college of medical genetics and genomics
- VUS:
-
Variant of uncertain significance
References
Tatton-Brown K, Loveday C, Yost S, Clarke M, Ramsay E, Zachariou A et al (2017) Mutations in epigenetic regulation genes are a major cause of overgrowth with intellectual disability. Am J Hum Genet 100(5):725–736
Burkardt DD, Zachariou A, Loveday C, Allen CL, Amor DJ, Ardissone A et al (2019) HIST1H1E heterozygous protein-truncating variants cause a recognizable syndrome with intellectual disability and distinctive facial gestalt: a study to clarify the HIST1H1E syndrome phenotype in 30 individuals. Am J Med Genet A 179(10):2049–2055
Burkardt D, Tatton-Brown K. HIST1H1E Syndrome. [Internet]. GeneReviews®. University of Washington, Seattle; 1993–2020. [cited 2021 Jan 10]. https://www.ncbi.nlm.nih.gov/books/NBK564966/
Kniffin C. Rahman Syndrome; RMNS. Online mendelian inheritance in man.
Duffney LJ, Valdez P, Tremblay MW, Cao X, Montgomery S, McConkie-Rosell A et al (2018) Epigenetics and autism spectrum disorder: a report of an autism case with mutation in H1 linker histone HIST1H1E and literature review. Am J Med Genet Part B, Neuropsychiatr Genet Off Publ Int Soc Psychiatr Genet 177(4):426–433
Takenouchi T, Uehara T, Kosaki K, Mizuno S (2018) Growth pattern of Rahman syndrome. Am J Med Genet A 176(3):712–714
Flex E, Martinelli S, Van Dijck A, Ciolfi A, Cecchetti S, Coluzzi E et al (2019) Aberrant function of the c-terminal tail of hist1h1e accelerates cellular senescence and causes premature aging. Am J Hum Genet 105(3):493–508
Ciolfi A, Aref-Eshghi E, Pizzi S, Pedace L, Miele E, Kerkhof J et al (2020) Frameshift mutations at the C-terminus of HIST1H1E result in a specific DNA hypomethylation signature. Clin Epigenetics 12(1):7
Wiśniowiecka-Kowalnik B, Nowakowska BA (2019) Genetics and epigenetics of autism spectrum disorder-current evidence in the field. J Appl Genet 60(1):37–47
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J et al (2015) Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 17(5):405–424
Tatton-Brown K, Seal S, Ruark E, Harmer J, Ramsay E, Del Vecchio DS et al (2014) Mutations in the DNA methyltransferase gene DNMT3A cause an overgrowth syndrome with intellectual disability. Nat Genet 46(4):385–388
Rentzsch P, Witten D, Cooper GM, Shendure J, Kircher M (2019) CADD: predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res 47(D1):D886–D894
National Center for Biotechnology Information (NCBI). U.S. National Library of Medicine. ClinVar database. 2020.
Leiden Open Variation Database (LOVD) v.3.0. [Internet]. [cited 2020 Dec 16]. Available from: https://www.lovd.nl/
Simple ClinVar [Internet]. [cited 2020 Dec 16]. Available from: http://simple-clinvar.broadinstitute.org/
Acknowledgements
To the patients and parents.
Funding
Not applicable.
Author information
Authors and Affiliations
Contributions
RGT and SIA drafted and critically reviewed the manuscript. ALL and JLPS was the principal physician in the patient’s case. ESR, NGR, MMG, ISN contributed to data collection, literature search and helped to draft the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Written informed consent was obtained from the patient for publication of case report and any accompanying images. A copy of the written consent is available for review by the Editor-in-Chief of this journal.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
González-Tarancón, R., Salvador-Rupérez, E., Goñi-Ros, N. et al. A novel likely pathogenic variant in the H1-4 gene c.139G > C p.(Ala47Pro) associated with Rahman syndrome: a clinical report. Egypt J Med Hum Genet 23, 50 (2022). https://doi.org/10.1186/s43042-022-00265-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s43042-022-00265-1