
Abstract
Immune thrombocytopenia (ITP) is a hematologic disorder characterized by a low platelet count, leading to an increased risk of bleeding. This review provides an overview of the historical milestones, pathophysiology, and treatment advances in ITP. Historical perspectives trace back to Avicenna's description in the eleventh century to pivotal Harrington-Hollingsworth experiment in 1950, laid the groundwork for understanding the immune-mediated platelet destruction intrinsic to ITP. Subsequent developments in investigation techniques, such as platelet-survival studies and antibody assays, contributed to diagnostic advancements. Treatment modalities have evolved significantly from the traditional approach of splenectomy to the use of corticosteroids, immunosuppressants, and novel targeted therapies. The efficacy and safety profiles of these treatments have been refined through clinical trials and consensus guidelines. Ongoing research continues to unravel the genetic and molecular underpinnings of ITP, offering insights into disease mechanisms and potential therapeutic targets. Emerging therapies, including immunomodulatory agents, hold promise for improving outcomes and quality of life for patients with ITP. In conclusion, this review provides a synthesis of historical insights, pathophysiological mechanisms, and treatment strategies in ITP. By elucidating the complex interplay between immune dysregulation and platelet destruction, this knowledge serves as a foundation for advancing the diagnosis, management, and future therapeutic innovations in ITP.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Development of knowledge
In 1025, Avicenna (980–1037) first described a patient with chronic purpura clinically resembling immune thrombocytopenia in the Canon of Medicine [1]. However, it took more than nine centuries until we knew exactly what the disease really is. In 1735, Paul Gottlieb Werlhof (1699–1767) reported the case of a 16-year-old girl with cutaneous and mucosal bleeding that occurred after an infectious disease. This condition was named “Werlhof’s disease” [2]. In the nineteenth century, some physicians also noted cases where individuals exhibited signs of easy bruising, petechiae (small red or purple spots on the skin), and bleeding tendencies. However, the understanding of the cellular pathophysiology was limited during that period.
In 1865, Max Schultze (1825–1874) was the first person who describe platelets as “little spherules of different sizes” which “are 6–8 times smaller than the red cells” [3]. It’s worth noting that he examined the living, mobile, and unstained blood cells. Indeed, the fact that platelets are colorless explains why most early studies, predating the discovery of staining technique, recognized only red and white cells. While he discovered what would later be known as platelets, the function of platelets was not discovered until 1882 when Giulio Bizzozzero described that platelets play an important role in hemostasis [4, 5].
The discovery of staining blood cells with aniline dyes by Ehrlich in 1877 led to explosive growth in blood morphology study. In 1896, Müller described small granules with a “very active molecular movement” and “unable to stain with osmic acid”. The significance of this new element was unknown at the time, so he termed them “haemoconia” or “blood dust” [6]. This term was famously used and was extensively studied over the following decades, with later observers identifying them as fragments of cells or free cell granules. It was not until the Romanowsky stains, discovered in 1891, and azure dyes began to be wildly used, that it became possible to separate the red-to-violet azurophilic granules of the platelet from other granular material in and out of cells [7]. In 1906, James Homer Wright (1869–1928)—who invented the Wright’s stain- proposed in Boston Medical and Surgical Journal, later becoming the New England Journal of Medicine, that megakaryocytes in the bone marrow produce platelets [8].
In the early twentieth century, ITP was first known by physicians as “Idiopathic Thrombocytopenic Purpura”. “Idiopathic” indicated that the cause was unknown, and the disease was diagnosed by ruling out other causes. “Thrombocytopenic” referred to the low platelet count, and “Purpura” was characteristic of patients who usually had skin discoloration caused by bleeding [9].
In 1916, Paul Kaznelson (1898–1959), then a medical student in Prague, proposed that platelets, similarly to red blood cells, were excessively destructed in the spleen. He encouraged his tutor to perform the first splenectomy in an ITP patient [10]. Remarkably, all three ITP patients recovered from bleeding [11]. Splenectomy later became the treatment of choice in refractory ITP for more than 30 years until the pathophysiology of ITP was discovered [9].
Harrington–Hollingsworth experiment
The Harrington experiment in 1950 marked a major change in understanding ITP. William J. Harrington (1924–1992) was a hematology fellow in St. Louis at that time when he discovered the pathophysiology of ITP by conducting an experiment on himself. His interest in ITP went back to the time when he was a medical student at Tufts Medical School taking care of a seventeen-year-old girl who later died from bleeding after splenectomy, the only treatment at that time [12]. Together with his colleague, James W. Hollingsworth, whose main interest was in the study of antibodies, they proposed that there is a “factor” in the blood that destroys platelets. This idea occurred after Harrington read a report that newborns from mothers with ITP had very transient thrombocytopenia. To test this hypothesis, he received 500 ml of blood from an ITP patient. (Of course, it was a different era when no committee approval had to be obtained and consent forms were needed.) Within a few hours, his platelets dropped and he experienced a major seizure. His platelet count recovered after four days. Moreover, to further test his hypothesis, he underwent bone marrow examination, which revealed unchanged megakaryocytes, indicating that this “thrombocytopenia factor”, later known to be IgG antibodies [13], affected only the circulating platelets [14].
To underscore the significance of the result, it’s important to note the prevailing theories prior to the groundbreaking experiment. Hematologists had long speculated that the pathophysiology of ITP mainly was abnormal bone marrow production. In 1915, Frank put forward the hypothesis that thrombocytopenia resulted from a toxin depressing megakaryocyte [15]. This idea gained traction among leading hematologists. Minot studied the bone marrow of severe ITP patients, and his findings were published in the Archives of Internal Medicine, now known as JAMA Internal Medicine, in 1917. His work suggested that while megakaryocytes were abundant, they couldn’t “allow platelets to be cut off from them in a normal fashion” [16]. William Dameshek (1900–1969) bolstered this concept through numerous papers, proposing that while megakaryocyte increased, platelet production significantly decreased. He also suggested that the spleen might produce either an abnormal substance or an excessive amount of an inhibitor of platelet production, citing evidence of increased platelet counts post-splenectomy [17, 18]. The experiment in the late twentieth century using platelet survival study proved, however, they weren’t entirely wrong. It turns out that their hypothesis regarding decreased platelet production holds some substantial truth.
The Harrington experiment significantly changed three important facts during the second half of the twentieth century. First, thrombocytopenia is the result of platelet destruction rather than a toxin depressing megakaryocytes, as previously hypothesized by Frank in 1915. Second, an antiplatelet factor with characteristics of antibodies is present in the plasma of the patients. Third, the disease is not always idiopathic [19]. By the late 1970s, physicians began to use ITP to stand for “Immune Thrombocytopenia Purpura” instead, while the cause of this immune process later to be clarified as “autoimmune” decades later [13].
In 2009, an International Working Group (IWG) standardized the term “idiopathic” by using “immune” instead, to emphasize the immune-mediated mechanism of the disease [20].
2 Development of investigation
Though ITP has always been a disease of exclusion, some scientists attempted to establish tools for diagnosis. In 1952, Hirsch and Gardner developed the first platelet-survival studies by transfusing normal fresh blood to the patient [21]. The studies showed rapid platelet destruction. This breakthrough led to the development of platelet kinetic studies, which measure platelet turnover (production) by dividing platelet count by platelet survival time. The use of radioisotope-labeled platelet, specifically sodium chromate (Cr51), later became standard for platelet-survival studies due to its practical, reproducible, and accurate characteristic [22]. In 1987, a groundbreaking study utilizing the platelet survival concept proved two key findings in ITP; 1) platelet clearance by liver and spleen increased, and 2) platelet production decreased [23].
Though platelet-survival studies have diminished in importance, their contribution to knowledge remains significant. In 1989, a landmark study involving sequential Radio-labeled autologous platelet studies pre- and post-interventions (e.g., steroids, splenectomy, and immunoglobulin therapy) revealed unchanged platelet survival with prednisolone treatment, suggesting increased platelet turnover as the cause of rising platelet counts. Splenectomized patients had an increase in platelet survival with unchanged production rates. Splenectomized responder demonstrated a lower reticuloendothelial uptake and a decreased liver clearance. Conversely, those who failed splenectomy showed increased liver uptake [24], prompting consideration for predicting splenectomy outcomes by some experts.
While targeting autoantibodies might be the case in many autoimmune diseases, this method proved to be difficult in ITP. Serum assays are not good tools because titers may be low or antibodies absent since most antibodies are bound to the platelets [13], detectable in only 60 percent of ITP patients [25]. Initial attempts involved mixing patient serum with normal platelets, resulting in low sensitivity and specificity [26]. While some antiplatelet antibodies triggered platelet activation, many did not. Interestingly, this method was later utilized in diagnosing heparin-induced thrombocytopenia (HIT).
The assumption that antiplatelet antibodies were platelet-bound IgG led to efforts measuring only immunoglobulin bound to platelets, both pathological and physiological binding. The “platelet-associated IgG (PAIgG) test”, utilized since the 1970s [27, 28], became a controversial test due to the discovery that nearly all platelet IgG resides within α-granules in platelets, with less than 1 percent present on the cell surface [29]. Several investigators measure both total PAIgG and surface PAIgG. Although total IgG level increases in ITP patients, it proved to be more sensitive for ITP yet less specific, as the total PAIgG levels could also rise in other platelet-destruction disorders. This is due to the fact that younger platelets contain more α-granule PAIgG than mature platelets [30]. Therefore, measuring total platelet IgG in patients with thrombocytopenia may be analogous to measuring reticulocyte count in anemic patients.
Flow cytometry was used to detect only platelet-surface IgG with higher specificity, yet its clinical adoption remains limited. Consequently, the American Society of Hematology did not recommend the PAIgG test in the 1996 practice guidelines [31].
According to recent guidelines, diagnosis of ITP remains by excluding other causes [32].
3 Pathophysiology of ITP
There are several different pathophysiologic mechanisms, mainly centre on peripheral clearance.
3.1 Antiplatelet antibody-mediated clearance
Our understanding of the targets of these antibodies began to form in 1982, with a study showing that the antibodies failed to bind platelets from Glanzmann thrombasthenia patients (who lack GPIIb/IIIa) [33]. Immunoassay studies in the 1990s confirmed and expanded on this, revealing that antibodies mostly target glycoprotein(GP) on platelet surface called “GPIIb/IIIa”, with some targeting GPIb-IX-V, GPIV, and GPIa/IIa [34, 35]. Antibody-coated platelets bind tissue macrophages through Fcγ receptors, leading to their clearance primarily in the spleen and to a lesser extent in the liver. The effectiveness of intravenous immunoglobulin (IVIG) treatment in 1981 provided evidence for this “Fcγ-dependent pathway”. By blocking macrophage Fcγ receptors, IVIG led to an elevation of platelet counts [36].
Furthermore, antiplatelet antibodies might not only bind but also activate the “classical complement pathway”, causing their lysis by assembling the membrane attack complex [37]. This is based on findings from the 1970s, which shown an increased number of platelet-associated C3, C4, and C9 on platelets in ITP [38, 39].
One unanswered question is why anti-GPIb-mediated ITP often refractory to treatments targeting Fcγ pathways. A study in 2015 showed that platelet clearance has another “Fcγ-independent pathway” via hepatocyte Ashwell-Morell receptors (AMR). Platelets lacking sialic acid, due to a neuraminidase-mediated process called “desialylation”, are removed by this AMR in the liver [40]. The AMR also plays a role in the thrombopoiesis feedback loop by regulating liver thrombopoietin production. A report in 2014 found that anti-GPIb antibodies could trigger platelet desialylation [41]. As a result, a neuraminidase inhibitor (e.g., oseltamivir) has emerged as a potential treatment.
3.2 Abnormalities in cell-mediated immunity
Normally, antigen-presenting cells recognize and process foreign antigens and then express these antigens on Major Histocompatibility Complex (MHC) molecules. The MHC-antigen complex activates naïve T cells to differentiate into distinct effector cells or cytotoxic T cells activation, depending on which MHC class (II or I) is presenting the antigen. CD4+ T cells response can be T-helper1(Th1), T-helper2(Th2), Th17, T follicular helper(Tfh), or T-regulatory(Treg) cells, while CD8+ T cells differentiate into cytotoxic T cells. Th1 cells are involved in macrophage activation and defense against intracellular pathogens. Th2 cells are involved in eosinophil and mast cell activation and defense against extracellular pathogens. Th17 cells are involved in neutrophil recruitment and activation and defense against extracellular bacteria and fungi. Tfh cells are involved in antibody production and defense against extracellular pathogens. Treg cells are involved in self-tolerance by inhibiting autoimmune response [42]. In ITP, pathophysiology might occur from three different components:
3.2.1 Abnormality in antigen-presenting cells (APCs) activity
Dendritic cells, the most potent APCs, play a significant role in ITP. Dendritic cells from ITP patients exhibit a higher ability to induce CD4+ CD25-T lymphocyte proliferation while reducing the induction of Tregs [43]. Abnormally high inflammatory cytokines, such as IL-17 and IFN-γ, can result in an autoreactive T cell response [44]. Additionally, abnormality in CD8+ T cell response in ITP can impact both platelet and megakaryocyte destruction [45, 46].
3.2.2 Decrease regulatory T cell activity
Decrease regulatory T cell activity, which normally inhibits autoimmune response, was first demonstrated by Liu in 2007 [47]. He showed that ITP patients have defective and reduced number of Treg cells. This mechanism is foundational to the pathophysiology of ITP, suggesting that the disease is due to a lack of peripheral T cell tolerance mechanisms [48].
3.2.3 Abnormality in B cell activity
Stimulation of T cells can result in IL-2 secretion, thus resulting in autoreactive B cell differentiation and antibody secretion [49]. The abnormally high Th1 and Th17 cell responses drive autoreactive B-cell clone and produce autoantibodies from both B-cells and plasma cells [44]. Early studies of PAIgG reported that antibodies in ITP were polyclonal [50]. Later studies using flow cytometry and DNA analysis for immunoglobulin heavy- and light-chain rearrangement, however, showed that some ITP patients had clonal B-cell proliferation [51]. This knowledge led to the usage of anti-CD20 and potentially anti-CD38 therapies.
3.3 Decrease platelet production from bone marrow
Potential mechanisms were provided by later studies in 2003 suggesting that autoantibodies against platelet surface glycoproteins might interfere with the maturation of megakaryocytes and suppress megakaryocyte production [52,53,54].
3.4 Suboptimal thrombopoietin (TPO) response
TPO was proposed and named by Kelemen in 1958 [55]. It was long speculated to regulate megakaryocytopoiesis, however, it was not proved until a milestone discovery in 1994 when TPO could be purified [56]. In 1996, study measuring TPO levels showed a markedly elevation in megakaryocytic hypoplasia, while in ITP, the TPO levels are normal or decreased [57]. This fact led to the successful usage of recombinant human thrombopoietin [58].
4 Treatment of ITP
In 1977, Harrington proposed that only patients with a platelet count below 30,000 per cubic millimeter need treatment, and a platelet count of 50,000 per cubic millimeter is sufficient to assure safety [59]. The first American Society of Hematology endorsed guideline in 1996 also stated that asymptomatic patients with a platelet count of more than 30,000 per cubic millimeter are not indicated for treatment unless there are risk factors for bleeding [31].
It has been generally accepted since 1977 that glucocorticoid should be the first-line treatment for ITP [60]. Once the diagnosis is made, treatment should be initiated immediately. Standard prednisolone 1 to 1½ mg per kg per day was administrated in 3 to 4 divided doses for 2 to 3 weeks at that time [59]. However, later studies recommended a morning single-dose or 2 divided doses to mimic the body’s natural cortisol circadian rhythm and reduce the risk of insomnia and other side effects [61, 62]. Other glucocorticoids can also be helpful in newly diagnosed ITP patients. A study in 2009 compared dexamethasone 40 mg/day for 4 days with prednisolone 60 mg/day showed that dexamethasone had a faster response [63]. According to ASH 2019, the current recommended dose for adults is prednisolone 1 mg/kg (maximum 80 mg) for 2 to 3 weeks, or dexamethasone 40 mg/d for 4 days, repeated up to 3 times [64].
Glucocorticoids should not be used long-term due to various side effects such as hyperglycemia, infection, glaucoma, osteoporosis, etc.[65] Guideline suggests that treatment should be switched to TPO-RA in patients who are corticosteroid-dependent or unresponsive to corticosteroids. TPO-RA is preferred over rituximab, and splenectomy should be delayed for 1 year after diagnosis due to potential spontaneous remission [32].
5 Treatment response evaluation
Treatment response evaluation varies among experts, with different criteria proposed for assessing patient outcomes. Ultimately, the choice of evaluation method depends on the preference of the treating physician.
According to the criteria of Reis [66] in 1976, responses were defined as follows (Table 1).
In 2009, an IWG of recognized expert clinicians convened a 2-day structured meeting (the Vicenza Consensus Conference) to define standard terminology and definitions. Responses were defined as follows (Table 2) [20, 67].
International experts updated the guidelines in 2019. This current guideline provides more detailed information and timeframe for different treatments with a consistent goal to maintain platelet target > 20–30 × 109/L [64].
In 2019, the American Society of Hematology defined response as follows (Table 3) [32].
6 Development of treatment
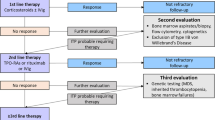
The timeline of treatment development can be seen in Fig. 1.

Timeline showed the development of treatment in ITP
6.1 Splenectomy
Although splenectomy was the first treatment discovered, a publication in 1966 concluded that it is reserved for chronic ITP cases or emergency (e.g. intracranial) bleeding only [19]. Splenectomy improves platelet counts primarily by prolonging platelet survival from 0.6 days to 4.7 days [24, 68]. The complete remission rate varies between studies, ranging between 70 to 80 percent in the pre-corticosteroid era [9, 69,70,71,72,73,74,75]. This number is lower to around two-thirds after that [76]. The rate of relapse is around 33 percent [77]. After the surgery, it is not unusual for the platelet count to increase with in the first 24 hour [9]. Patients who failed to respond after splenectomy should be evaluated for accessory spleen. Accessory spleen is found in about 20 percent of autopsy cases [78, 79].
6.2 Glucocorticoids
Splenectomy was the treatment of choice for more than 30 years [9]. However, in 1950, the landscape shifted when glucocorticoids, specifically at that time ACTH and cortisone, were first revealed to be effective treatments [80]. Severe bleeding showed improvement within one or two days, while platelet increment was observed within 1 week. The improvement in bleeding was primarily attributed to the rise in platelet count by increased platelet production [24]. Other factors such as improvement of vascular resistance [81] without stimulation of platelet production [82] is not entirely convincing by the evidence. Glucocorticoids later became the first-line of treatment in ITP since the late 1950s [83].
6.3 Immunosuppressants
Since 1960, immunosuppressive drugs have been used in severe chronic ITP patients who failed corticosteroids. Cytotoxic agents (e.g. azathioprine), which have been found to be useful in the treatment of autoimmune disease, are thought to help ITP patients as well [19]. Azathioprine is used in a dosage of 1.2–2.4 mg/kg (50–250 mg/day) for at least 3 months [84].
Vinca alkaloids usage started during the 1970s when several physicians started using vincristine in ITP patients [66, 85,86,87,88]. Vincristine was given by weekly intravenous injections (2 mg for adults and 1 mg for children). Overall response rate was 73 percent, 34.6 percent achieved complete response, and 38.4 percent achieved partial response. The platelet increase was within one or two days [89]. Vinblastine also show as effective as vincristine response with dosage of 10 mg weekly [90].
Cyclophosphamide usage has been reported since 1971 [91]. The dosage is between 50–200 mg/day [92]. Response seen in 2 to 4 weeks [93].
6.4 Combination chemotherapy
Combination chemotherapy was reported to be beneficial in refractory immune thrombocytopenia patients. Patients received from 3 to 8 cycles of chemotherapy consisting of cyclophosphamide and prednisolone combined with either vincristine, vincristine and procarbazine, or etoposide. Six in ten patients achieved complete response [94].
6.5 Danazol
Danazol, a steroid with weak androgenic activity, was reported in 1983 to be effective in patients who did not respond to splenectomy [95]. The mechanism is believed to be associated with a decrease in the number of monocyte binding sites for IgG without interfering with the level of platelet-associated IgG [96].
6.6 Intravenous immunoglobulin (IVIG)
First reported in adult in 1982 [97] after a report in children 1 year before [36]. Dosage between 1 to 1.5 g per kg was used with a striking result of platelet increase of more than 100,000 per cubic millimeter within five to seven days [97]. The mechanism is to interfere with phagocyte Fc-receptor-mediated immune clearance [97, 98] and acceleration of antiplatelet antibody elimination [99]. Single high-dose immunoglobulin followed by platelet transfusion would increase platelet counts faster than without platelet transfusion [100]. A regimen of 1 g per kg per day for two days will increase the platelet count in most patients within 3 days [76, 101]. IVIG is safe to use in pregnancy [102]. Later study in 2016 also supported the efficacy and safety in pregnancy [103].
6.7 Cyclosporine
In the 1980s, concern about long-term side effects of vincristine prompted physicians to seek alternative treatments. Cyclosporine usage was first reported in refractory ITP patient in 1985. The dosage used is 10 mg/kg/day divided into 2 daily doses [104]. Cyclosporine plasma level ranged between 200 to 250 ng/ml.
6.8 Colchicine
Colchicine was used for refractory ITP patients in 1984. The dosage used is 1.2 mg/day for at least 2 weeks, with response rate of 29 percent [105].
6.9 Dapsone
Dapsone was used for refractory ITP patients in 1991 [106]. The dosage used is 75 mg/day for 2 to 48 months, with significant increase in platelet count. The overall response rate ranged between 40 to 50 percent [107, 108]. The mechanism is believed to increase red blood cell destruction, hence interfering with platelet sequestration by the reticuloendothelial system [106].
6.10 Rituximab
Started using around the 2000s. Rituximab, a chimeric monoclonal antibody directed against the CD20 antigen, dose of 375 mg/meter square once weekly for 4 weeks, showed an overall response rate of 52 percent in chronic ITP patients [109]. Meta-analysis also shows a similar over all response rate at 57 percent [110]. Although rituximab was reported as an alternative treatment for ITP in pregnancy, [111, 112] it should be reserved for very severe cases due to immunosuppression and subsequent infection [64].
6.11 Thrombopoietin-receptor agonist (TPO-RA)
Not long before TPO-RAs became widely studied, two recombinant TPOs—recombinant human TPO (rhTPO) and pegylated recombinant human megakaryocyte growth and development factor (PEG-rHuMGDF)—were in use. Recombinant TPO was first used in cancer patients undergoing chemotherapy in 1995 prior to its study in ITP patients in 2001 [113]. The first-generation usage was a daily injection drug [114, 115]. Unfortunately, data in 2001 revealed the development of antibodies that could cross-react with endogenous TPO after PEG-rHuMMGDF use [116]. Consequently, the development of both PEG-rHuMGDF and rhTPO was eventually stopped, paving the way for the second-generation TPOs-romiplostim and eltrombopag—which bind and activate the TPO receptors.
Thrombopoietin-stimulating protein, AMG 531, known as romiplostim, was reported to be effective in relapsed or refractory ITP in 2007 [58]. Thrombopoietin-receptor agonists increased platelet counts by boosting platelet production in a dose-dependent manner. The overall response rate is 88 percent. Mean time to respond is 13.8 weeks [117]. Eltrombopag, an oral thrombopoietin-receptor agonist, also showed similar responses with response rate between 70 to 81 percent [118].
6.12 Mycophenolate mofetil
In the 2000s, physicians started using mycophenolate mofetil as an immunosuppressant in SLE patients and solid organ transplant patients. Hematologists started using mycophenolate mofetil in refractory ITP with efficacy between 38 to 83 percent [119,120,121,122,123]. Dosage use is 1.5 to 2 g/day orally for at least 12 weeks. A study in 2021 showed that the addition of mycophenolate mofetil to glucocorticoid for first-line treatment resulted in a greater response [124].
7 Current novel ITP treatment
During the last decade, newer novel treatments for ITP targeting different pathways in the pathophysiology have been developed, mainly focusing on chronic ITP patients.
7.1 Increase platelet production
Avatrombopag (formerly AKR-501), an oral TPO-receptor agonist, has been studied since 2009 [125]. In 2014, phase II studies using avatrombopag revealed very good results in ITP patients without serious adverse effects [126, 127]. Phase III RCT showed that avatrombopag was effective in ITP patients with chronic liver disease who were scheduled to undergo procedures [128], leading to FDA approval in 2018 [129]. Unlike other TPO-receptor agonists, avatrombopag does not require a 4-h food restriction window and is not associated with hepatotoxicity [130]. However, further studies are needed to compare its efficacy with eltrombopag [131].
7.2 Decrease platelet destruction
Fostamatinib (formerly R406), the first spleen tyrosine kinase (Syk) inhibitor, has been studied since 2009 [132]. The mechanism involves inhibiting Syk activation in splenic macrophages [133, 134]. Fostamatinib, given orally at 100 mg twice daily for 24 weeks, showed an overall response rate of 43 percent in chronic ITP patients compared with 14 percent in the placebo group [135].
Rilzabrutinib (formerly PRN 1008), an oral BTK inhibitor, has been studied since 2020 [136]. The mechanisms involve decreasing macrophage (Fcγ receptor)-mediated platelet destruction and reducing the production of autoantibodies [137]. Rilzabrutinib, dosed at 400 mg twice daily, showed a 40 percent response rate in refractory ITP patients without major adverse events [138].
7.3 Interfere with autoantibodies
Efgartigimod, a human IgG1 Fc fragment engineered for increased affinity to FcRn, has been studied since 2019 [139]. The mechanisms involve blocking FcRn, preventing IgG1 recycling, and causing targeted IgG degradation. Results from the phase II study showed that efgartigimod, dosed at 5 mg/kg intravenous weekly four times, has an overall response rate of 43 percent in primary ITP patients.
Daratumumab, an anti-CD38 antibody targeting plasma cells, was studied recently for its potential to treat autoimmune diseases. It was believed that B-cell depletion by anti-CD20 was insufficient since antibody can be secreted by long-lived plasma cells. A Phase II study conducted in 2021 reported promising data with responses in 2 out of 3 patients receiving eight weekly injection doses [140]. Additionally, two case reports on refractory ITP supported its potential as a novel treatment by providing lasting clinical stabilization [141, 142].
8 Ongoing research and advancements
The field of ITP research continues to evolve. Scientists are exploring the genetic and molecular factors contributing to the development of the disease. Novel therapeutic approaches, including targeted therapies, are being investigated to improve treatment outcomes and minimize side effects. This comprehensive review consolidates findings from various studies conducted between 2014 and 2023, shedding light on different aspects of ITP.
In 2014, a pivotal investigation in a canine antibody-mediated immune thrombocytopenia (ITP) model elucidated platelets as a source of serum interleukin-8, echoing congruence with analogous studies in humans. This observation implicates platelet IL-8 as a putative modulator in the intricate interplay between platelets and neutrophils [143]. Concurrently, novel microaggregation and platelet reactivity assays were employed in ITP patients with severely diminished platelet counts, revealing a discernible correlation between platelet functionality and the clinical heterogeneity observed in these patients. In this study, ITP patients were classified into mild or severe bleeding phenotypes based on the overall Buchanan bleeding score. Then, the mild bleeding phenotype, with scores ranging from grade 0–3, was compared to the severe bleeding phenotype, with scores ranging from grade 4–5. Patients with severe ITP showed significant decreases in platelet microaggregation and platelet reactivity [144].
Addressing the absence of specific markers for differential diagnosis in 2016, a comprehensive study identified 161 differentially expressed proteins, encompassing oncoproteins, tumor suppressors, and anti-nuclear autoantibodies within a subset of ITP patients. This intricate protein landscape suggests potential associations between ITP, malignancies, and autoimmune disorders. Furthermore, a plausible pathogenic link to ITP involving RUNX1 expression was proposed [145]. In 2017, an investigation utilizing mass spectrometry-based antibody-mediated identification of autoantigen (MS-AMIDA), a new proteomic approach, helped identify novel autoantigens associated with ITP, expanding insights into the disease's immunological profile [146].
A 2019 review underscored the therapeutic potential of low levels of CD4+ CD25+ Foxp3+ regulatory T cells in ITP, proposing low-dose Histone Deacetylase Inhibitors (HDACi's), notably chidamide, as a viable therapeutic avenue. Administration of chidamide in animal and human models demonstrated noteworthy improvements in regulatory T cell counts and positive clinical outcomes. The proposed mechanism involves the attenuation of macrophage phagocytosis of antibody-coated platelets, akin to the action of splenic tyrosine kinase inhibitors. This study also highlighted the pivotal role of CTLA4 in ITP pathogenesis, with diminished expression noted in ITP patients relative to controls. Following treatment with low-dose chidamide, enhanced CTLA4 expression on T cells was observed, correlated with increased histone acetylation. This implies a plausible relationship between CTLA4 impairment in ITP and histone acetylation. HDACi's emerge as a promising therapeutic paradigm, influencing macrophage phagocytosis and reinstating Treg and CTLA4 expression in animal models. Nonetheless, the specific triggers of ITP in human pathology remain elusive, necessitating further investigation [147, 148].
In 2019, a case report underscored a 2-year-old girl presenting symptomatic ITP associated with DADA2 [149]. In 2022, the identification of long noncoding RNAs IFNG-AS1 and Gas5 as novel prognostic genetic markers for childhood ITP, exhibiting overexpression in chronic and persistent cases, offered valuable prognostic insights [150]. Finally, in 2023, the development of a fragment crystallizable modified anti-haptoglobin for ITP in animal models demonstrated non-inferiority at lower doses than IVIG, proposing a potentially efficacious alternative treatment strategy [151]. These cumulative findings enrich our comprehensive understanding of ITP, delineating avenues for future research and therapeutic innovation.
9 Conclusion
The history of ITP treatment has been marked by significant advances in understanding its pathophysiology and developing effective therapies. From the early identification of antiplatelet antibodies and the spleen's role in platelet clearance to the introduction of therapies targeting specific pathways, such as Fcγ receptors and B cell clonality, the field has made remarkable progress. Recent insights into alternative mechanisms of platelet clearance via the Ashwell-Morell receptor and the emergence of neuraminidase inhibitors like oseltamivir highlight the evolving nature of ITP treatment strategies. Furthermore, innovative therapies targeting plasma cells, such as daratumumab, offer promising new avenues for managing refractory cases. We hope this article provides general medical practitioners, students, and others interested in ITP with valuable insights into how the field has evolved and where it is heading. Additionally, we hope this article will inspire further research and advancements in the future.
Data availability
No datasets were generated or analysed during the current study.
References
Bolton-Maggs PHB, George JN. Immune thrombocytopenia treatment. N Engl J Med. 2021;385(10):948–50.
Werlhof PG. Disquisitio medica et philologica de variolis et anthracibus. 1735: Sumptibus haeredum Nicolai Foersteri et filii.
Schultze M. Ein heizbarer Objecttisch und seine Verwendung bei Untersuchungen des Blutes. Arch Mikrosk Anat. 1865;1(1):1–42.
Brewer DB. Max Schultze (1865), G. Bizzozero (1882) and the discovery of the platelet. Br J Haematol. 2006;133(3):251–8.
Bizzozero J. Ueber einen neuen Formbestandtheil des Blutes und dessen Rolle bei der Thrombose und der Blutgerinnung: Untersuchungen. Archiv für pathologische Anatomie und Physiologie und für klinische Medicin. 1882;90(2):261–332.
Gesellschaft DP. Zentralblatt für allgemeine Pathologie und pathologische Anatomie. G. Fischer: Schaffhausen; 1896.
Tocantins LM. Historical notes on blood platelets. Blood. 1948;3(10):1073–82.
Wright JH. The origin and nature of the blood plates. Boston Med Surg J. 1906;154(23):643–5.
Elliott RH. Diagnostic and therapeutic considerations in the management of idiopathic thrombocytopenic purpura. Bull N Y Acad Med. 1939;15(3):197–210.
Kaznelson P. Verschwinden der hamorrhagischen diathese bei einem falle von essentieller thrombopenie (frank) nach milzexstiparation: Splenogene thrombolytische purpura. Wien Klin Wochenschr. 1916;29:1451.
Yoshida Y. Historical review. The light and shadow of Paul Kaznelson: his life and contribution to hematology. Ann Hematol. 2008;87(11):877–9.
Altman LK. Who goes first?: The story of self-experimentation in medicine. Berkeley: University of California Press; 1998.
McMillan R. Chronic idiopathic thrombocytopenic purpura. N Engl J Med. 1981;304(19):1135–47.
Harrington WJ, et al. Demonstration of a thrombocytopenic factor in the blood of patients with thrombocytopenic purpura. J Lab Clin Med. 1951;38(1):1–10.
Frank E. Die essentielle Thrombopenie. (Konstitutionelle Purpura-Pseudohamophilie), I: Klinische Bild. Berl Klin Wochenschr. 1915;52:454–8.
Minot GR. Diminished blood platelets and marrow insufficiency: a classification and differential diagnosis of purpura hemorrhagica, aplastic anemia, and allied conditions. Arch Intern Med. 1917;19(6):1062–84.
Dameshek W, Miller EB. The megakaryocytes in idiopathic thrombocytopenic purpura, a form of hypersplenism. Blood. 1946;1:27–50.
Dameshek W. New forms of idiopathic thrombocytopenic purpura. Blood. 1947;2(6):597.
Baldini M. Idiopathic thrombocytopenic purpura. N Engl J Med. 1966;274(24):1360–7.
Rodeghiero F, et al. Standardization of terminology, definitions and outcome criteria in immune thrombocytopenic purpura of adults and children: report from an international working group. Blood. 2009;113(11):2386–93.
Hirsch E, Gardner F. The transfusion of human blood platelets with a note on the transfusion of granulocytes. J Lab Clin Med. 1952;39(4):556–69.
Aas KA, Gardner FH. Survival of blood platelets labeled with chromium. J Clin Invest. 1958;37(9):1257–68.
Ballem PJ, et al. Mechanisms of thrombocytopenia in chronic autoimmune thrombocytopenic purpura. Evidence of both impaired platelet production and increased platelet clearance. J Clin Invest. 1987;80(1):33–40.
Gernsheimer T, et al. Mechanisms of response to treatment in autoimmune thrombocytopenic purpura. N Engl J Med. 1989;320(15):974–80.
Brighton TA, et al. Prospective evaluation of the clinical usefulness of an antigen-specific assay (MAIPA) in idiopathic thrombocytopenic purpura and other immune thrombocytopenias. Blood. 1996;88(1):194–201.
Kelton JG, et al. A prospective comparison of four techniques for measuring platelet-associated IgG. Br J Haematol. 1989;71(1):97–105.
Dixon R, Rosse W, Ebbert L. Quantitative determination of antibody in idiopathic thrombocytopenic purpura. Correlation of serum and platelet-bound antibody with clinical response. N Engl J Med. 1975;292(5):230–6.
Cines DB, Schreiber AD. Immune thrombocytopenia. Use of a coombs antiglobulin test to detect IgG and C3 on platelets. N Engl J Med. 1979;300(3):106–11.
Sinha RK, Kelton JG. Current controversies concerning the measurement of platelet-associated IgG. Transfus Med Rev. 1990;4(2):121–35.
George JN. Platelet immunoglobulin G: its significance for the evaluation of thrombocytopenia and for understanding the origin of alpha-granule proteins. Blood. 1990;76(5):859–70.
George JN, et al. Idiopathic thrombocytopenic purpura: a practice guideline developed by explicit methods for the American Society of Hematology. Blood. 1996;88(1):3–40.
Neunert C, et al. American Society of Hematology 2019 guidelines for immune thrombocytopenia. Blood Adv. 2019;3(23):3829–66.
van Leeuwen EF, et al. Specificity of autoantibodies in autoimmune thrombocytopenia. Blood. 1982;59(1):23–6.
He R, et al. Spectrum of Ig classes, specificities, and titers of serum antiglycoproteins in chronic idiopathic thrombocytopenic purpura. Blood. 1994;83(4):1024–32.
Kiefel V, et al. Autoantibodies against platelet glycoprotein Ib/IX: a frequent finding in autoimmune thrombocytopenic purpura. Br J Haematol. 1991;79(2):256–62.
Imbach P, et al. High-dose intravenous gammaglobulin for idiopathic thrombocytopenic purpura in childhood. Lancet. 1981;1(8232):1228–31.
Tsubakio T, et al. Complement activation in vitro by antiplatelet antibodies in chronic immune thrombocytopenic purpura. Br J Haematol. 1986;63(2):293–300.
Hauch TW, Rosse WF. Platelet-bound complement (C3) in immune thrombocytopenia. Blood. 1977;50(6):1129–36.
Kurata Y, et al. Platelet-associated complement in chronic ITP. Br J Haematol. 1985;60(4):723–33.
Li J, et al. Desialylation is a mechanism of Fc-independent platelet clearance and a therapeutic target in immune thrombocytopenia. Nat Commun. 2015;6:7737.
Li J, et al. Severe platelet desialylation in a patient with glycoprotein Ib/IX antibody-mediated immune thrombocytopenia and fatal pulmonary hemorrhage. Haematologica. 2014;99(4):e61–3.
Abbas AK, Lichtman AH, Pillai S. Cellular and molecular immunology, 10e, South Asia Edition-E-Book. 2021: Elsevier Health Sciences.
Zhang X, et al. CD70-silenced dendritic cells induce immune tolerance in immune thrombocytopenia patients. Br J Haematol. 2020;191(3):466–75.
Zhang J, et al. Elevated profile of Th17, Th1 and Tc1 cells in patients with immune thrombocytopenic purpura. Haematologica. 2009;94(9):1326–9.
Zhang F, et al. Cell-mediated lysis of autologous platelets in chronic idiopathic thrombocytopenic purpura. Eur J Haematol. 2006;76(5):427–31.
Li S, et al. CD8+ T cells suppress autologous megakaryocyte apoptosis in idiopathic thrombocytopenic purpura. Br J Haematol. 2007;139(4):605–11.
Liu B, et al. Abnormality of CD4(+)CD25(+) regulatory T cells in idiopathic thrombocytopenic purpura. Eur J Haematol. 2007;78(2):139–43.
Ji L, et al. The ratio of Treg/Th17 cells correlates with the disease activity of primary immune thrombocytopenia. PLoS ONE. 2012;7(12): e50909.
Semple JW, Freedman J. Increased antiplatelet T helper lymphocyte reactivity in patients with autoimmune thrombocytopenia. Blood. 1991;78(10):2619–25.
Hymes K, Schur PH, Karpatkin S. Heavy-chain subclass of round antiplatelet IgG in autoimmune thrombocytopenic purpura. Blood. 1980;56(1):84–7.
van der Harst D, et al. Clonal B-cell populations in patients with idiopathic thrombocytopenic purpura. Blood. 1990;76(11):2321–6.
Chang M, et al. Immune thrombocytopenic purpura (ITP) plasma and purified ITP monoclonal autoantibodies inhibit megakaryocytopoiesis in vitro. Blood. 2003;102(3):887–95.
McMillan R, et al. Antibody against megakaryocytes in idiopathic thrombocytopenic purpura. JAMA. 1978;239(23):2460–2.
McMillan R, et al. Suppression of in vitro megakaryocyte production by antiplatelet autoantibodies from adult patients with chronic ITP. Blood. 2004;103(4):1364–9.
Kelemen E, Cserhati I, Tanos B. Demonstration and some properties of human thrombopoietin in thrombocythaemic sera. Acta Haematol. 1958;20(6):350–5.
McDonald TP. Regulation of megakaryocytopoiesis by thrombopoietin. Ann N Y Acad Sci. 1987;509:1–24.
Emmons RV, et al. Human thrombopoietin levels are high when thrombocytopenia is due to megakaryocyte deficiency and low when due to increased platelet destruction. Blood. 1996;87(10):4068–71.
Bussel JB, et al. AMG 531, a thrombopoiesis-stimulating protein, for chronic ITP. N Engl J Med. 2006;355(16):1672–81.
Ahn YS, Harrington WJ. Treatment of idiopathic thrombocytopenic purpura (ITP). Annu Rev Med. 1977;28:299–309.
Lacey JV, Penner JA. Management of idiopathic thrombocytopenic purpura in the adult. Semin Thromb Hemost. 1977;3(3):160–74.
Turhan AB, Ozdemir ZC, Bor O. Use of single- or two-dose pulse methylprednisolone in the treatment of acute immune thrombocytopenic purpura. Sisli Etfal Hastan Tip Bul. 2018;52(4):279–84.
Ferrara G, et al. Clinical use and molecular action of corticosteroids in the pediatric age. Int J Mol Sci. 2019;20(2):444.
Praituan W, Rojnuckarin P. Faster platelet recovery by high-dose dexamethasone compared with standard-dose prednisolone in adult immune thrombocytopenia: a prospective randomized trial. J Thromb Haemost. 2009;7(6):1036–8.
Provan D, et al. Updated international consensus report on the investigation and management of primary immune thrombocytopenia. Blood Adv. 2019;3(22):3780–817.
Grennan D, Wang S. Steroid side effects. JAMA. 2019;322(3):282.
Ries CA. Vincristine for treatment of refractory autoimmune thrombocytopenia. N Engl J Med. 1976;295(20):1136.
Song F, Al-Samkari H. Management of adult patients with immune thrombocytopenia (ITP): a review on current guidance and experience from clinical practice. J Blood Med. 2021;12:653–64.
Branehog I. Platelet kinetics in idiopathic thrombocytopenic purpura (ITP) before and at different times after splenectomy. Br J Haematol. 1975;29(3):413–26.
Watson-Williams EJ, Macpherson AI, Davidson S. The treatment of idiopathic thrombocytopenic purpura; a review of ninety-three cases. Lancet. 1958;2(7040):221–6.
Carpenter AF, et al. Treatment of idiopathic thrombocytopenic purpura. J Am Med Assoc. 1959;171:1911–6.
Meyers MC. Results of treatment in 71 patients with idiopathic thrombocytopenic purpura. Am J Med Sci. 1961;242:295–302.
Charlesworth D, Torrance HB. Splenectomy in idiopath0cthrombocytopenic purpura. Br J Surg. 1968;55(6):437–9.
Thompson RL, et al. Idiopathic thrombocytopenic purpura. Long-term results of treatment and the prognostic significance of response to corticosteroids. Arch Intern Med. 1972;130(5):730–4.
JiJi RM, Firozvi T, Spurling CL. Chronic idiopathic thrombocytopenic purpura. Treatment with steroids and splenectomy. Arch Intern Med. 1973;132(3):380–3.
Macpherson AI, Richmond J. Planned splenectomy in treatment of idiopathic thrombocytopenic purpura. Br Med J. 1975;1(5949):64–6.
George JN, El-Harake MA, Raskob GE. Chronic idiopathic thrombocytopenic purpura. N Engl J Med. 1994;331(18):1207–11.
Vianelli N, et al. Splenectomy as a curative treatment for immune thrombocytopenia: a retrospective analysis of 233 patients with a minimum follow up of 10 years. Haematologica. 2013;98(6):875–80.
Wadham BM, Adams PB, Johnson MA. Incidence and location of accessory spleens. N Engl J Med. 1981;304(18):1111.
Rudowski WJ. Accessory spleens: clinical significance with particular reference to the recurrence of idiopathic thrombocytopenic purpura. World J Surg. 1985;9(3):422–30.
Bethell FH, et al. Effects of ACTH and cortisone on idiopathic thrombocytopenic purpura. Trans Assoc Am Physicians. 1951;64:199–203.
Robson HN, Duthie JJ. Capillary resistance and adrenocortical activity. Br Med J. 1950;2(4686):971–7.
Faloon WW, Greene RW, Lozner EL. The hemostatic defect in thrombocytopenia as studied by the use of ACTH and cortisone. Am J Med. 1952;13(1):12–20.
Dameshek W, et al. Treatment of idiopathic thrombocytopenic purpura (ITP) with prednisone. J Am Med Assoc. 1958;166(15):1805–15.
Bouroncle BA, Doan CA. Refractory idiopathic thrombocytopenic purpura treated with azathioprine. N Engl J Med. 1966;275(12):630–5.
Hicsonmez G, Ozsoylu S. Vincristine for treatment of chronic thrombocytopenia in children. N Engl J Med. 1977;296(8):454–5.
Steinherz PG, et al. Platelet dysfunction in vincristine treated patients. Br J Haematol. 1976;32(3):439–50.
Ahn YS, et al. Vincristine therapy of idiopathic and secondary thrombocytopenias. N Engl J Med. 1974;291(8):376–80.
Letter: Vincristine therapy of thrombocytopenias. N Engl J Med, 1975;292(2): 108–9.
Tangun Y, Atamer T. More on vincristine in treatment of ITP. N Engl J Med. 1977;297(16):894–5.
Marmont AM, Damasio EE, Gori E. Vinblastine sulphate in idiopathic thrombocytopenic purpura. Lancet. 1971;2(7715):94.
Laros RK Jr, Penner JA. “Refractory” thrombocytopenic purpura treated successfully with cyclophosphamide. JAMA. 1971;215(3):445–9.
Verlin M, Laros RK Jr, Penner JA. Treatment of refractory thrombocytopenic purpura with cyclophosphamine. Am J Hematol. 1976;1(1):97–104.
Finch SC, et al. Immunosuppressive therapy of chronic idiopathic thrombocytopenic purpura. Am J Med. 1974;56(1):4–12.
Figueroa M, et al. Combination chemotherapy in refractory immune thrombocytopenic purpura. N Engl J Med. 1993;328(17):1226–9.
Ahn YS, et al. Danazol for the treatment of idiopathic thrombocytopenic purpura. N Engl J Med. 1983;308(23):1396–9.
Schreiber AD, et al. Effect of danazol in immune thrombocytopenic purpura. N Engl J Med. 1987;316(9):503–8.
Fehr J, Hofmann V, Kappeler U. Transient reversal of thrombocytopenia in idiopathic thrombocytopenic purpura by high-dose intravenous gamma globulin. N Engl J Med. 1982;306(21):1254–8.
Bussel JB, et al. Intravenous gammaglobulin treatment of chronic idiopathic thrombocytopenic purpura. Blood. 1983;62(2):480–6.
Jin F, Balthasar JP. Mechanisms of intravenous immunoglobulin action in immune thrombocytopenic purpura. Hum Immunol. 2005;66(4):403–10.
Baumann MA, et al. Urgent treatment of idiopathic thrombocytopenic purpura with single-dose gammaglobulin infusion followed by platelet transfusion. Ann Intern Med. 1986;104(6):808–9.
Blanchette VS, Kirby MA, Turner C. Role of intravenous immunoglobulin G in autoimmune hematologic disorders. Semin Hematol. 1992;29(3 Suppl 2):72–82.
Newland AC, Boots MA, Patterson KG. Intravenous IgG for autoimmune thrombocytopenia in pregnancy. N Engl J Med. 1984;310(4):261–2.
Sun D, et al. Corticosteroids compared with intravenous immunoglobulin for the treatment of immune thrombocytopenia in pregnancy. Blood. 2016;128(10):1329–35.
Kelsey PR, Schofield KP, Geary CG. Refractory idiopathic thrombocytopenic purpura (ITP) treated with cyclosporine. Br J Haematol. 1985;60(1):197–8.
Strother SV, Zuckerman KS, LoBuglio AF. Colchicine therapy for refractory idiopathic thrombocytopenic purpura. Arch Intern Med. 1984;144(11):2198–200.
Durand JM, et al. Dapsone for idiopathic autoimmune thrombocytopenic purpura in elderly patients. Br J Haematol. 1991;78(3):459–60.
Hernandez F, et al. Dapsone for refractory chronic idiopathic thrombocytopenic purpura. Br J Haematol. 1995;90(2):473–5.
Godeau B, et al. Dapsone for chronic autoimmune thrombocytopenic purpura: a report of 66 cases. Br J Haematol. 1997;97(2):336–9.
Stasi R, et al. Rituximab chimeric anti-CD20 monoclonal antibody treatment for adults with chronic idiopathic thrombocytopenic purpura. Blood. 2001;98(4):952–7.
Chugh S, et al. Rituximab plus standard of care for treatment of primary immune thrombocytopenia: a systematic review and meta-analysis. Lancet Haematol. 2015;2(2):e75-81.
Donohoe F, et al. Rituximab - a novel therapy for severe ITP in pregnancy: a case report. Obstet Med. 2019;12(4):196–8.
Gall B, et al. Rituximab for management of refractory pregnancy-associated immune thrombocytopenic purpura. J Obstet Gynaecol Can. 2010;32(12):1167–71.
Basser RL, et al. Thrombopoietic effects of pegylated recombinant human megakaryocyte growth and development factor (PEG-rHuMGDF) in patients with advanced cancer. Lancet. 1996;348(9037):1279–81.
Rice L, et al. Cyclic immune thrombocytopenia responsive to thrombopoietic growth factor therapy. Am J Hematol. 2001;68(3):210–4.
Nomura S, et al. Effects of pegylated recombinant human megakaryocyte growth and development factor in patients with idiopathic thrombocytopenic purpura. Blood. 2002;100(2):728–30.
Li J, et al. Thrombocytopenia caused by the development of antibodies to thrombopoietin. Blood. 2001;98(12):3241–8.
Kuter DJ, et al. Efficacy of romiplostim in patients with chronic immune thrombocytopenic purpura: a double-blind randomised controlled trial. Lancet. 2008;371(9610):395–403.
Bussel JB, et al. Eltrombopag for the treatment of chronic idiopathic thrombocytopenic purpura. N Engl J Med. 2007;357(22):2237–47.
Provan D, et al. Efficacy of mycophenolate mofetil as single-agent therapy for refractory immune thrombocytopenic purpura. Am J Hematol. 2006;81(1):19–25.
Hou M, et al. Mycophenolate mofetil (MMF) for the treatment of steroid-resistant idiopathic thrombocytopenic purpura. Eur J Haematol. 2003;70(6):353–7.
Colovic M, et al. Mycophenolate mophetil therapy for chronic immune thrombocytopenic purpura resistant to steroids, immunosuppressants, and/or splenectomy in adults. Platelets. 2011;22(2):153–6.
Zhang WG, et al. Mycophenolate mofetil as a treatment for refractory idiopathic thrombocytopenic purpura. Acta Pharmacol Sin. 2005;26(5):598–602.
Howard J, et al. Mycophenolate mofetil for the treatment of refractory auto-immune haemolytic anaemia and auto-immune thrombocytopenia purpura. Br J Haematol. 2002;117(3):712–5.
Bradbury CA, et al. Mycophenolate mofetil for first-line treatment of immune thrombocytopenia. N Engl J Med. 2021;385(10):885–95.
Fukushima-Shintani M, et al. AKR-501 (YM477) a novel orally-active thrombopoietin receptor agonist. Eur J Haematol. 2009;82(4):247–54.
Terrault NA, et al. Phase II study of avatrombopag in thrombocytopenic patients with cirrhosis undergoing an elective procedure. J Hepatol. 2014;61(6):1253–9.
Bussel JB, et al. A randomized trial of avatrombopag, an investigational thrombopoietin-receptor agonist, in persistent and chronic immune thrombocytopenia. Blood. 2014;123(25):3887–94.
Terrault N, et al. Avatrombopag before procedures reduces need for platelet transfusion in patients with chronic liver disease and thrombocytopenia. Gastroenterology. 2018;155(3):705–18.
Shirley M. Avatrombopag: first global approval. Drugs. 2018;78:1163–8.
Cheloff AZ, Al-Samkari H. Avatrombopag for the treatment of immune thrombocytopenia and thrombocytopenia of chronic liver disease. J Blood Med. 2019;10:313–21.
Tarantino MD, et al. A phase 3, randomized, double-blind, active-controlled trial evaluating efficacy and safety of avatrombopag versus eltrombopag in ITP. Br J Haematol. 2023;202(4):897–9.
Podolanczuk A, et al. Of mice and men: an open-label pilot study for treatment of immune thrombocytopenic purpura by an inhibitor of Syk. Blood. 2009;113(14):3154–60.
Hughes DM, et al. Transitioning from thrombopoietin agonists to the novel SYK inhibitor fostamatinib: a multicenter, real-world case series. J Adv Pract Oncol. 2021;12(5):508–17.
Tungjitviboonkun S, et al. Efficacy and safety of fostamatinib in refractory immune thrombocytopenia: a meta-analysis from randomized controlled trials. Ann Hematol. 2024. https://doi.org/10.1007/s00277-024-05824-7.
Bussel J, et al. Fostamatinib for the treatment of adult persistent and chronic immune thrombocytopenia: results of two phase 3, randomized, placebo-controlled trials. Am J Hematol. 2018;93(7):921–30.
Kuter DJ, et al. Oral rilzabrutinib, a Bruton tyrosine kinase inhibitor, showed clinically active and durable platelet responses and was well-tolerated in patients with heavily pretreated immune thrombocytopenia. Blood. 2020;136:13–4.
Langrish CL, et al. Preclinical efficacy and anti-inflammatory mechanisms of action of the bruton tyrosine kinase inhibitor rilzabrutinib for immune-mediated disease. J Immunol. 2021;206(7):1454–68.
Kuter DJ, et al. Rilzabrutinib, an oral BTK, in inhibitor immune thrombocytopenia. N Engl J Med. 2022;386(15):1421–31.
Newland AC, et al. Phase 2 study of efgartigimod, a novel FcRn antagonist, in adult patients with primary immune thrombocytopenia. Am J Hematol. 2020;95(2):178–87.
Tsykunova G, et al. PB2313: daratumumab as a treatment for adult immune thrombocytopenia: a phase II study with safety run-in (the dart study). HemaSphere. 2022;6:2182–3.
Vernava I, Schmitt CA. Daratumumab as a novel treatment option in refractory ITP. Blood Cells Mol Dis. 2023;99: 102724.
Strussmann T, et al. Long-term complete remission of refractory severe idiopathic immune thrombocytopenia (ITP) treated with daratumumab. Ann Hematol. 2023;102(1):245–7.
LeVine DN, et al. A novel canine model of immune thrombocytopenia: has immune thrombocytopenia (ITP) gone to the dogs? Br J Haematol. 2014;167(1):110–20.
van Bladel ER, et al. Functional platelet defects in children with severe chronic ITP as tested with 2 novel assays applicable for low platelet counts. Blood. 2014;123(10):1556–63.
Bal G, et al. Identification of novel biomarkers in chronic immune thrombocytopenia (ITP) by microarray-based serum protein profiling. Br J Haematol. 2016;172(4):602–15.
Kamhieh-Milz J, et al. Identification of novel autoantigens via mass spectroscopy-based antibody-mediated identification of autoantigens (MS-AMIDA) using immune thrombocytopenic purpura (ITP) as a model disease. J Proteomics. 2017;157:59–70.
Neunert CE, Lambert MP. More than one pathway: novel treatment for ITP. Blood. 2019;133(7):629–30.
Zhao HY, et al. Low-dose chidamide restores immune tolerance in ITP in mice and humans. Blood. 2019;133(7):730–42.
Sundin M, et al. “Immune” thrombocytopenia as key feature of a novel ADA2 deficiency variant: implication on differential diagnostics of ITP in children. J Pediatr Hematol Oncol. 2019;41(2):155–7.
Ali MA, et al. The Ifng antisense RNA 1 (IFNG-AS1) and growth arrest-specific transcript 5 (GAS5) are novel diagnostic and prognostic markers involved in childhood ITP. Front Mol Biosci. 2022;9:1007347.
Nakajima-Kato Y, et al. A novel monoclonal antibody with improved FcgammaR blocking ability demonstrated non-inferior efficacy compared to IVIG in cynomolgus monkey ITP model at considerably lower dose. Clin Exp Immunol. 2023;211(1):23–30.
Acknowledgements
We would like to thank the reviewers for their time, insightful comments, and invaluable guidance. Their thoughtful suggestions have significantly improved the quality and clarity of this manuscript. We greatly appreciate their dedication and expertise.
Funding
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
Author information
Authors and Affiliations
Contributions
Songphol Tungjitviboonkun wrote the first draft of the manuscript. Naharuthai Bumrungratanayos revised the manuscript. All authors reviewed and edited the manuscript and approved the final version of the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Competing interests
The authors declare no conflicts of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Tungjitviboonkun, S., Bumrungratanayos, N. Immune thrombocytopenia (ITP): historical perspectives, pathophysiology, and treatment advances. Discov Med 1, 7 (2024). https://doi.org/10.1007/s44337-024-00008-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s44337-024-00008-8