
Abstract
Mikulicz's disease (MD) has been included within the diagnosis of primary Sjögren's syndrome (SS), but it represents a unique condition involving persistent enlargement of the lacrimal and salivary glands characterized by few autoimmune reactions and good responsiveness to glucocorticoids, leading to the recovery of gland function. Mikulicz's disease was recently reported to be associated with elevated immunoglobulin G4 (IgG4) concentrations in the serum and prominent infiltration of plasmacytes expressing IgG4 into the lacrimal and salivary glands. The following features were used for diagnosis: (1) visual confirmation of symmetrical and persistent swelling in more than two lacrimal and major salivary glands; (2) prominent mononuclear cell infiltration of lacrimal and salivary glands; and (3) exclusion of other diseases that present with glandular swelling, such as sarcoidosis and lymphoproliferative disease. These features are not observed in most SS cases. The complications of MD include autoimmune pancreatitis, retroperitoneal fibrosis, tubulointerstitial nephritis, autoimmune hypophysitis, and Riedel's thyroiditis, all of which show IgG4 involvement in their pathogenesis. Mikulicz's disease thus differs from SS and may be a systemic IgG4-related plasmacytic disease.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction to Mikulicz's disease
Mikulicz's disease (MD) refers to idiopathic, bilateral, painless, and symmetrical swelling of the lacrimal, parotid, and submandibular glands. Because MD and Sögren's syndrome (SS) are histologically similar, MD has been considered as a subtype of SS. However, there are some clinical differences between MD and typical SS. In MD, the enlargement of lacrimal and salivary glands is persistent and secretory dysfunction is either not detectable or slight. Further, MD shows good responsiveness to steroids. Serologically, MD patients exhibit normal or hypergammaglobulinemia and normal or hypocomplementemia, but lacks anti-SS-A and anti-SS-B antibodies. Recently, it has also been confirmed that MD patients show elevated serum immunoglobulin G4 (IgG4) concentrations and infiltration of plasmacytes expressing IgG4 into the lacrimal and salivary glands. Thus, MD apparently differs from SS. Attention has also been paid to the pathological similarities between MD and autoimmune pancreatitis (AIP), particularly the role of IgG4, and new developments are expected to clarify the pathogenesis of both diseases. Here, we summarize the results of recent studies and provide an overview of MD.
Historical details and the concept of Mikulicz's disease
In 1888, Johann von Mikulicz-Radecki reported a case that exhibited bilateral, painless, and symmetrical swelling of the lacrimal, parotid, and submandibular glands.1 Napp later reported that these symptoms presented with leukemia, lymphoma, sarcoidosis, tuberculosis, and lues.2 To clarify this, in 1927 Schaffer linked cases with obvious diseases presenting these symptoms as Mikulicz's syndrome and idiopathic cases as MD.3 On the other hand, in 1933, Sjögren analyzed 19 cases with keratoconjunctivitis sicca, 2 of which displayed swelling of the major salivary glands.4 The concept of SS was established after this report. In 1953, Morgan and Castleman examined specimens from 18 cases diagnosed with MD. After finding histological similarities between MD and SS, they announced that most cases reported as MD could be considered as SS.5,6 Subsequently, MD has been considered as a subtype of SS, and case reports of MD have disappeared.
However, when we resurveyed the thesis by Morgan and Castleman,5 we identified several problems, such as the inclusion of the many cases that lack swelling of the lacrimal glands or only show swelling of one side of the glands. This led us to question whether these cases were actually MD. The histological characteristics that Morgan reported as being common among the 18 cases were as follows: (1) degeneration and disappearance of acini due to severe mononuclear cell infiltration, (2) proliferation of ductal epithelial cells and duct stenosis, (3) formation of myoepithelial islands, and (4) cystic dilatation of peripheral ducts. However, it is reported that these characteristics were observed not only in SS but also in persistent and destructive pathogenesis such as sialolithiasis in the glands.7 Thus, the exclusion of MD from SS, which Morgan defined is under increasing scrutiny, and numerous studies on the relationships between MD and SS have been undertaken in Japan.
