
Abstract
The Wnt/β-catenin signaling pathway is associated with the pathogenesis of steroid-induced osteonecrosis. Our investigation studied whether aberrant CpG island hypermethylation of the FZD1 gene was present in patients with osteonecrosis of the femoral head (ONFH), which results in Wnt/β-catenin signaling inactivation and subsequent cell dysfunction. Bone marrow was collected from the proximal femurs of patients with steroid-associated ONFH (n = 21) and patients with new femoral neck fractures (n = 22), and then mesenchymal stem cells (MSCs) were isolated. We investigated cell viability, the transcription and translation levels of Wnt/β-catenin signaling-related genes, the extent of methylation at CpG islands of the FZD1 promoter, and the osteogenic and adipogenic differentiation abilities of MSCs from the control group and from the ONFH group treated with or without 5′-Aza-dC. According to the results, MSCs from the ONFH group showed a reduced proliferation ability, low transcription and translation levels of FZD1, inhibition of the Wnt/β-catenin signaling pathway, weakened osteogenesis and enhanced adipogenesis ability. Aberrant CpG island hypermethylation of FZD1 was observed in the ONFH group. Treatment with 5’-Aza-dC resulted in de novo FZD1 expression, reactivation of the Wnt/β-catenin signaling pathway and promotion of osteogenesis. Taken together, our study not only provides novel insights into the regulation of the Wnt/β-catenin signaling pathway in this disease but also reveals potential for the use of demethylating agents for the treatment of GC-associated ONFH.
Similar content being viewed by others


Introduction
Since glucocorticoids (GC) are widely used in daily clinical practice, their exogenous usage ranks first among the known risk factors for nontraumatic osteonecrosis of the femoral head, called glucocorticoid-induced osteonecrosis of femoral head (ONFH). Currently, several alternative mechanisms have been postulated for GC-induced ONFH, such as fat embolisation1, intramedullary pressure changes2, modified arterial constriction3,4, circulatory impairment5, coagulation disorders6, and cell dysfunction7,8,9. However, none of these can explain the underlying mechanism. Our previous study demonstrated that the abnormal differentiation of bone marrow mesenchymal stem cells (MSCs) is a principal mechanism involved in the onset and progression of this disease10,11.
MSCs, originally identified in adult bone marrow, can proliferate and differentiate into multiple mesodermal lineages, such as osteoblasts12, cardiocytes13,chondrocytes14, and adipocytes15. In GC-induced ONFH, decreased numbers, hypoproliferative activity and abnormal differentiation of MSCs of the femoral head, neck, and metaphysis result in poor self-repair and a negative prognosis. When the adipocyte population becomes more numerous than the osteoblast population, the intraosseous pressure may rise, ultimately causing ONFH. Therefore, to treat GC-associated ONFH, it is important to inhibit MSCs from undergoing adipogenesis and promote their osteogenesis.
The canonical Wnt signaling pathway plays a vital role in the regulation of bone homeostasis, which inhibits the differentiation of MSCs into chondrocytes and adipocytes while promoting osteoblastic differentiation16,17,18. This pathway controls both the proliferation and differentiation of osteoblastic precursors and maintains mature osteoblasts19,20. As a receptor for the Wnt signaling pathway, Frizzled1 (FZD1) plays an important role in osteoblast mineralization. Its promoter is regulated by several transcription factors, including Sp1, E2F1, and AP221,22,23. Recent studies showed that FZD1 is closely related to bone mineral density (BMD)24,25. However, its expression in the MSCs of patients with steroid-induced ONFH has not been investigated yet.
In this study, we focused on FZD1 in MSCs and investigated whether FZD1-promoter hypermethylation is present in patients with ONFH and results in Wnt/β-catenin signaling inactivation and subsequent cell dysfunction. We performed methylation analysis of the promoter CpG islands of FZD1 by bisulfite sequencing (BSP) and investigated whether demethylation of FZD1 exerts a beneficial influence on cell viability and osteogenic differentiation. We attempted to confirm that FZD1-promoter hypermethylation is related to Wnt/β-catenin signaling inactivation in the development of GC-induced ONFH.
Materials and Methods
Patient recruitment
This study was approved by the Ethics Committee of Renmin Hospital. All methods were performed in accordance with the relevant guidelines and regulations of the authors’ institution. Between June 2016 and May 2017, 21 patients (10 men, 11 women; mean age 51.6, ranging from 38–63 years) with GC-induced ONFH were selected at the authors’ institution (Renmin Hospital, Wuhan, China), and 22 subjects with femoral neck fractures (11 men and 11 women; mean age 54.3, ranging from 37 to 70 years) were enrolled as controls. Clinical characteristics for all participants are summarized summarized in Table 1. For GC-induced ONFH, the steroid exposure threshold is 1800 mg GC or its equivalent over 4 weeks26. After written informed consent was obtained from patients, bone marrow aspirates (5 mL) were procured from the proximal ends of femurs while inserting a tapered awl into the femoral canal during hip replacement surgery.
Cell culture
hMSCs were isolated from bone marrow aspirates and cultured as described previously27. Adherent cells were cultured for 12 to 14 days until they attained a confluence of greater than 80%. The cells were then digested with a solution of 0.25% trypsin and 0.02% EDTA (Invitrogen, Carlsbad, CA, USA) and replated at a 1:2 dilution for initial subculture. hMSCs underwent this treatment three times before they were collected for further use.
Cell viability measurement
As described previously, MSCs were plated in 96-well plates at a density of 2 × 103 cells per well28. After adherence to the plates, the initial defining medium was aspirated away and replaced with complete medium supplemented with 5’-Aza-dC (Sigma-Aldrich, St. Louis, MO) in the treatment group. At 24, 48, and 72 h, cell proliferation was assayed by MTT according to the manufacturer’s instructions.
Immunofluorescent staining
Cells were fixed in 4% paraformaldehyde for 24 h, permeated with 0.2% Triton X-100 (Sigma), blocked, and then finally incubated with primary antibodies at a dilution of 1:100 at 4 °C overnight. Cells selected for DKK1, FZD1, and β-catenindetection were incubated with FITC- and Cy3-conjugated secondary antibodies at 37 °C for an additional one hour using standard concentrations from the supplier. Then, the slides were washed and mounted with CitiFluormountant (Agar Scientific, UK).
Western blot
Cells were washed twice with ice-cold PBS, scraped into 0.2 mL of buffer (50 mM Tris with pH 7.4, 150 mM NaCl, 1% Triton X-100, 1% sodium deoxycholate, 0.1% SDS, and 0.05 mM EDTA), and incubated on ice for 20 min, followed by centrifugation at 12,000 rpm for 10 min. Protein concentrations were quantified by a BCA Protein Assay Kit (Beyotime Institute of Biotechnology, China). Afterwards, proteins were diluted to equal concentrations, boiled for 5 min, separated by 10% SDS-PAGE, and then blotted onto polyvinylidene fluoride (PVDF) membranes (Millipore, USA), which were probed with a FZD1 antibody (1:100, Abcam #ab71342), β-catenin antibody (1:1000, Abcam #ab16051), Runx2 antibody (1:1000, CST #12556) and PPARγ antibody (1:1000, CST #2435) overnight at 4 ℃. Membranes were incubated with horseradish peroxidase-conjugated secondary antibodies for 1 h at room temperature (Boster Biosciences, China). GAPDH was used to normalize for protein loading.
Adipogenic and osteogenic differentiation
Adipogenic and osteogenic differentiation of human MSCs were performed as previously described29. For Oil red O staining, the cells were washed twice with PBS and fixed with 4% formaldehyde in PBS for 30 min at room temperature. Subsequently, they were stained for 1 h at room temperature with filtered Oil red O solution, washed twice with PBS, visualized under light microscopy and photographed. To extract the incorporated Oil red O, 1 mL of isopropanol was added to each well followed by 15 min of shaking at room temperature. After appropriate dilution, the absorbance of triplicate samples was read at 490 nm.
For ALP staining, the cells were washed twice with PBS and fixed with 4% formaldehyde in PBS for 30 min at room temperature. After three washes with PBS, ALP staining was performed by the addition of 5 ml of staining buffer (100 mM Tris-HCl, 150 mM NaCl, 1 mM MgCl2) containing chromogen substrate solution composed of 33 μl of 50 mg/ml nitro blue tetrazolium (NBT) and 16.5 μl of 50 mg/ml 5-bromo-4-chloro-3-indolyl phosphate (BCIP). Cells were stained with BCIP/NBT substrate for 30 min. Finally, the substrate solution was removed, and the cells were rinsed with deionized water, visualized under light microscopy and photographed.
For Alizarin Red S (ARS) staining, the cells were washed twice with PBS and fixed with 4% formaldehyde in PBS for 30 min at room temperature. After a brief wash with PBS, they were stained for 20 min with 40 mM ARS solution (pH 4.2). Next, they were rinsed five times with PBS to reduce nonspecific staining. Using Meta Morph imaging software (Universal Imaging, Downingtown, PA), osteogenic differentiation was quantified by measuring the area stained with Alizarin Red S. Measurements were performed in duplicate for each experiment, and experiments were repeated three times.
Quantitative real-time PCR
Total RNA was extracted via standard protocols using standard commercial kits (TRIZOL® Reagent, Invitrogen, USA). Real-time PCR was performed using SYBR Green Master Mix according to the protocols of the supplier (Invitrogen, Carlsbad, CA, USA). The primer sequences are shown in Table 2. The SYBR Green signal was detected by a StepOne™ real-time PCR machine (ABI, USA). The relative levels of transcript expression were quantified using the ΔΔCt method. All real-time PCR was run in triplicate, and gene expression was analyzed using an ABI PRISM 7900HT Sequence Detection System (Applied Biosystems, USA).
Bisulfite sequencing
Bisulfite conversion was performed as previously described30. Briefly, total genomic DNA was isolated from MSCs using a DNeasy Tissue Kit (Cwbiotech). Two micrograms of genomic DNA were denatured in a volume of 50 μL by freshly prepared 0.3 M NaOH for 30 min at 42 ℃. After denaturation, 30 μL of freshly prepared hydroquinone (10 mM) and 510 μL of sodium bisulfite (3.6 M, pH 5.0) were added and incubated at 50 ℃ for 16 h. Modified DNA was purified using a DNeasy spin column (Qiagen) and eluted in a volume of 50 μL. This was followed by desulfonification by adding 5.5 μL of 3 M NaOH for 15 min at 37 ℃. Samples were neutralized by adding 33 μL of ammonium acetate (10 M, pH 7.0), followed by ethanol precipitation and resuspension in water.
PCR was performed at 95 ℃ for 5 min followed by 40 cycles of 95 ℃ for 30 s, 55 ℃ for 30 s and 72 ℃ for 1 min with a final extension at 72 ℃ for 7 min. Primer pairs were designed using a custom interface with primer3. The PCR products were tested in 2% agarose gel and then cloned into the pEASY-T1 vector (TransGen Biotech, Beijing, China). Colony PCR was undertaken to screen the positive colonies. Clones with correctly sized PCR products were sequenced on an ABI sequencer with dye terminators (Applied Biosystems, Foster City, CA, USA). Based on the sequencing results for ten clones, the methylation frequency was determined for each CpG site.
Statistical analysis
Statistical analysis was carried out via SPSS version 13.0 for Windows (SPSS, Chicago, IL, USA). Significant differences were determined using either the Mann-Whitney U test or the Wilcoxon signed-rank test. Data are presented as the mean ± standard deviation. Probabilities lower than 5% (P < 0.05) were considered statistically significant. All experiments were repeated three or more times.
Results
Measurement of hMSC viability
According to Fig. 1a, cellular viability reached its peak at the dosage of 15 μM, and obvious decreases were seen at dosages of 30 μM and above. Hence, the concentration of 15 μM was considered moderate and chosen for use in subsequent experiments.

a Cellular viability reached its peak at the dosage of 15 μM at t = 72 h. b The proliferation capacity of MSCs at t = 24 h, 48 h and 72 h. *P < 0.05 vs. ONFH group, 5’-Aza-dC group at t = 24 h; **P < 0.01 vs. ONFH group at t = 48 h; ***P < 0.001 vs. ONFH group at t = 72 h; #P > 0.05 vs. ONFH group at t = 24 h, 48 h; ##P > 0.05 vs. 5’-Aza-dC group at t = 72 h; $P < 0.01 vs. ONFH group at t = 72 h
As shown in Fig. 1b, the cellular viability in the control group was remarkably stronger than that in the GC-induced ONFH group at three different time points (P < 0.05). Following treatment with 15 μM 5’-Aza-dC, the cellular viability in the GC-induced ONFH group increased by more than one fourth but was still a little lower than that in the control group at t = 72 h (P < 0.05).
Immunofluorescent staining
Compared to the control group, FZD1 and β-catenin showed obviously weak staining and Dkk1showed strong staining in the ONFH group. Following treatment with 5’-Aza-dC, β-catenin and FZD1 staining was obviously increased but was still weaker than that of the control group (Fig. 2a–f). However, 5′-Aza-dC treatment obviously decreased Dkk1 staining. The control group had weak Dkk1 staining (Fig. 2g–i). In addition, compared to the ONFH group, we found that 5’-Aza-dC increased the accumulation of β-catenin-Cy3 complexes in the nucleus, but this accumulation was still lower than that of the control group.

MSCs were stained with β-catenin (red), FZD1 (green) and DKK1 (green) antibodies (green), and the cells were co-stained with DAPI to indicate the nuclei (blue). a–c β-catenin showed obviously weak staining in the ONFH group. Following treatment with 5′-Aza-dC, β-catenin staining was obviously increased but was still weaker than that of the control group. d–f FZD1 showed obviously weak staining in the ONFH group. Following treatment with 5’-Aza-dC, FZD1 staining was obviously increased but was also weaker than that of the control group. g–i Dkk1 had strong staining in the ONFH group. Treatment with 5′-Aza-dC obviously decreased Dkk1 staining. The control group had weak Dkk1 staining (bars 50 μm)
Adipogenic and osteogenic differentiation
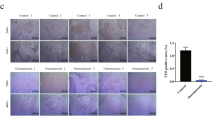
As shown in Fig. 3a–d, compared to control group, oil red O showed strong deposition in the GC-associated ONFH group after induction for 3weeks (P < 0.05). Treatment with 15 μM 5’-Aza-dC resulted in weakened staining, which was almost the same level as that of the control group (P > 0.05).

MSCs from all groups were induced to adipocytes and osteoblasts for 21 days. a Oil red O staining of the ONFH group. b Oil red O staining of the 5’-Aza-dC group. c Oil red O staining of the control group. d Incorporated Oil red O was extracted with isopropanol, and quantification was performed as described in the “Methods” section. e BCIP/NBT staining of the ONFH group. f BCIP/NBT staining of the 5′-Aza-dC group. g BCIP/NBT staining of the control group. h Alizarin red S staining of the ONFH group. (i) Alizarin red S staining of the 5’-Aza-dC group. j Alizarin red S staining of the control group. k The area stained with alizarin red S was measured as described in the Methods. Data are represented as the mean value ± standard deviation. *P < 0.01 vs. 5’-Aza-dC group, control group. #P < 0.05 vs. control group (bars 100 μm)
As shown in Fig. 3e–g, widespread blue-purple staining could be detected in the 5’-Aza-dC and control groups with light microscopy after conversion of the substrate BCIP/NBT by ALP. However, positive staining was hardly observed in the ONFH group.
As shown in Fig. 3h–k, ARS staining of the GC-associated ONFH group was weakest among all the groups. Treatment with 5’-Aza-dC resulted in alizarin red S staining with deeper intensity, which indicated enhanced calcium mineralization compared with that of the GC-associated ONFH group (P < 0.05).
Western blot
Our results showed that 5’-Aza-dC markedly promoted FZD1, β-catenin and Runx2 translation while inhibiting PPARγ translation. It was observed that MSCs in the ONFH group showed obviously low expression of FZD1 and β-catenin compared to that of the control group. When treated with 5’-Aza-dC for 72 h, this expression correspondingly increased (Fig. 4). Cells in the ONFH group had strong PPARγ expression and weak Runx2 expression compared to that of the control group. Following pretreatment with 5’-Aza-dC for 72 h, this expression reached nearly the same levels as those of the control group.

a Western blot analysis of FZD1, β-catenin, Runx2 and PPARγ. b Gray level analysis of western blot. *P < 0.05 vs. 5’-Aza-dC group. #P < 0.001 vs. ONFH group.$P < 0.01 vs. 5′-Aza-dC group, 5′-Aza-dC group. **P < 0.001 vs. ONFH group
Quantitative real-time PCR
Our results showed that 15 μM5’-Aza-dC markedly promoted the transcription of FZD1, CyclinD1, Axin2, and osteogenesis-related genes (RUNX2, osteocalcin) while inhibiting adipogenesis-related gene (PPARγ) transcription. As shown in Fig. 5, the amount of FZD1 transcripts in the ONFH group was the lowest among all groups (P < 0.01), reaching a level nearly 5-fold higher at 72 h after treatment with 5’-Aza-dC, which was almost the same level as that of the control group (P > 0.05). Meantime, CyclinD1 and Axin2 transcription was elevated to varying degrees by treatment with 5’-Aza-dC. Pretreatment with 5’-Aza-dC also helped to promote RUNX2 and osteocalcin transcription and decrease PPARγ transcription.

The transcript levels of osteogenesis, adipogenesis and Wnt/β-catenin signaling pathway-related genes in the three groups. Data are represented as the mean value ± standard deviation. *P < 0.01 vs. ONFH group
Bisulfite sequencing
To determine whether the increased expression of FZD1 in MSCs had an underlying epigenetic basis, DNA methylation status in the FZD1 promoter was examined by bisulfite sequencing. A schematic overview of the promoter structure is shown in Fig. 6. Two regions in the promoter were selected: region 1, -350 to -112, and region 2, -115 to + 165. CpG hypermethylation was detected within the CpG island in the GC-associated ONFH group. After treatment with 15 μM 5′-Aza-dC for 72 h, the methylation ratio of region 1 decreased from 33.0 to 1.8%, while the methylation level of region 2 was clearly reduced from 38.6 to 4.6%. The methylation ratios of the control group were 0 (region 1) and 3.6% (region 2) (Fig. 6).

Region 1 (−350 to −112 bp): The methylation ratios were 33.0% in the ONFH group, 1.8% in the 5’-Aza-dC group and 0 in the control group. Region 2 (−115 to + 165 bp): The methylation ratios were 38.6% in the ONFH group, 4.6% in the 5’-Aza-dC group and 3.6% in the control group
Discussion
Currently, we see an increased incidence of ONFH due to the widespread use of exogenous glucocorticoids. At the present time, the precise mechanism underlying steroid-associated ONFH has not yet been elucidated, although numerous studies have been done. Epigenetics, an effective method to study the interplay between environmental signals and the genome, has received a great deal of recent attention. Our previous studies demonstrated that DNA hypermethylation plays an important role in the pathophysiology of GC-associated ONFH. It is beneficial for dysfunctional MSCs to reverse DNA hypermethylation modifications10,11. However, the exact mechanism has not yet been clarified.
The Wnt/β-catenin signaling pathway is reported to be involved in the pathogenesis of early-stage ONFH31. Mutations in components of the Wnt/β-catenin pathway are associated with decreased bone mineral density and increased fracture incidence as well as other skeletal disorders32. Dickkopf-1 (DKK1), one of the Wnt inhibitors, showed increased expression in ONFH patients with histories of corticosteroid and alcohol intake33. Some studies demonstrated that FZD1 is very important in osteoblast differentiation and mineralization, and its promoter can be regulated by several transcription factors21,22,23. Our pilot study showed that FZD1 was obviously decreased at the transcription and translation levels in GC-associated ONFH. Therefore, we hypothesized that FZD1 promoter hypermethylation resulted in low expression of FZD1, downregulation of the Wnt/β-catenin signaling pathway and MSC dysfunction in the MSCs of patients with GC-associated ONFH. Reversal of the hypermethylation status of the FZD1 promoter is helpful for reactivation of the Wnt/β-catenin signaling pathway and improvement of MSC osteogenic differentiation.
In this study, we assessed methylation at CpG islands of the FZD1 gene in MSCs from 43 patients with GC-induced ONFH and femoral neck fractures. We found that the methylation ratio of the FZD1 promoter in the GC-induced ONFH group was obviously higher than that in femoral neck fractures group. Additionally, FZD1 mRNA and protein levels in the GC-induced ONFH group were one sixth and one tenth of those in femoral neck fractures group, respectively. Apparently, expression of the FZD1 gene was partly inhibited due to the promoter hypermethylation in the GC-induced ONFH group.
5′-Aza-dC, an inhibitor of DNA methyltransferase (DNMT), was demonstrated to be involved in cell proliferation and differentiation34. DNA methylation is a key mechanism associated with pluripotency and self-renewal of stem cells. It was reported that 5-Aza induced the direct conversion of adult gingival MSCs into cells of three embryonic lineages, suggesting their potential use for autologous cell therapy35. Cho et al. demonstrated that epigenetic modification induced by 5’-Aza-dC permitted direct programming of adipocytes into osteoblasts in a mouse model of osteoporosis, indicating that 5’-Aza-dC is a promising treatment for osteoporosis36.
Notably, we observed that cellular viability in the GC-induced ONFH group was remarkably lower than that in the control group at three different time points. Following treatment with 15 μM 5′-Aza-dC, cellular viability in the ONFH group increased by more than one fourth, while methylation ratios decreased to 1.8% (region 1) and 4.6% (region 2). This finding is consistent with our previous article10,11 and the recent report from Katarzyna Kornicka et al.37, who found that 5-azacytidine reduced reactive oxygen species accumulation, ameliorated superoxide dismutase activity, increased cellular proliferation and the BCL-2/BAX ratio, and ultimately slowed down and even reversed aged-related degenerative changes in MSCs. Our previous data showed the similar effects of 5-azacytidine on the MSCs of patients with GC-associated ONFH.
From an immunofluorescence analysis, we found that the MSCs of patients with GC-associated ONFH demonstrated hampered intranuclear translocation of β-catenin compared to that of the control group. 5’-Aza-dC can promote the intranuclear translocation of β-catenin. FZD1 was weakly expressed in the MSCs of patients with GC-associated ONFH, and this expression was increased by treatment with 5’-Aza-dC. However, expression of the DKK1 protein exhibited an opposite pattern. The PCR results showed that 5’-Aza-dC also increased the mRNA expression of FZD1, Axin 2, Cyclin D1 and Runx2, which were decreased in GC-associated ONFH. Moreover, MSCs treated with 5’-Aza-dC showed weakened adipogenesis and enhanced osteogenesis according to the oil red O, ALP and ARS staining results. Taken together, there is a good possibility that the beneficial effects of 5’-Aza-dC on GC-associated ONFH are attributable to FZD1-promoter demethylation of MSCs and reactivation of the Wnt/β-catenin signaling pathway. However, the underlying mechanism must be studied further.
In summary, this study has demonstrated that aberrant CpG island hypermethylation of the FZD1 gene is present in patients with ONFH, resulting in Wnt/β-catenin signaling inactivation and subsequent cell dysfunction. By comparative observation, we confirmed that 5’-Aza-dC, at a suitable concentration, benefits the MSCs of patients with GC-associated ONFH by inducing de novo FZD1 expression. This study has provided new information regarding treatment targeting epigenetic changes in ONFH. However, more specific in vivo studies are needed to further elucidate the molecular pathomechanism underlying the development of ONFH.
References
Murata, M., Kumagai, K., Miyata, N., Osaki, M. & Shindo, H. Osteonecrosis in stroke-prone spontaneously hypertensive rats: effect of glucocorticoid. J. Orthop. Sci. 12, 289–295 (2007).
Yeh, C. H., Chang, J. K., Wang, Y. H., Ho, M. L. & Wang, G. J. Ethanol may suppress Wnt/beta-catenin signaling on human bone marrow stroma cells: a preliminary study. Clin. Orthop. Relat. Res. 466, 1047–1053 (2008).
Drescher, W. et al. Enhanced constriction of supplying arteries-a mechanism of femoral head necrosis in Wistar rats? Ann. Anat. 192, 58–61 (2010).
Drescher, W. et al. Femoral artery constriction by norepinephrine is enhanced by methylprednisolone in a rat model. J. Bone Jt. Surg. Am. 88, 162–166 (2006).
Chan, W. P. et al. MRI of joint fluid in femoral head osteonecrosis. Skelet. Radiol. 31, 624–630 (2002).
Cuadrado, M. J. & Lopez-Pedrera, C. Antiphospholipid syndrome. Clin. Exp. Med 3, 129–139 (2003).
Yun, S. I., Yoon, H. Y., Jeong, S. Y. & Chung, