
Abstract
Fingolimod is an approved treatment for relapsing–remitting multiple sclerosis (MS), and its properties in different pathways have raised interest in therapy research for other neurodegenerative diseases. Fingolimod is an agonist of sphingosine-1-phosphate (S1P) receptors. Its main pharmacologic effect is immunomodulation by lymphocyte homing, thereby reducing the numbers of T and B cells in circulation. Because of the ubiquitous expression of S1P receptors, other effects have also been described. Here, we review preclinical experiments evaluating the effects of treatment with fingolimod in neurodegenerative diseases other than MS, such as Alzheimer’s disease or epilepsy. Fingolimod has shown neuroprotective effects in different animal models of neurodegenerative diseases, summarized here, correlating with increased brain-derived neurotrophic factor and improved disease phenotype (cognition and/or motor abilities). As expected, treatment also induced reductions in different neuroinflammatory markers because of not only inhibition of lymphocytes but also direct effects on astrocytes and microglia. Furthermore, fingolimod treatment exhibited additional effects for specific neurodegenerative disorders, such as reduction of amyloid-β production, and antiepileptogenic properties. The neuroprotective effects exerted by fingolimod in these preclinical studies are reviewed and support the translation of fingolimod into clinical trials as treatment in neurodegenerative diseases beyond neuroinflammatory conditions (MS).
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Drugs acting on several targets may be promising for the treatment of neurodegeneration. |
Fingolimod had positive effects in neurodegeneration. |
The reviewed data support the repurposing of fingolimod for other neurology diseases. |
1 Introduction
Fingolimod is a substrate of sphingosine kinases. It binds to sphingosine-1-phosphate (S1P) receptors in its phosphorylated state. Fingolimod is an agonist of several S1P receptor subtypes except for S1P2 receptors [1]. However, in lymphocytes, fingolimod acts as a functional antagonist by inducing internalization of S1P receptors after binding [2]. The main pharmacologic effect of fingolimod is immunomodulation by lymphocyte sequestration and reducing the numbers of T and B cells in circulation [3]. In addition, other effects of fingolimod treatment can be expected because of the ubiquitous expression of S1P receptors and their participation in several pathways.
S1P1 receptors are mainly expressed by immune, neural, and endothelial cells [4]. The main function of this receptor is regulation of immune cell trafficking, but it is also involved in angiogenesis and neurogenesis. The S1P1 receptor is involved in T cell migration across endothelial barriers. Fingolimod induces lymphocyte retention in lymph nodes by inhibiting the function of S1P1 receptors in lymphocyte egression [5]. In addition, the S1P1 receptor is also involved in diverse functions in the central nervous system (CNS) such as neurogenesis, astrocytic activation and proliferation, and communication between astrocytes and neurons and the blood–brain barrier (BBB) [4]. Furthermore, fingolimod can also bind to the S1P5 receptor, which is predominantly expressed in oligodendrocytes and brain endothelial cells [4, 6, 7]. S1P5 receptor activation protects adult oligodendrocytes from apoptosis and contributes to the maintenance of BBB integrity [7, 8]. In addition, fingolimod treatment regulates the biosynthesis of sphingolipids, which play important roles in neurodegenerative diseases [9].
Fingolimod can be administered orally and has high bio-availability and a half-life of 6–9 days [10]. It was first clinically tested in allograft rejection after kidney transplantation. However, its effects were not superior to those of standard therapy in these kidney-transplanted patients. On the other hand, because of its effects on inflammatory cells, fingolimod was also tested in different animal models of multiple sclerosis (MS), with promising results [11,12,13]. These preclinical animal studies encouraged clinical trials using fingolimod as a therapeutic agent in relapsing–remitting MS (RRMS). The 5-year FREEDOMS trial in 2010 was a placebo-controlled trial that showed a > 50% reduction in relapse rate compared with placebo treatment, together with a reduction of magnetic resonance (MR) lesions and brain volume loss [14]. Furthermore, the 6-month TRANSFORMS trial in 2010 compared the effects of fingolimod with those of the standard RRMS therapy, intramuscular interferon-β1a. Oral administration of fingolimod showed superior efficacy in terms of relapse rates and MR outcomes [15]. In 2010, the US FDA and the European Medicines Agency (EMA) approved fingolimod as a treatment for RRMS.
In the preclinical and clinical studies performed to date in MS, fingolimod has been proven to exert different protective effects, including inhibition of lymphocyte egress, inhibition of microglial and astroglial activation, reduction of neuronal death, restoration of lost synapsis, and reduction of dendritic spine loss, among others [14, 16,17,18,19]. In addition, preclinical and clinical data have shown that fingolimod promotes remyelination in optic neuritis [20, 21] and Krabbe disease [22]. The effects seen in MS include several potential targets shared by many neurodegenerative diseases, such as neuronal loss and neuroinflammation. The multiple pathways targeted by fingolimod suggest that this drug may be a promising therapy for neurodegenerative diseases such as Alzheimer’s disease (AD) and Parkinson’s disease (PD). Here, we summarize the reported effects of fingolimod treatment in animal models of neurodegenerative diseases that support the positive expectations of fingolimod repurposing for several conditions with neurodegeneration.
2 Search Strategy
All literature searches were conducted using the PubMed database. The search terms used were “neurodegenerative disease,” “Alzheimer’s disease,” “Parkinson’s disease,” “epilepsy,” “Huntington’s disease,” “Rett syndrome” OR “neuronal ceroid lipofuscinoses,” AND “fingolimod” or “FTY720.” Only publications written in English were considered. Additionally, other studies were identified from citations in review articles and reference lists from the search results. All references were screened to ensure relevant studies were included.
3 Neurodegenerative Diseases
3.1 Amyotrophic Lateral Sclerosis
Amyotrophic lateral sclerosis (ALS) is an incurable disease characterized by progressive degeneration of motor neurons in the spinal cord and the motor cortex, resulting in global muscle weakness, paralysis, and ultimately death due to respiratory dysfunction [23, 24]. Currently, the only FDA-approved treatments for this neurodegenerative disease (riluzole and edaravone) have limited effects [25]. Therefore, new therapeutic approaches to halt or slow down this pathology are needed. Patients with ALS and animal models both present an increased number of macrophages and T cells in the spinal cord as well as activated microglia [26,27,28]. Because of the effects of fingolimod on inflammatory cell infiltration and the inhibition of pro-inflammatory cytokines, it has been proposed as a potential therapy and has been tested both in an animal model and in patients with ALS [29, 30].
Potenza et al. [29] described the disease-modifying effects of fingolimod treatment in an animal model of ALS. They reported a modulation of neuroinflammation in treated SOD mice, along with improvements in phenotype and survival rate [29]. In addition, Berry et al. [30] tested the safety of fingolimod in patients with ALS in a phase IIa trial in 2017 to rule out potential side effects in this population, which differ from those described in patients with MS. The authors reported no serious adverse effects from the treatment and concluded that fingolimod administration in patients with ALS was safe and well-tolerated [30]. However, this study did not evaluate long-term safety.
3.2 Alzheimer’s Disease
AD is currently the most common cause of dementia and affects more than 30 million people worldwide [31]. AD is characterized by the accumulation of neurotoxic proteins (amyloid-β [Aβ] plaques and tau tangles), leading to neurodegeneration and irreversible cognitive decline [32, 33]. The precise mechanisms responsible for the accumulation of Aβ in the brain remain unknown, impeding the development of successful therapeutic strategies. Several molecular mechanisms have been identified as involved in AD pathogenesis, including Aβ overproduction and impaired Aβ clearance, dysregulated tau protein phosphorylation, altered glutamatergic neurotransmission, and astrocyte and microglia activation, starting before the first clinical signs appear [34,35,36]. Recently, it has been shown that ceramide-enriched exosomes are increased in the serum and brain tissue in mouse models of AD and that this may drive Aβ neurotoxicity [37, 38]. Fingolimod treatment downregulates the expression of ceramide metabolism genes [38]. Thus, targeting these mechanisms during the preclinical stage may reduce AD prevalence and/or delay its onset [39].
As described, fingolimod targets several mechanisms that have been proposed to play a key role in AD pathogenesis, such as neuroinflammation and neuronal loss. In addition, fingolimod has shown promising inhibitory effects on Aβ toxicity and synthesis in vitro. Fingolimod ameliorated Aβ neurotoxicity in neuronal cultures, reducing neuronal death, probably by increasing concentrations of brain-derived neurotrophic factor (BDNF) [40, 41]. Moreover, Takasugi et al. [42] reported that fingolimod decreased Aβ production in neuronal cells. Thus, in recent years, various groups have tested fingolimod in preclinical models of AD.
Fingolimod exerted neuroprotective effects in intracerebral Aβ injection models, reducing neuronal loss in parallel with a reduction in the activation of the Caspase-3 pathway and Aβ concentration. This neuroprotection was accompanied by attenuation of memory and learning deficits [43,44,45,46]. In the 5xFAD transgenic mouse model of AD, fingolimod also showed a reduction of Aβ load and attenuation of neuroinflammatory signs (astrocyte and microglia activation) and cognitive improvement [47, 48]. In addition, McManus et al. [49] reported attenuation of the effects of infection on Aβ accumulation and astrocyte activation after fingolimod treatment in APPswe/PS1dE9 mice.
In combination, the results of these in vitro and in vivo studies indicate that fingolimod reduced neuronal loss by inhibiting the Caspase-3 pathway, increasing the concentration of BDNF and enhancing pro-survival signaling. In addition, fingolimod decreased microglial and astroglial activation, reducing leukocyte infiltration and pro-inflammatory cytokine production. Furthermore, fingolimod treatment also reduced Aβ load in mice by inhibiting beta-secretase (BACE) and ceramide and, possibly, by modulating the transport of Aβ through the BBB [49, 50]. In summary, fingolimod treatment in these animal models suggests a potential benefit of this treatment in patients with mild cognitive impairment and patients with AD.
3.3 Parkinson’s Disease
PD is a neurological disease characterized by the loss of dopaminergic neurons in the substantia nigra and dysregulation of the extrapyramidal network caused by aggregation of misfolded toxic α-synuclein [51]. In addition, neuroinflammation occurs in areas with neurodegeneration in patients with PD, although it remains unclear whether activated astrocytes and microglia have beneficial or deleterious effects on the progress of PD [52]. Patients with PD experience tremor, postural instability, cognitive impairment, fatigue, and psychiatric symptoms, among other alterations [52]. Current therapies for PD effectively treat symptoms but do not exert disease-modifying effects that stop or delay disease progression. Potential pharmacological targets for disease modification in PD include neuroinflammation, mitochondrial dysfunction, calcium channel activity, LRRK2 kinase activity, and α-synuclein aggregation, as well as regeneration of lost dopamine neurons [52, 53]. Thus, targeting several mechanisms is thought to be more effective than targeting single targets. A successful disease-modifying treatment could transform PD into a chronic disease with modest nondisabling symptoms.
The anti-inflammatory and neuroprotective properties of fingolimod may be advantageous in the treatment of PD. Effects of fingolimod treatment has been studied in different animal models of PD. In the 6-hydroxydompamine (6-OHDA) intracerebral injection mouse model, animals receiving fingolimod showed attenuated neuroinflammation and motor deficits and reduced neuronal loss compared with vehicle-treated mice [54, 55]. Similarly, fingolimod administration was reported to attenuate motor dysfunction in the MPTP mouse model [56, 57]. In addition, mice receiving fingolimod showed increased dopamine release compared with vehicle-treated PD mice [55, 56]. Finally, genetic models of PD (A53T Tg and GM2 ± mice) showed an attenuated phenotype and increased BDNF expression when receiving fingolimod [58, 59]. However, Komnig et al. [60] reported no beneficial effects of fingolimod pretreatment in the MPTP mouse model.
Some animal studies suggest that fingolimod may improve PD therapy by targeting neuroinflammation. In addition, fingolimod treatment has shown effects on BDNF expression, which may recover lost dopaminergic neurons in early phases of the disease. However, the positive effects of this treatment in PD are unclear, as some animal studies have shown no improvement [60].
3.4 Epilepsy
Epilepsies are a group of neurological diseases characterized by the occurrence of unprovoked seizures [61]. Idiopathic epilepsy, i.e., unknown cause, represents about 40% of cases globally [62]. Currently, only symptomatic treatments are available. Furthermore, 30–40% of patients with epilepsy are partially or fully resistant to the therapy [63]. In recent years, research has focused on understanding the processes leading to epilepsy, i.e., epileptogenesis, to better understand the pathophysiology of this disorder and has identified neuroinflammation, neurotransmitter dysregulation, and neuronal loss, among others, as potential key actors in epilepsy pathogenesis [64,65,66,67,68].
Fingolimod has previously shown effects in neuroinflammation, neuronal loss, impaired neurogenesis, and BBB integrity. All these mechanisms have been described as altered during epileptogenesis and/or in patients with chronic epilepsy. Thus, fingolimod could exert anti-epileptogenic effects or, at least, slow the progression of epileptogenesis by altering key mechanisms in the epileptogenic process. Various groups have tested the potential of this drug in animal models of epileptogenesis. Gao et al. [69] showed that fingolimod treatment after status epilepticus reduced neuroinflammation, neuronal loss, and mossy fiber sprouting in the early phase of epileptogenesis in the rat lithium-pilocarpine model. More importantly, fingolimod also had an anti-epileptogenic effect, shown by a reduction of spontaneous convulsions in treated rats [69]. The same group reported an attenuation of the ABCB1 overexpression described in this model, which is thought to be partially responsible for the high rate of drug-resistant epilepsy [70]. Similarly, fingolimod showed antiepileptic effects during the chronic epileptic phase of the mouse kainate model. Pitsch et al. [71] reported decreased seizure frequency in animals receiving fingolimod compared with the vehicle group. In addition, treated animals showed reduced neuroinflammation and attenuated neuronal loss. Fingolimod also exerted beneficial effects in the pentylenetetrazol mouse-kindling model. Treated animals showed reduced epileptic activity when applied before pentylenetetrazol and in kindled animals, suggesting anticonvulsant effects from fingolimod. In addition, treatment reduced neuroinflammation signs and neuronal loss [72]. Finally, fingolimod exerted positive effects in WAG/Rij rats, an animal model of genetic epilepsy. Rats treated with fingolimod showed a transitory reduction of the frequency of absence seizures and long-lasting reductions in depression-like behavior [73].
In line with other animal models of neurodegenerative diseases, fingolimod showed neuroprotective and anti-inflammatory effects in animal models of epileptogenesis. Neuroinflammation and neurodegeneration are hypothesized to be key players in the development of epilepsy. The ameliorating effects of fingolimod shown in these studies support the further evaluation of this drug as anti-epileptogenic treatment, whether alone or in combination with other drugs, in patients at risk of developing epilepsy (e.g., status epilepticus, stroke, etc.). In addition, fingolimod showed anticonvulsant properties, which were not explored in other animal or clinical studies. This effect warrants the investigation of fingolimod treatment during chronic epilepsy.
3.5 Huntington’s Disease
Huntington’s disease (HD) is a genetic neurodegenerative disease that causes involuntary movements, cognitive impairment, and severe neuropsychological alterations. It is caused by a CAG repeat expansion in the huntingtin protein, leading to neuronal death and synaptic maladjustments, among other alterations [74, 75]. Patients with HD show neuronal loss, most prominently in the striatum and cerebral cortex, loss of neurotransmitters and glutamate, γ-amino-hydroxy-butyric acid (GABA) and dopamine receptors, reactive gliosis, metabolic alterations, and huntingtin aggregates [76]. Despite the identification of the genetic cause of the disease in 1993 [75], only symptomatic treatment of HD is available, and not all symptoms respond to the available treatments. Currently, no treatment can stop or reverse the disease [77].
Fingolimod effects such as neuroprotection and inhibition of neuroinflammation may be of interest for the therapy of HD [78]. In addition, research has shown promising effects of fingolimod treatment in animal models of HD. Di Pardo et al. [79] found that low-dose fingolimod (0.1 mg/kg) improved motor function, reduced brain atrophy, and extended survival of R6/2 mice, a mouse model of HD expressing a portion of the human HD gene [79]. In addition, fingolimod induced phosphorylation of huntingtin in vitro and reduction of huntingtin aggregates in vivo [79]. Miguez et al. [80] also showed beneficial effects on R6/1 mice after fingolimod treatment. Treated animals showed ameliorated memory deficits and neuroinflammation. Fingolimod also prevented the loss of dendritic spines and increased BDNF, which might lead to the cognitive improvement observed in these animals [80].
Preclinical research has shown that fingolimod treatment has effects on huntingtin aggregation, neuroinflammation, and neurogenesis. Based on the effects on HD pathology shown in in vitro and animal research, fingolimod may be an interesting option to be tested in patients with HD in combination with current therapies.
3.6 Rett Syndrome
Rett syndrome is a severe neurological disorder that affects mainly females. Patients with Rett syndrome experience compromised brain function, mental retardation, language and learning disabilities, and development regression [81]. The most prevalent cause is a mutation in the X-linked methyl-CpG binding protein 2 (Mecp2) gene [82]. Mecp2 controls the expression of a large set of genes, including BDNF. Dysfunctional Mecp2 decreases BDNF levels in patients and in animal models. Increasing BDNF levels, one of the described effects of fingolimod, may be an important approach to generate new therapies in Rett syndrome [83].
Deogracias et al. [84] showed beneficial effects with fingolimod treatment in Mecp2-deficient mice, an animal model of Rett syndrome. After showing increased BDNF secretion, together with reduced N-methyl-d-aspartate (NMDA)-induced neuronal loss and an increased network activity in vitro induced by fingolimod, the authors treated Mecp2-deficient mice with fingolimod and showed higher BDNF levels in the cortex and recovery of striatal weight in parallel with improvements in motor deficits [84]. A clinical trial is currently assessing the safety and efficacy of oral fingolimod (FTY720) in children aged > 6 years with Rett syndrome (FINGORETT; NCT02061137).
3.7 Neuronal Ceroid Lipofuscinoses
Neuronal ceroid lipofuscinoses (NCL) are rare genetic lysosomal storage diseases characterized by progressive axonal degeneration and neurodegeneration. Patients with NCL present diverse neurological manifestations, including seizures, intellectual and motor deterioration, sleep problems, visual loss, anxiety, and psychosis [85,86,87]. NCLs are grouped together because of the common presence of neuronal and extraneural autofluorescent pigment accumulations, although they have diverse genetic etiologies. NCLs were traditionally classified according to the age at onset: infantile, late infantile, juvenile, and adult NCL. However, NCLs are now classified according to the affected gene combined with the age at onset. To date, 14 genetically distinct NCLs have been identified (represented by the genes CLN1–14), with CLN3 being the most prevalent form [86, 88].
Groh et al. [89] showed beneficial effects from fingolimod treatment in an animal model of NCL. The authors described a reduction in neuroinflammatory signs, brain atrophy, and frequency of myoclonic jerks in CLN3 animals treated with fingolimod compared with the untreated group [89], which suggests that fingolimod treatment could alleviate or postpone some of the symptoms of these rare genetic diseases in patients.
4 Discussion
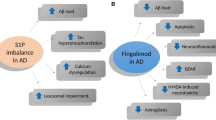
Fingolimod has shown promising effects in the treatment of RRMS in several clinical trials, showing better results than placebo and the traditional therapy, interferon-β1a [14, 15]. However, fingolimod has not shown such effects on primary progressive MS, in which neurodegenerative processes might have more importance than inflammatory infiltrates in the CNS [90]. Nevertheless, the diverse anti-inflammatory and neuroprotective effects exerted by fingolimod in different animal models suggest that this drug may also be an interesting medication for neurodegenerative/neuroinflammatory conditions other than RRMS. Besides the anti-inflammatory effects of fingolimod shown in RRMS, its regulation of sphingolipid biosynthesis may be of interest in the treatment of neurodegenerative diseases [9]. In recent years, preclinical research has focused on the therapeutic effects of fingolimod in other diseases in addition to MS. The studies summarized here have reported common neuroprotective and anti-inflammatory properties of fingolimod treatment in different preclinical models of neurological diseases (Table 1). In addition to the effects already described in MS models and patients with MS, these authors have shown additional mechanisms of fingolimod that may increase the therapeutic potential of this drug in specific diseases such as AD or epilepsy. Fingolimod has been shown to inhibit BACE and Aβ production and to have anticonvulsant properties, which are of interest in the treatment of these neurodegenerative disorders, respectively. In addition, fingolimod has modulatory effects on diverse pathways that are not targeted by current conventional therapies in the neurodegenerative diseases described (Fig. 1).

Summary of reported effects of fingolimod relevant to neurological disorders. Effects are specifically shown in different cell types, blood–brain barrier and brain deposits. These effects include different pathways involved in neuroinflammation and neurodegeneration as well as disease-specific effects. APP amyloid precursor protein, Aβ amyloid-beta, BACE beta-secretase, BBB blood–brain barrier, BDNF brain-derived neurotrophic factor, Cas-3 caspase-3, Htt huntingtin, α-syn alpha-synuclein
Several treatments that worked in animal models of neurodegenerative diseases have failed in their translation to human patients. Thus, it is important to test the effects described in preclinical models in clinical studies to confirm that the action of fingolimod is similar in human patients. The pathways targeted by fingolimod in the animal models used in these studies need to be confirmed as affected in human neuropathology and that fingolimod can induce similar changes in patients as seen in the animal models. In this regard, a significant advantage of fingolimod is that it is already approved by the FDA and the EMA as a treatment for MS. Thus, translation of the results from animal models to clinical assays potentially requires less time than would newly developed drugs, which need years before the first test in humans.
However, fingolimod has also shown side effects in human patients, such as bradycardia after the first dose, lymphopenia, increased liver transaminases, herpes virus infections, or hypertension [91]. Nevertheless, the symptoms derived from the different neurological diseases reviewed here are severe and, in most cases, incapacitating and potentially lethal. Therefore, the benefits described in these models are expected to outweigh the potential risks of the use of fingolimod in patients with neurodegeneration. In addition, fingolimod has already been shown to be safe in MS [91, 92] and ALS [30]. However, the target populations of some of the diseases included here differ significantly from those used in these studies as some include children or the elderly. Although no further side effects are expected in other neurodegenerative diseases, some of the side effects previously described may be of importance in specific patient populations. New S1P modulators such as ozanimod have shown higher receptor specificity and fewer side effects than fingolimod [93]. These new drugs might provide new opportunities in treatment of neurodegenerative diseases that focus on neuroprotective mechanisms instead of anti-inflammatory effects.
The positive effects offered by fingolimod in the treatment of patients with RRMS and the numerous pathways in which S1P receptors are involved have made this drug an attractive option for treating other neurodegenerative diseases. The animal experiments reviewed here showed positive effects on different neurological diseases together with additional alterations in pathways not identified in MS research. These promising results support the implementation of new clinical trials repurposing fingolimod to treat other neurodegenerative diseases in addition to MS.
References
Mandala S, Hajdu R, Bergstrom J, Quackenbush E, Xie J, Milligan J, et al. Alteration of lymphocyte trafficking by sphingosine-1-phosphate receptor agonists. Science. 2002;296(5566):346–9. https://doi.org/10.1126/science.1070238.
Matloubian M, Lo CG, Cinamon G, Lesneski MJ, Xu Y, Brinkmann V, et al. Lymphocyte egress from thymus and peripheral lymphoid organs is dependent on S1P receptor 1. Nature. 2004;427(6972):355–60. https://doi.org/10.1038/nature02284.
Chiba K, Yanagawa Y, Masubuchi Y, Kataoka H, Kawaguchi T, Ohtsuki M, et al. FTY720, a novel immunosuppressant, induces sequestration of circulating mature lymphocytes by acceleration of lymphocyte homing in rats. I. FTY720 selectively decreases the number of circulating mature lymphocytes by acceleration of lymphocyte homing. J Immunol. 1998;160(10):5037–44.
Brinkmann V. Sphingosine 1-phosphate receptors in health and disease: mechanistic insights from gene deletion studies and reverse pharmacology. Pharmacol Ther. 2007;115(1):84–105. https://doi.org/10.1016/j.pharmthera.2007.04.006.
Pham TH, Okada T, Matloubian M, Lo CG, Cyster JG. S1P1 receptor signaling overrides retention mediated by G alpha i-coupled receptors to promote T cell egress. Immunity. 2008;28(1):122–33. https://doi.org/10.1016/j.immuni.2007.11.017.
Jaillard C, Harrison S, Stankoff B, Aigrot MS, Calver AR, Duddy G, et al. Edg8/S1P5: an oligodendroglial receptor with dual function on process retraction and cell survival. J Neurosci. 2005;25(6):1459–69. https://doi.org/10.1523/jneurosci.4645-04.2005.
van Doorn R, Lopes Pinheiro MA, Kooij G, Lakeman K, van het Hof B, van der Pol SM, et al. Sphingosine 1-phosphate receptor 5 mediates the immune quiescence of the human brain endothelial barrier. J Neuroinflamm. 2012;9:133. https://doi.org/10.1186/1742-2094-9-133.
Miron VE, Jung CG, Kim HJ, Kennedy TE, Soliven B, Antel JP. FTY720 modulates human oligodendrocyte progenitor process extension and survival. Ann Neurol. 2008;63(1):61–71. https://doi.org/10.1002/ana.21227.
Czubowicz K, Jesko H, Wencel P, Lukiw WJ, Strosznajder RP. The role of ceramide and sphingosine-1-phosphate in Alzheimer’s disease and other neurodegenerative disorders. Mol Neurobiol. 2019;56(8):5436–55. https://doi.org/10.1007/s12035-018-1448-3.
Huwiler A, Zangemeister-Wittke U. The sphingosine 1-phosphate receptor modulator fingolimod as a therapeutic agent: recent findings and new perspectives. Pharmacol Ther. 2018;185:34–49. https://doi.org/10.1016/j.pharmthera.2017.11.001.
Brinkmann V, Lynch KR. FTY720: targeting G-protein-coupled receptors for sphingosine 1-phosphate in transplantation and autoimmunity. Curr Opin Immunol. 2002;14(5):569–75. https://doi.org/10.1016/s0952-7915(02)00374-6.
Kataoka H, Sugahara K, Shimano K, Teshima K, Koyama M, Fukunari A, et al. FTY720, sphingosine 1-phosphate receptor modulator, ameliorates experimental autoimmune encephalomyelitis by inhibition of T cell infiltration. Cell Mol Immunol. 2005;2(6):439–48.
Webb M, Tham CS, Lin FF, Lariosa-Willingham K, Yu N, Hale J, et al. Sphingosine 1-phosphate receptor agonists attenuate relapsing-remitting experimental autoimmune encephalitis in SJL mice. J Neuroimmunol. 2004;153(1–2):108–21. https://doi.org/10.1016/j.jneuroim.2004.04.015.
Kappos L, Radue EW, O’Connor P, Polman C, Hohlfeld R, Calabresi P, et al. A placebo-controlled trial of oral fingolimod in relapsing multiple sclerosis. N Engl J Med. 2010;362(5):387–401. https://doi.org/10.1056/nejmoa0909494.
Cohen JA, Barkhof F, Comi G, Hartung HP, Khatri BO, Montalban X, et al. Oral fingolimod or intramuscular interferon for relapsing multiple sclerosis. N Engl J Med. 2010;362(5):402–15. https://doi.org/10.1056/nejmoa0907839.
Rossi S, Lo Giudice T, De Chiara V, Musella A, Studer V, Motta C, et al. Oral fingolimod rescues the functional deficits of synapses in experimental autoimmune encephalomyelitis. Br J Pharmacol. 2012;165(4):861–9. https://doi.org/10.1111/j.1476-5381.2011.01579.x.
Smith PA, Schmid C, Zurbruegg S, Jivkov M, Doelemeyer A, Theil D, et al. Fingolimod inhibits brain atrophy and promotes brain-derived neurotrophic factor in an animal model of multiple sclerosis. J Neuroimmunol. 2018;318:103–13. https://doi.org/10.1016/j.jneuroim.2018.02.016.
Zhang J, Zhang ZG, Li Y, Ding X, Shang X, Lu M, et al. Fingolimod treatment promotes proliferation and differentiation of oligodendrocyte progenitor cells in mice with experimental autoimmune encephalomyelitis. Neurobiol Dis. 2015;76:57–66. https://doi.org/10.1016/j.nbd.2015.01.006.
Groves A, Kihara Y, Chun J. Fingolimod: direct CNS effects of sphingosine 1-phosphate (S1P) receptor modulation and implications in multiple sclerosis therapy. J Neurol Sci. 2013;328(1–2):9–18. https://doi.org/10.1016/j.jns.2013.02.011.
Albert C, Mikolajczak J, Liekfeld A, Piper SK, Scheel M, Zimmermann HG, et al. Fingolimod after a first unilateral episode of acute optic neuritis (MOVING)—preliminary results from a randomized, rater-blind, active-controlled, phase 2 trial. BMC Neurol. 2020;20(1):75. https://doi.org/10.1186/s12883-020-01645-z.
An X, Kezuka T, Usui Y, Matsunaga Y, Matsuda R, Yamakawa N, et al. Suppression of experimental autoimmune optic neuritis by the novel agent fingolimod. J Neuroophthalmol. 2013;33(2):143–8. https://doi.org/10.1097/wno.0b013e31828ea2fc.
Bechet S, O’Sullivan SA, Yssel J, Fagan SG, Dev KK. Fingolimod rescues demyelination in a mouse model of Krabbe’s disease. J Neurosci. 2020;40(15):3104–18. https://doi.org/10.1523/jneurosci.2346-19.2020.
Mitchell JD, Callagher P, Gardham J, Mitchell C, Dixon M, Addison-Jones R, et al. Timelines in the diagnostic evaluation of people with suspected amyotrophic lateral sclerosis (ALS)/motor neuron disease (MND)–a 20-year review: can we do better? Amyotroph Lateral Scler. 2010;11(6):537–41. https://doi.org/10.3109/17482968.2010.495158.
van den Bos MAJ, Geevasinga N, Higashihara M, Menon P, Vucic S. Pathophysiology and diagnosis of ALS: insights from advances in neurophysiological techniques. Int J Mol Sci. 2019. https://doi.org/10.3390/ijms20112818.
Lutz C. Mouse models of ALS: past, present and future. Brain Res. 2018;1693(Pt A):1–10. https://doi.org/10.1016/j.brainres.2018.03.024.
Alexianu ME, Kozovska M, Appel SH. Immune reactivity in a mouse model of familial ALS correlates with disease progression. Neurology. 2001;57(7):1282–9. https://doi.org/10.1212/wnl.57.7.1282.
Lewis CA, Manning J, Rossi F, Krieger C. The neuroinflammatory response in ALS: the roles of microglia and T cells. Neurol Res Int. 2012;2012:803701. https://doi.org/10.1155/2012/803701.
Troost D, van den Oord JJ, de Jong JM, Swaab DF. Lymphocytic infiltration in the spinal cord of patients with amyotrophic lateral sclerosis. Clin Neuropathol. 1989;8(6):289–94.
Potenza RL, De Simone R, Armida M, Mazziotti V, Pezzola A, Popoli P, et al. Fingolimod: a disease-modifier drug in a mouse model of amyotrophic lateral sclerosis. Neurotherapeutics. 2016;13(4):918–27. https://doi.org/10.1007/s13311-016-0462-2.
Berry JD, Paganoni S, Atassi N, Macklin EA, Goyal N, Rivner M, et al. Phase IIa trial of fingolimod for amyotrophic lateral sclerosis demonstrates acceptable acute safety and tolerability. Muscle Nerve. 2017;56(6):1077–84. https://doi.org/10.1002/mus.25733.
World Health Organization. AD report 2019. 2019. https://www.who.int/news-room/fact-sheets/detail/dementia. Accessed 17 Jun 2019.
Thinakaran G, Koo EH. Amyloid precursor protein trafficking, processing, and function. J Biol Chem. 2008;283(44):29615–9. https://doi.org/10.1074/jbc.r800019200.
Haass C, Selkoe DJ. Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer’s amyloid beta-peptide. Nat Rev Mol Cell Biol. 2007;8(2):101–12. https://doi.org/10.1038/nrm2101.
Pahnke J, Langer O, Krohn M. Alzheimer’s and ABC transporters–new opportunities for diagnostics and treatment. Neurobiol Dis. 2014;72(Pt A):54–60. https://doi.org/10.1016/j.nbd.2014.04.001.
Pahnke J, Walker LC, Scheffler K, Krohn M. Alzheimer’s disease and blood-brain barrier function—why have anti-beta-amyloid therapies failed to prevent dementia progression? Neurosci Biobehav Rev. 2009;33(7):1099–108. https://doi.org/10.1016/j.neubiorev.2009.05.006.
Lane CA, Hardy J, Schott JM. Alzheimer’s disease. Eur J Neurol. 2018;25(1):59–70. https://doi.org/10.1111/ene.13439.
Elsherbini A, Kirov AS, Dinkins MB, Wang G, Qin H, Zhu Z, et al. Association of Abeta with ceramide-enriched astrosomes mediates Abeta neurotoxicity. Acta Neuropathol Commun. 2020;8(1):60. https://doi.org/10.1186/s40478-020-00931-8.
Jesko H, Wencel PL, Wojtowicz S, Strosznajder J, Lukiw WJ, Strosznajder RP. Fingolimod affects transcription of genes encoding enzymes of ceramide metabolism in animal model of Alzheimer’s disease. Mol Neurobiol. 2020;57(6):2799–811. https://doi.org/10.1007/s12035-020-01908-3.
Briggs R, Kennelly SP, O’Neill D. Drug treatments in Alzheimer’s disease. Clin Med (Lond). 2016;16(3):247–53. https://doi.org/10.7861/clinmedicine.16-3-247.
Doi Y, Takeuchi H, Horiuchi H, Hanyu T, Kawanokuchi J, Jin S, et al. Fingolimod phosphate attenuates oligomeric amyloid beta-induced neurotoxicity via increased brain-derived neurotrophic factor expression in neurons. PLoS One. 2013;8(4):e61988. https://doi.org/10.1371/journal.pone.0061988.
Ruiz A, Joshi P, Mastrangelo R, Francolini M, Verderio C, Matteoli M. Testing Abeta toxicity on primary CNS cultures using drug-screening microfluidic chips. Lab Chip. 2014;14(15):2860–6. https://doi.org/10.1039/c4lc00174e.
Takasugi N, Sasaki T, Ebinuma I, Osawa S, Isshiki H, Takeo K, et al. FTY720/fingolimod, a sphingosine analogue, reduces amyloid-beta production in neurons. PLoS One. 2013;8(5):e64050. https://doi.org/10.1371/journal.pone.0064050.
Asle-Rousta M, Kolahdooz Z, Dargahi L, Ahmadiani A, Nasoohi S. Prominence of central sphingosine-1-phosphate receptor-1 in attenuating abeta-induced injury by fingolimod. J Mol Neurosci. 2014;54(4):698–703. https://doi.org/10.1007/s12031-014-0423-3.
Hemmati F, Dargahi L, Nasoohi S, Omidbakhsh R, Mohamed Z, Chik Z, et al. Neurorestorative effect of FTY720 in a rat model of Alzheimer’s disease: comparison with memantine. Behav Brain Res. 2013;252:415–21. https://doi.org/10.1016/j.bbr.2013.06.016.
Asle-Rousta M, Kolahdooz Z, Oryan S, Ahmadiani A, Dargahi L. FTY720 (fingolimod) attenuates beta-amyloid peptide (Abeta42)-induced impairment of spatial learning and memory in rats. J Mol Neurosci. 2013;50(3):524–32. https://doi.org/10.1007/s12031-013-9979-6.
Fukumoto K, Mizoguchi H, Takeuchi H, Horiuchi H, Kawanokuchi J, Jin S, et al. Fingolimod increases brain-derived neurotrophic factor levels and ameliorates amyloid beta-induced memory impairment. Behav Brain Res. 2014;268:88–93. https://doi.org/10.1016/j.bbr.2014.03.046.
Aytan N, Choi JK, Carreras I, Brinkmann V, Kowall NW, Jenkins BG, et al. Fingolimod modulates multiple neuroinflammatory markers in a mouse model of Alzheimer’s disease. Sci Rep. 2016;6:24939. https://doi.org/10.1038/srep24939.
Carreras I, Aytan N, Choi JK, Tognoni CM, Kowall NW, Jenkins BG, et al. Dual dose-dependent effects of fingolimod in a mouse model of Alzheimer’s disease. Sci Rep. 2019;9(1):10972. https://doi.org/10.1038/s41598-019-47287-1.
McManus RM, Finucane OM, Wilk MM, Mills KHG, Lynch MA. FTY720 attenuates infection-induced enhancement of abeta accumulation in APP/PS1 mice by modulating astrocytic activation. J Neuroimmune Pharmacol. 2017;12(4):670–81. https://doi.org/10.1007/s11481-017-9753-6.
van Doorn R, Nijland PG, Dekker N, Witte ME, Lopes-Pinheiro MA, van het Hof B, et al. Fingolimod attenuates ceramide-induced blood-brain barrier dysfunction in multiple sclerosis by targeting reactive astrocytes. Acta Neuropathol. 2012;124(3):397–410. https://doi.org/10.1007/s00401-012-1014-4.
Kinoshita K, Tada Y, Muroi Y, Unno T, Ishii T. Selective loss of dopaminergic neurons in the substantia nigra pars compacta after systemic administration of MPTP facilitates extinction learning. Life Sci. 2015;137:28–36. https://doi.org/10.1016/j.lfs.2015.07.017.
Kalia LV, Lang AE. Parkinson’s disease. Lancet. 2015;386(9996):896–912. https://doi.org/10.1016/s0140-6736(14)61393-3.
AlDakheel A, Kalia LV, Lang AE. Pathogenesis-targeted, disease-modifying therapies in Parkinson disease. Neurotherapeutics. 2014;11(1):6–23. https://doi.org/10.1007/s13311-013-0218-1.
Ren M, Han M, Wei X, Guo Y, Shi H, Zhang X, et al. FTY720 attenuates 6-OHDA-associated dopaminergic degeneration in cellular and mouse Parkinsonian models. Neurochem Res. 2017;42(2):686–96. https://doi.org/10.1007/s11064-016-2125-4.
Zhao P, Yang X, Yang L, Li M, Wood K, Liu Q, et al. Neuroprotective effects of fingolimod in mouse models of Parkinson’s disease. FASEB J. 2017;31(1):172–9. https://doi.org/10.1096/fj.201600751r.
Yao S, Li L, Sun X, Hua J, Zhang K, Hao L, et al. FTY720 inhibits MPP(+)-induced microglial activation by affecting NLRP3 inflammasome activation. J Neuroimmune Pharmacol. 2019;14(3):478–92. https://doi.org/10.1007/s11481-019-09843-4.
Motyl J, Przykaza L, Boguszewski PM, Kosson P, Strosznajder JB. Pramipexole and Fingolimod exert neuroprotection in a mouse model of Parkinson’s disease by activation of sphingosine kinase 1 and Akt kinase. Neuropharmacology. 2018;135:139–50. https://doi.org/10.1016/j.neuropharm.2018.02.023.
Vidal-Martinez G, Najera K, Miranda JD, Gil-Tommee C, Yang B, Vargas-Medrano J, et al. FTY720 improves behavior, increases brain derived neurotrophic factor levels and reduces alpha-synuclein pathology in parkinsonian GM2 ± mice. Neuroscience. 2019;411:1–10. https://doi.org/10.1016/j.neuroscience.2019.05.029.
Vidal-Martinez G, Vargas-Medrano J, Gil-Tommee C, Medina D, Garza NT, Yang B, et al. FTY720/Fingolimod Reduces Synucleinopathy and Improves Gut Motility in A53T Mice: CONTRIBUTIONS OF PRO-BRAIN-DERIVED NEUROTROPHIC FACTOR (PRO-BDNF) AND MATURE BDNF. J Biol Chem. 2016;291(39):20811–21. https://doi.org/10.1074/jbc.m116.744029.
Komnig D, Dagli TC, Habib P, Zeyen T, Schulz JB, Falkenburger BH. Fingolimod (FTY720) is not protective in the subacute MPTP mouse model of Parkinson’s disease and does not lead to a sustainable increase of brain-derived neurotrophic factor. J Neurochem. 2018;147(5):678–91. https://doi.org/10.1111/jnc.14575.
Fisher RS, Acevedo C, Arzimanoglou A, Bogacz A, Cross JH, Elger CE, et al. ILAE official report: a practical clinical definition of epilepsy. Epilepsia. 2014;55(4):475–82. https://doi.org/10.1111/epi.12550.
Shorvon SD. The etiologic classification of epilepsy. Epilepsia. 2011;52(6):1052–7. https://doi.org/10.1111/j.1528-1167.2011.03041.x.
Kobau R, Zahran H, Thurman DJ, Zack MM, Henry TR, Schachter SC, et al. Epilepsy surveillance among adults–19 states, Behavioral Risk Factor Surveillance System, 2005. MMWR Surveill Summ. 2008;57(6):1–20.
Brackhan M, Bascunana P, Postema JM, Ross TL, Bengel FM, Bankstahl M, et al. Serial quantitative TSPO-targeted PET reveals peak microglial activation up to 2 weeks after an epileptogenic brain insult. J Nucl Med. 2016;57(8):1302–8. https://doi.org/10.2967/jnumed.116.172494.
Brackhan M, Bascunana P, Ross TL, Bengel FM, Bankstahl JP, Bankstahl M. [(18) F]GE180 positron emission tomographic imaging indicates a potential double-hit insult in the intrahippocampal kainate mouse model of temporal lobe epilepsy. Epilepsia. 2018;59(3):617–26. https://doi.org/10.1111/epi.14009.
Bascunana P, Gendron T, Sander K, Jahreis I, Polyak A, Ross TL, et al. Ex vivo characterization of neuroinflammatory and neuroreceptor changes during epileptogenesis using candidate positron emission tomography biomarkers. Epilepsia. 2019;60(11):2325–33. https://doi.org/10.1111/epi.16353.
Dingledine R, Varvel NH, Dudek FE. When and how do seizures kill neurons, and is cell death relevant to epileptogenesis? Adv Exp Med Biol. 2014;813:109–22. https://doi.org/10.1007/978-94-017-8914-1_9.
Webster KM, Sun M, Crack P, O’Brien TJ, Shultz SR, Semple BD. Inflammation in epileptogenesis after traumatic brain injury. J Neuroinflamm. 2017;14(1):10. https://doi.org/10.1186/s12974-016-0786-1.
Gao F, Liu Y, Li X, Wang Y, Wei D, Jiang W. Fingolimod (FTY720) inhibits neuroinflammation and attenuates spontaneous convulsions in lithium-pilocarpine induced status epilepticus in rat model. Pharmacol Biochem Behav. 2012;103(2):187–96. https://doi.org/10.1016/j.pbb.2012.08.025.
Gao F, Gao Y, Meng F, Yang C, Fu J, Li Y. The sphingosine 1-phosphate analogue FTY720 alleviates seizure-induced overexpression of P-glycoprotein in rat hippocampus. Basic Clin Pharmacol Toxicol. 2018;123(1):14–20. https://doi.org/10.1111/bcpt.12973.
Pitsch J, Kuehn JC, Gnatkovsky V, Muller JA, van Loo KMJ, de Curtis M, et al. Anti-epileptogenic and anti-convulsive effects of fingolimod in experimental temporal lobe epilepsy. Mol Neurobiol. 2019;56(3):1825–40. https://doi.org/10.1007/s12035-018-1181-y.
Gol M, Ghorbanian D, Hassanzadeh S, Javan M, Mirnajafi-Zadeh J, Ghasemi-Kasman M. Fingolimod enhances myelin repair of hippocampus in pentylenetetrazol-induced kindling model. Eur J Pharm Sci. 2017;96:72–83. https://doi.org/10.1016/j.ejps.2016.09.016.
Leo A, Citraro R, Amodio N, De Sarro C, Gallo Cantafio ME, Constanti A, et al. Fingolimod exerts only temporary antiepileptogenic effects but longer-lasting positive effects on behavior in the WAG/Rij rat absence epilepsy model. Neurotherapeutics. 2017;14(4):1134–47. https://doi.org/10.1007/s13311-017-0550-y.
Novak MJ, Tabrizi SJ. Huntington’s disease: clinical presentation and treatment. Int Rev Neurobiol. 2011;98:297–323. https://doi.org/10.1016/b978-0-12-381328-2.00013-4.
Group HsDCR. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. The Huntington’s Disease Collaborative Research Group. Cell. 1993;72(6):971–83. https://doi.org/10.1016/0092-8674(93)90585-e.
Waldvogel HJ, Kim EH, Tippett LJ, Vonsattel J-PG, Faull RL. The neuropathology of Huntington’s disease. In: Nguyen HHP, Cenci MA, editors. Behavioral neurobiology of Huntington’s disease and Parkinson’s disease. Heidelberg: Springer; 2015. p. 33–80.
Wyant KJ, Ridder AJ, Dayalu P. Huntington’s disease-update on treatments. Curr Neurol Neurosci Rep. 2017;17(4):33. https://doi.org/10.1007/s11910-017-0739-9.
Wood H. Neurodegenerative disease: Could fingolimod provide cognitive benefits in patients with Huntington disease? Nat Rev. 2015;11(8):426. https://doi.org/10.1038/nrneurol.2015.117.
Di Pardo A, Amico E, Favellato M, Castrataro R, Fucile S, Squitieri F, et al. FTY720 (fingolimod) is a neuroprotective and disease-modifying agent in cellular and mouse models of Huntington disease. Hum Mol Genet. 2014;23(9):2251–65. https://doi.org/10.1093/hmg/ddt615.
Miguez A, Garcia-Diaz Barriga G, Brito V, Straccia M, Giralt A, Gines S, et al. Fingolimod (FTY720) enhances hippocampal synaptic plasticity and memory in Huntington’s disease by preventing p75NTR up-regulation and astrocyte-mediated inflammation. Hum Mol Genet. 2015;24(17):4958–70. https://doi.org/10.1093/hmg/ddv218.
Liyanage VR, Rastegar M. Rett syndrome and MeCP2. Neuromol Med. 2014;16(2):231–64. https://doi.org/10.1007/s12017-014-8295-9.
Bienvenu T, Carrie A, de Roux N, Vinet MC, Jonveaux P, Couvert P, et al. MECP2 mutations account for most cases of typical forms of Rett syndrome. Hum Mol Genet. 2000;9(9):1377–84. https://doi.org/10.1093/hmg/9.9.1377.
Li W, Pozzo-Miller L. BDNF deregulation in Rett syndrome. Neuropharmacology. 2014;76(Pt C):737–46. https://doi.org/10.1016/j.neuropharm.2013.03.024.
Deogracias R, Yazdani M, Dekkers MP, Guy J, Ionescu MC, Vogt KE, et al. Fingolimod, a sphingosine-1 phosphate receptor modulator, increases BDNF levels and improves symptoms of a mouse model of Rett syndrome. Proc Natl Acad Sci USA. 2012;109(35):14230–5. https://doi.org/10.1073/pnas.1206093109.
Nita DA, Mole SE, Minassian BA. Neuronal ceroid lipofuscinoses. Epileptic Disord. 2016;18(S2):73–88. https://doi.org/10.1684/epd.2016.0844.
Mole SE, Williams RE. Neuronal Ceroid-Lipofuscinoses. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Stephens K et al, editors. GeneReviews((R)). Seattle; 1993.
Cooper JD, Tarczyluk MA, Nelvagal HR. Towards a new understanding of NCL pathogenesis. Biochim Biophys Acta. 2015;1852(10 Pt B):2256–61. https://doi.org/10.1016/j.bbadis.2015.05.014.
Chabrol B, Caillaud C, Minassian B. Neuronal ceroid lipofuscinoses. Handb Clin Neurol. 2013;113:1701–6. https://doi.org/10.1016/b978-0-444-59565-2.00038-1.
Groh J, Berve K, Martini R. Fingolimod and teriflunomide attenuate neurodegeneration in mouse models of neuronal ceroid lipofuscinosis. Mol Ther. 2017;25(8):1889–99. https://doi.org/10.1016/j.ymthe.2017.04.021.
Lublin F, Miller DH, Freedman MS, Cree BAC, Wolinsky JS, Weiner H, et al. Oral fingolimod in primary progressive multiple sclerosis (INFORMS): a phase 3, randomised, double-blind, placebo-controlled trial. Lancet. 2016;387(10023):1075–84. https://doi.org/10.1016/s0140-6736(15)01314-8.
Calabresi PA, Radue EW, Goodin D, Jeffery D, Rammohan KW, Reder AT, et al. Safety and efficacy of fingolimod in patients with relapsing-remitting multiple sclerosis (FREEDOMS II): a double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Neurol. 2014;13(6):545–56. https://doi.org/10.1016/s1474-4422(14)70049-3.
Kappos L, O’Connor P, Radue EW, Polman C, Hohlfeld R, Selmaj K, et al. Long-term effects of fingolimod in multiple sclerosis: the randomized FREEDOMS extension trial. Neurology. 2015;84(15):1582–91. https://doi.org/10.1212/wnl.0000000000001462.
Grassi S, Mauri L, Prioni S, Cabitta L, Sonnino S, Prinetti A, et al. Sphingosine 1-phosphate receptors and metabolic enzymes as druggable targets for brain diseases. Front Pharmacol. 2019;10:807. https://doi.org/10.3389/fphar.2019.00807.
Acknowledgements
The work of JP was supported by the following grants: Deutsche Forschungsgemeinschaft, Germany (DFG PA930/12); Ministerium für Wirtschaft und Wissenschaft Sachen-Anhalt, Germany (ZS/2016/05/78617); Leibniz Gemeinschaft, Germany (SAW-2015-IPB-2); Latvian Council of Science, Latvia (lzp-2018/1-0275); Nasjonalforeningen (16154), HelseSØ, Norway (2016062, 2019054, 2019055); Norges forskningsrådet, Norway (251290, 260786); and the European Commission (643417).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of Interest
Pablo Bascuñana, Luisa Möhle, Mirjam Brackhan, and Jens Pahnke have no conflicts of interest that are directly relevant to the content of this review/study.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Bascuñana, P., Möhle, L., Brackhan, M. et al. Fingolimod as a Treatment in Neurologic Disorders Beyond Multiple Sclerosis. Drugs R D 20, 197–207 (2020). https://doi.org/10.1007/s40268-020-00316-1
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40268-020-00316-1