Differences between Mikulicz's disease and Sjögren's syndrome
Our classification of Mikulicz's disease
We based our classification of MD on the following features: (1) visual confirmation of symmetrical and persistent swelling in more than two lacrimal and major salivary glands, (2) prominent mononuclear cell infiltration of lacrimal and salivary glands, and (3) exclusion of other diseases that present with glandular swelling such as sarcoidosis and lymphoproliferative disease.7 The aim of this classification is to establish disease independence and clarify the serological and histopathological differences between MD and SS.8 Analyses were performed in 16 MD patients (4 men and 12 women) who consulted doctors at Sapporo Medical University and its related facilities between April 1997 and October 2005; these cases were compared with primary SS cases. Another 16 patients in whom SS was diagnosed on the basis of the revised Japanese criteria were also studied.9 We defined cases as MD when they satisfied both our classification and the revised Japanese criteria for SS.
Clinical characteristics of Mikulicz's disease
Our MD patients were mainly middle-aged or elderly females. The average age was 58.6 years (range 25–73 years) with a sex ratio of approximately 3 : 1 in favor of females. However, male patients predominated when reports from other facilities were analyzed together with our data.10 The enlargement of the lacrimal and salivary glands in MD patients was found to be elastic, painless, and persistent (occurring for more than 3 months). Half of the MD patients did not exhibit keratoconjunctivitis sicca. With regard to salivary gland function, secretion by these glands in MD was normal or slightly decreased, and this improved with steroid treatment. On the other hand, the 16 patients with SS were all females in their forties and fifties. The sex ratio for SS is usually approximately 20 :1 (female:male).11 Our results did not contradict the general consensus. The lacrimal and salivary swelling in SS presented repeatedly and disappeared without treatment. The sicca symptoms were severe in SS. We prescribed glucocorticoids to SS patients for treating the swelling of the lateral submandibular gland, but the amount of saliva did not increase. We do not expect improvements in glandular secretion in advanced stages of SS since SS is an autoimmune disease that irreversibly damages glands.12 There were a few reports on the efficacy of a low dose of prednisolone on saliva production in patients with primary SS; however, in the thesis, it was not clear how long these cases had suffered from SS.12,13 It was then mentioned that in authoritative textbooks, systemic corticosteroids were used for treating only severe extraglandular diseases, including diffuse interstitial pneumonia, glomerulonephritis, vasculitis, and peripheral neuropathy.14–16 The impact of steroids on the natural course of SS is not well established. The clinical figures of MD are thus quite different from those of typical SS.
Diagnostic imaging of Mikulicz's disease
Computed tomography (CT) from the head to the neck in MD patients revealed severe enlargement of symmetrical lacrimal and submandibular or parotid glands. The size of the neck lymph nodes was less than 1cm. Ultrasonography of the enlarged glands revealed that the foci were irregular, hypoechoic, and multilocular, while the power Doppler method showed the presence of hypervascular masses. Sialography was normal, and the “apple-tree sign,” which is typical of SS, was not observed in MD.
Serological tests in Mikulicz's disease
Half of the MD patients exhibited hypergammaglobulinemia. A few patients had antinuclear antibodies in MD, and with the exception of one patient, did not have anti-SS-A or anti-SS-B antibodies. On the other hand, SS patients exhibited hypergammaglobulinemia and were largely positive for anti-SS-A (69%) and anti-SS-B (31%) antibodies.
Mikulicz's disease and IgG4
We focused on hypergammaglobulinemia, which was observed in MD. Serum IgG subclasses in each group of MD and SS (each 16 patients) were measured by nephelometry. Mean total IgG levels were 3226.9 mg/dl and 2398.1 mg/dl in MD and SS, respectively. There were no significant differences between the groups. Serum IgG subclasses in MD were 1256.4 mg/dl and 1111.0mg/dl for IgG1 and IgG4, respectively, and those in SS were 1624.9 mg/dl and 88.8mg/dl for IgG1 and IgG4 (P < 0.05, P < 0.0001), respectively. The IgG1/total IgG and IgG4/total IgG ratios were 41.5% and 28.6%, respectively, in MD and 73.0% (P < 0.0001) and 2.8% (P < 0.0001), respectively, in SS (Fig. 1). In healthy controls, these ratios for each IgG subclass were as follows: IgG1, 65%; IgG2, 25%; IgG3, 6%; and IgG4, 4%. IgG1 usually accounts for most of the total IgG.17 Generally, the amount of IgG4 does not vary with sex or age, and the quantity of IgG4 as well as the IgG4/total IgG ratio tends to remain constant.18 Our serological analysis revealed that all MD patients had elevated concentrations of IgG4, which has not been observed in any other connective tissue disease, including SS, systemic lupus erythematosus, rheumatoid arthritis, and polymyositis.19 Elevated serum IgG4 concentrations were very specific to MD patients.

Ratio of each IgG subclass/total IgG. The IgG4/total IgG ratio increased significantly in Mikulicz's disease, when compared with that in Sjögren syndrome
As described above, Morgan and Castleman reported that MD and SS were histopathologically identical.5,6 Upon hematoxylin-eosin staining, lacrimal or salivary gland specimens from MD showed severe mononuclear cell infiltration and lymphoid follicles around the ductal and acinar cells. We were not able to identify any difference between MD and SS using typical histopathological analyses. However, Tsubota et al. reported that the frequency of apoptosis of lacrimal gland cells was significantly lower in MD.20,21 We also observed this phenomenon in salivary glands by using the terminal deoxynucleotidyl transferase-mediated dUTP digoxigenin nick-end labeling (TUNEL) method.22 This phenomenon may be related to the reversibility of lacrimal and salivary functions by glucocorticoid treatment. The reasons apoptosis is not widely induced in MD are unclear, but abnormalities in the Fas/Fas ligand system in lymphocytes, acinar and ductal cells may be involved.22
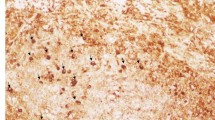
We then examined the lacrimal and salivary glands for anti-IgG4 antibody staining since elevated concentrations of IgG4 were detected in MD patients. The infiltration of numerous IgG4-positive plasmacytes near acinar and ductal cells and around lymphoid follicles was confirmed in MD;8 however, the specimens from SS patients showed no plasma cells with IgG4 (Fig. 2). Thus, this finding clearly differentiates between MD and SS (Table 1). It is possible that MD is a systemic IgG4-related plasmacytic disease because in MD, we also detected abundant IgG4-bearing plasma cells in the stomach, colon, and kidney as well as in lymphoid tissues such as the cervical lymph nodes and bone marrow.7,8

Specimens of labial salivary glands in patients with Mikulicz's disease (MD) and Sjögren's syndrome (anti-IgG4 monoclonal antibody staining, magnified 1 : 200). a Mikulicz's disease, b Sjögren's syndrome. The MD specimen exhibits abundant infiltration of plasmacytes with IgG4
Treatment of Mikulicz's disease
Mikulicz's disease is mainly treated by the administration of steroids. We initiate prednisolone at 30–40 mg/day against MD without organ failure. This leads to rapid improvement in glandular swelling as well as in lacrimal and salivary secretion. Prescription of glucocorticoids for 2 months led to an increase from 6.9mm/5min to 15.7mm/5min (P < 0.05) in Schirmer's test (16 eyes), which reflects improved lacrimal secretion, and an increase from 1.98 g/2min to 3.66 g/2min (P < 0.05) in Saxon's test (8 MD patients), which reflects improved salivary secretion.23 Glucocorticoid administration also improved hypergammaglobulinemia. A prescription for 2 months induced decreases in serum IgG levels (from 4270.0 mg/dl to 1440.1 mg/dl; P < 0.005) in 8 MD patients. The serum IgG1 levels decreased from 1632.9 mg/dl to 681.0 mg/dl (P < 0.005) and those of serum IgG4 decreased from 1556.4mg/dl to 234.7mg/dl (P < 0.005). However, the ratio of IgG1/total IgG increased from 38.3% to 46.8% (P < 0.01) while that of IgG4/total IgG decreased from 34.9% to 16.7% (P < 0.005).23 When steroids were discontinued, swelling of the lacrimal and salivary glands was observed, and the serum IgG4 levels increased. It is thus necessary to continue prednisolone administration at 5–10mg/day or combine it with an immunosuppressant such as azathioprine.
Controversial points regarding Mikulicz's disease
Prognosis and relationship with lymphoproliferative diseases
The long-term prognosis of MD is unknown. Among SS patients, the incidence of non-Hodgkin's lymphoma is 43.8-fold higher than the prevailing incidence in the general population.24 Sugai classified the severity of SS into three stages: stage I, having only sicca symptoms; stage II, having systemic organ involvement; and stage III, is complicated by lymphoma. Glandular and extraglandular SS may thus be considered as an early stage of lymphoma.25 There have also been reports on malignant transformation in MD.26 Recently, Ihrler and Harrison speculated that the first MD case reported by Mikulicz in 1892 was MALT lymphoma based on Mikulicz's sketch of the specimen.27 However, none of the present cases exhibited IgH gene rearrangement. It is necessary to continue the analysis of complicating lymphoma in MD as there is no data on its long-term prognosis.
Interpretation of asymmetrical or individual lacrimal or salivary gland swelling
In MD, enlargement of only the lacrimal or submandibular glands sometimes leads to other glandular swelling. Küttner's tumor is chronic sclerosing sialadenitis that presents with asymmetrical firm swelling of the submandibular glands.28 It is predominantly found in males. The histological characteristics of Küttner's tumor are severe fibrosis, atrophic acinus, and mononuclear cell infiltration. Kitagawa et al. analyzed 12 patients with Küttner's tumor and found that five cases were associated with sclerosing lesions in the extrasalivary glandular tissues. Immunohistochemically, the proportion of IgG4/IgG-positive plasmacytes was more than 45% in Küttner's tumor, while it was less than 5% in SS.29 We experienced a case of Küttner's tumor presenting with enlargement of only the right submandibular gland. We did not evaluate the gland by immunostaining but found elevated concentrations of serum IgG4 (33.1%). We also experienced a case of chronic dacryadenitis presenting with bilateral swelling of only the lacrimal glands. This case also exhibited elevated concentrations of serum IgG4 (26.0%) and prominent infiltration of IgG4-bearing plasma cells into the glands. It is difficult to assess whether Küttner's tumor and chronic dacryadenitis will develop in MD or whether these differ substantially from MD. We cannot resolve this problem at this point because the opinions of otorhinolaryngologists, ophthalmologists, and pathologists must be considered. However, we have the impression that some of the pathogenic features diagnosed in Küttner's tumor or chronic dacryadenitis are the same as those of MD. Thus, we must also analyze and resolve the relationships between MD and other diseases.
We regard symmetrical glandular swelling to be an indispensable characteristic for diagnosing MD. We use the name MD out of respect for the first reporter of this condition — Johann von Mikulicz-Radecki. When it is proved that Küttner's tumor and chronic dacryadenitis are identical to MD, we will call them by another common name. It is possible that bilateral glandular swelling occurs in SS; however, it usually disappears within a few weeks without any treatment. Persistence of the condition for a period of more than 3 months is very rare.
Relationships with other IgG4-related diseases, particularly autoimmune pancreatitis
Autoimmune pancreatitis is an IgG4-related disease. It also exhibits hypergammaglobulinemia, particularly with regard to IgG4, and severe infiltration of IgG4-positive plasmacytes into the pancreas.30,31 Autoimmune pancreatitis was proposed by Yoshida et al. in 199532 because it was presumed that autoimmunity was a part of the mechanism of idiopathic chronic pancreatitis. The diagnostic criteria for AIP were designed by the Japan Pancreas Society.33,34 Obstructive jaundice with narrowing at the lower parts of the common bile duct and impaired glucose tolerance due to pancreatic dysfunction are the clinical characteristics of AIP. Autoimmune pancreatitis patients are predominantly males with the male to female ratio reported to be 2–5 in Japan33–36 and 2 in Europe.37 The mean age at diagnosis is over 55 years.35,36 Autoimmune pancreatitis patients generally show serum hypergammaglobulinemia and increased IgG levels as well as the presence of antibodies such as antinuclear antibodies and rheumatoid factor. Computed tomography or ultrasonography typically reveals a diffusely enlarged pancreas, and endoscopic retrograde cholangiopancreatography (ERCP) images show segmental or diffuse narrowing of the main pancreatic duct. Steroid therapy is usually effective for improving pancreatic swelling and pancreatic endocrine and exocrine secretion.38,39 These characteristics are similar to those of MD, i.e., elevated concentrations of serum IgG4, infiltration of IgG4-bearing plasma cells into the glands, and recovery of secretion by glucocorticoid treatment. Furthermore, AIP is sometimes complicated by MD.10,40 Thus, MD and AIP are considered to be related. Nakamura et al. reported an AIP case in which IgG4-positive mononuclear cell infiltration was observed not only in the pancreas but also in the salivary gland and kidney.41 Other complications associated with MD and AIP include autoimmune hypophysitis,42 Riedel's thyroiditis, 43 interstitial pneumonia,44,45 sclerosing cholangitis,46,47 retroperitoneal fibrosis31,44,47,48 and interstitial tubular nephritis. 10,41,49 In our analysis, the concentrations of serum IgG4 tended to be proportional to the frequency of these complications. It is possible that serum IgG4 concentrations reflect disease activity in systemic IgG4-related plasmacytic diseases, which include MD and AIP.
Conclusion
We discussed the differences between MD and SS, such as the elevated concentrations of serum IgG4 and the prominent infiltration of IgG4-bearing plasma cells into lacrimal and salivary glands observed in MD. Rheumatologists should initially administer corticosteroids to MD patients in order to treat glandular swelling and improve exocrine and endocrine secretions. However, further analysis of the pathogenesis of MD at the cellular and molecular levels is required. Based on the evidence presented here, MD should be considered as a part of a larger concept, i.e., IgG4-related palasmacytic diseases.
References
J Mikulicz (1892) Über eine eigenartige symmetrishe Erkrankung der Thränen- und Mundspeicheldrüsen Beitr. z. Chir. Festschr. f. Theodor Billroth Stuttgart 610–30
O Napp (1907) ArticleTitleÜber die Beziehungen der Mikuliczschen Erkrankung Tuberkulose Stschr Augenheilk 17 513
AJ Schaffer AW Jacobsen (1927) ArticleTitleMikulicz's syndrome: a report of ten cases Am J Dis Child 34 327–46
H Sjögren (1933) ArticleTitleZur Kenntnis der Keratoconjunctivitis Sicca (Keratitis filiformis bei Hypofunktion der Tränendrüsen) Acta Ophthalmol Suppl II 1–151
WS Morgan B Castleman (1953) ArticleTitleA clinicopathologic study of “Mikulicz's disease.” Am J Pathol 29 471–503 Occurrence Handle1:STN:280:DyaG3s%2FmsFagtw%3D%3D Occurrence Handle13040489
WS Morgan (1954) ArticleTitleThe possible systemic nature of Mikulicz's disease and its relation to Sjögren's syndrome N Engl J Med 251 5–10 Occurrence Handle1:STN:280:DyaG2c%2Fps1Oqsg%3D%3D Occurrence Handle13176645 Occurrence Handle10.1056/NEJM195407012510102
M Yamamoto H Takahashi S Sugai K Imai (2005) ArticleTitleClinical and pathological characteristics of Mikulicz's disease (IgG4-related plasmacytic exocrinopathy) Autoimmunity Rev 4 195–200 10.1016/j.autrev.2004.10.005 Occurrence Handle10.1016/j.autrev.2004.10.005
M Yamamoto S Harada M Ohara C Suzuki Y Naishiro H Yamamoto et al. (2005) ArticleTitleClinical and pathological differences between Mikulicz's disease and Sjögren's syndrome Rheumatology (Oxf) 44 227–34 10.1093/rheumatology/keh447 Occurrence Handle10.1093/rheumatology/keh447 Occurrence Handle1:STN:280:DC%2BD2M%2FlsVSlsw%3D%3D
T Fujibayashi S Sugai N Miyasaka Y Hayashi K Tsubota (2004) ArticleTitleRevised Japanese criteria for Sjögren syndrome (1999): availability and validity Mod Rheumatol 14 425–34 10.1007/s10165-004-0338-x Occurrence Handle10.1007/s10165-004-0338-x
T Saeki A Saito T Hiura H Yamazaki I Emura M Ueno et al. (2006) ArticleTitleLymphoplasmacytic infiltration of multiple organs with immunoreactivity for IgG4:IgG4-related systemic disease Intern Med 45 163–7 10.2169/internalmedicine.45.1431 Occurrence Handle10.2169/internalmedicine.45.1431 Occurrence Handle16508232
CC Whitacre (2001) ArticleTitleSex differences in autoimmune disease Nat Immunol 2 777–80 10.1038/ni0901-777 Occurrence Handle10.1038/ni0901-777 Occurrence Handle1:CAS:528:DC%2BD3MXmsFeiu7w%3D Occurrence Handle11526384
PC Fox M Datiles JC Atkinson AA Macynski J Scott D Fletcher et al. (1993) ArticleTitlePrednisolene and piroxicam for treatment of primary Sjögren's syndrome Clin Exp Rheumatol 11 149–56 Occurrence Handle1:STN:280:DyaK3s3osVOmuw%3D%3D Occurrence Handle8508556
S Miyawaki S Nishiyama K Matoba (1999) ArticleTitleEfficacy of low dose prednisolone maintenance for saliva production and serological abnormalities in patients with primary Sjögren's syndrome Intern Med 38 938–43 Occurrence Handle10.2169/internalmedicine.38.938 Occurrence Handle1:CAS:528:DC%2BD3cXhtlyksbY%3D Occurrence Handle10628930
AG Tzioufas HM Moutsopoulos (1998) Sjögren's syndrome JH Klippel PA Dieppe (Eds) Rheumatology EditionNumber2nd ed Mosby London 7.32.1–7.32.12
HM Moutsopoulos (2001) Sjögren syndrome E Braunwald AS Fauci DL Kasper SL Hauser DL Longo JL Jameson (Eds) et al. Harrison's principles of internal medicine EditionNumber15th ed McGraw-Hill New York 1947–9
S Naguwa ME Gershwin (2004) Sjögren syndrome L Goldman D Ausiello (Eds) Cecil textbook of internal medicine EditionNumber22nd ed Saunders Philadelphia 1677–80
IM Roitt (1997) Antibodies IM Roitt (Eds) Roitt's essential immunology EditionNumber9th ed Blackwell Science London 43–62
RJ Lock DJ Unsworth (2003) ArticleTitleImmunoglobulins and immunoglobulin subclass in the elderly Ann Clin Biochem 40 143–8 10.1258/000456303763046067 Occurrence Handle10.1258/000456303763046067 Occurrence Handle1:CAS:528:DC%2BD3sXjsVWhtr8%3D Occurrence Handle12662402
M Yamamoto M Ohara C Suzuki Y Naishiro H Yamamoto H Takahahi et al. (2004) ArticleTitleElevated IgG4 concentrations in serum of patients with Mikulicz's disease Scand J Rheumatol 33 432–3 10.1080/03009740410006439 Occurrence Handle10.1080/03009740410006439 Occurrence Handle1:CAS:528:DC%2BD2MXmt1Gmsg%3D%3D Occurrence Handle15794205
K Tsubota H Fujita K Tsuzaka T Takeuchi (2000) ArticleTitleMikulicz's disease and Sjögren's syndrome Invest Ophthalmol Vis Sci 41 1666–73 Occurrence Handle1:STN:280:DC%2BD3czgt1ajtA%3D%3D Occurrence Handle10845583
K Tsubota H Fujita K Tadano N Onoda K Tsuzaka T Takeuchi (2002) ArticleTitleAbnormal expression and function of Fas ligand of lacrimal glands and peripheral blood in Sjögren's syndrome patients with enlarged exocrine glands Clin Exp Immunol 129 177–82 10.1046/j.1365-2249.2002.01882.x Occurrence Handle10.1046/j.1365-2249.2002.01882.x Occurrence Handle1:CAS:528:DC%2BD38XmtlWrtL0%3D Occurrence Handle12100038
Yamamoto M, Miyamoto N, Yamamoto H, Takahashi H, Imai K. Clinical and histopathological study of differences between Sjögren syndrome and Mikulicz's disease in frequency of apoptosis in salivary glands. The 8th International Symposium on Sjögren syndrome; 2002
M Yamamoto S Harada M Ohara C Suzuki Y Naishiro H Yamamoto et al. (2005) ArticleTitleBeneficial effects of steroid therapy for Mikulicz's disease Rheumatology (Oxf) 44 1322–3 10.1093/rheumatology/kei026 Occurrence Handle10.1093/rheumatology/kei026 Occurrence Handle1:STN:280:DC%2BD2MrgtFSisQ%3D%3D
SS Kassan TL Thomas HM Moutsopoulos R Hoover RP Kimberly DR Budman et al. (1978) ArticleTitleIncreased risk of lymphoma in sicca syndrome Ann Intern Med 89 888–92 Occurrence Handle1:STN:280:DyaE1M%2FmtVGrsg%3D%3D Occurrence Handle102228
S Sugai (1999) ArticleTitleSjögren syndrome (in Japanese) Nippon Naika Gakkai Zasshi 88 1896–903 Occurrence Handle1:CAS:528:DyaK1MXmslCntLY%3D Occurrence Handle10581778
E Lopez-Enriquez L Ceballos J Cintron E Gonzalez (1980) ArticleTitleMalignant transformation in Mikulicz's disease Bol Asoc Med P R 72 1–6 Occurrence Handle1:STN:280:DyaL3c7ltlKksg%3D%3D Occurrence Handle6928774
S Ihrler JD Harrison (2005) ArticleTitleMikulicz's disease and Mikulicz's syndrome: analysis of the original case report of 1892 in the light of current knowledge identifies a MALT lymphoma Oral Surg Oral Med Pathol Oral Radiol Endod 100 334–9 10.1016/j.tripleo.2005.01.016 Occurrence Handle10.1016/j.tripleo.2005.01.016
H Küttner (1896) ArticleTitleÜber entzündliche tumoren der submaxillar-speicheldrüse Beitr Klin Chir 15 815–34
S Kitagawa Y Zen K Harada M Sasaki Y Sato H Minato et al. (2005) ArticleTitleAbundant IgG4-positive plasma cell infiltration characterizes chronic sclerosing sialadenitis (Küttner's tumor) Am J Surg Pathol 29 783–91 10.1097/01.pas.0000164031.59940.fc Occurrence Handle10.1097/01.pas.0000164031.59940.fc Occurrence Handle15897744
H Hamano S Kawa A Horiuchi H Unno N Furuya T Akamatsu et al. (2001) ArticleTitleHigh serum IgG4 concentrations in patients with sclerosing pancreatitis N Engl J Med 344 732–8 10.1056/NEJM200103083441005 Occurrence Handle10.1056/NEJM200103083441005 Occurrence Handle1:CAS:528:DC%2BD3MXitFamt7k%3D Occurrence Handle11236777
H Hamano S Kawa Y Ochi H Unno N Shiba M Wajiki et al. (2002) ArticleTitleHydronephrosis associated with retroperitoneal fibrosis and sclerosing pancreatitis Lancet 359 1403–4 10.1016/S0140-6736(02)08359-9 Occurrence Handle10.1016/S0140-6736(02)08359-9 Occurrence Handle11978339
K Yoshida F Toki T Takeuchi S Watanabe K Shiratori N Hayashi (1995) ArticleTitleChronic pancreatitis caused by an autoimmune abnormality. Proposal of the concept of autoimmune pancreatitis Dig Dis Sci 40 1561–8 10.1007/BF02285209 Occurrence Handle10.1007/BF02285209 Occurrence Handle1:STN:280:DyaK2MzlsVCqtg%3D%3D Occurrence Handle7628283
K Okazaki K Uchida M Matsushita M Takaoka (2005) ArticleTitleAutoimmune pancreatitis Intern Med 44 1215–23 10.2169/internalmedicine.44.1215 Occurrence Handle10.2169/internalmedicine.44.1215 Occurrence Handle1:CAS:528:DC%2BD28XhslOiu7g%3D Occurrence Handle16415540
KP Kim MH Kim JC Kim SS Lee DW Seo SK Lee (2006) ArticleTitleDiagnostic criteria for autoimmune chronic pancreatitis revisited World J Gastroenterol 12 2487–96 Occurrence Handle16688792
K Okazaki (2005) ArticleTitleAutoimmune pancreatitis: etiology, pathogenesis, clinical findings and treatment. The Japanese experience JOP 6 IssueID1 Suppl 89–96 Occurrence Handle15650291
T Kamisawa M Yoshuike N Egawa H Nakajima K Tsuruta A Okamoto (2005) ArticleTitleTreating patients with autoimmune pancreatitis: results from a long-term follow-up study Pancreatology 5 234–40 10.1159/000085277 Occurrence Handle10.1159/000085277 Occurrence Handle15855821
G Zamboni J Luttges P Capelli L Frulloni G Cavallini P Pederzoli et al. (2004) ArticleTitleHistopathological features of diagnostic and clinical relevance in autoimmune pancreatitis: a study on 53 resection specimens and 9 biopsy specimens Virchows Arch 445 552–63 10.1007/s00428-004-1140-z Occurrence Handle10.1007/s00428-004-1140-z Occurrence Handle15517359
S Tanaka T Kobayashi K Nakanishi M Okubo T Murase M Hashimoto et al. (2000) ArticleTitleCorticosteroid-responsive diabetes mellitus associated with autoimmune pancreatitis Lancet 356 910–1 10.1016/S0140-6736(00)02684-2 Occurrence Handle10.1016/S0140-6736(00)02684-2 Occurrence Handle1:STN:280:DC%2BD3cvos1Cmsw%3D%3D Occurrence Handle11036899
S Tanaka T Kobayashi K Nakanishi M Okubo M Odawara T Murase et al. (2002) ArticleTitleCorticosteroid-responsive diabetes mellitus associated with autoimmune pancreatitis: pathological examinations of the endocrine and exocrine pancreas Ann N Y Acad Sci 958 152–9 Occurrence Handle12021096 Occurrence Handle10.1111/j.1749-6632.2002.tb02959.x
M Yamamoto M Ohara C Suzuki T Oka Y Naishiro H Yamamoto et al. (2005) ArticleTitleA case of Mikulicz's disease complicated by autoimmune pancreatitis, in which impaired glucose tolerance was improved by glucocorticoid treatment (in Japanese) Nihon Rinsho Meneki Gakkai Kaishi 28 349–56 Occurrence Handle16276049
H Nakamura H Wada T Origuchi A Kawakami N Taura T Aramaki et al. (2006) ArticleTitleA case of IgG4-related autoimmune disease with multiple organ involvement Scand J Rheumatol 35 69–71 10.1080/03009740500499484 Occurrence Handle10.1080/03009740500499484 Occurrence Handle1:STN:280:DC%2BD28%2FotFOluw%3D%3D Occurrence Handle16467047
M Yamamoto M Ohara C Suzuki Y Naishiro H Yamamoto H Takahashi et al. (2006) ArticleTitleCase of Mikulicz's disease (IgG4-related plasmacytic disease) complicated by autoimmune hypophysitis Scand J Rheumatol 35 410–1 10.1080/03009740600758110 Occurrence Handle10.1080/03009740600758110 Occurrence Handle1:STN:280:DC%2BD28nivFyhsw%3D%3D Occurrence Handle17062446
M Yamamoto C Suzuki Y Naishiro H Takahashi Y Shinomura K Imai (2006) ArticleTitleThe significance of disease-independence in Mikulicz’s disease — revival interests in Mikulicz’s disease (in Japanese) Nihon Rinsho Meneki Gakkai Kaishi 29 1–7 Occurrence Handle16505597
C Duvic J Desrame C Leveque G Nedelec (2004) ArticleTitleRetroperitoneal fibrosis, sclerosing pancreatitis and bronchiolitis obliterans with organizing pneumonia Nephrol Dial Transplant 19 2397–9 10.1093/ndt/gfh050 Occurrence Handle10.1093/ndt/gfh050 Occurrence Handle15299101 Occurrence Handle1:CAS:528:DC%2BD2MXht1entL3J
Y Zen S Kitagawa H Minato H Kurumaya K Katayanagi S Masuda et al. (2005) ArticleTitleIgG4-positive plasma cells in inflammatory pseudotumor (plasma cell granuloma) of the lung Hum Pathol 36 710–7 10.1016/j.humpath.2005.05.011 Occurrence Handle10.1016/j.humpath.2005.05.011 Occurrence Handle1:CAS:528:DC%2BD2MXntVOisrg%3D Occurrence Handle16084938
H Hamano S Kawa T Uehara Y Ochi M Takayama K Komatsu et al. (2005) ArticleTitleImmunoglobulin G4-related lymphoplasmacytic sclerosing cholangitis that mimics infiltrating hilar cholangiocarcinoma: part of a spectrum of autoimmune pancreatitis? Gastrointest Endosc 62 152–7 10.1016/S0016-5107(05)00561-4 Occurrence Handle10.1016/S0016-5107(05)00561-4 Occurrence Handle15990840
T Fukui K Okazaki H Yoshizawa S Ohashi H Tamaki K Kawasaki et al. (2005) ArticleTitleA case of autoimmune pancreatitis associated with sclerosing cholangitis, retroperitoneal fibrosis and Sjögren syndrome Pancreatology 5 86–91 10.1159/000084494 Occurrence Handle10.1159/000084494 Occurrence Handle15775703
H Ohara T Nakazawa H Sano T Ando T Okamoto H Takada et al. (2005) ArticleTitleSystemic extrapancreatic lesions associated with autoimmune pancreatitis Pancreas 31 232–7 10.1097/01.mpa.0000175178.85786.1d Occurrence Handle10.1097/01.mpa.0000175178.85786.1d Occurrence Handle16163054
M Yamamoto H Takahashi T Mizukoshi R Murakami T Tokuno Y Makiguchi et al. (2000) ArticleTitleA male case of Sjögren's syndrome presenting with generalized lymphadenopathy and swelling of both lacrimal and salivary glands which responded remarkably well to an administration of corticosteroids (in Japanese) Nihon Rinsho Meneki Gakkai Zasshi 23 22–9 Occurrence Handle1:STN:280:DC%2BD3c3jsVSltg%3D%3D
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article
Cite this article
Yamamoto, M., Takahashi, H., Ohara, M. et al. A new conceptualization for Mikulicz's disease as an IgG4-related plasmacytic disease. Mod Rheumatol 16, 335–340 (2006). https://doi.org/10.1007/s10165-006-0518-Y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10165-006-0518-Y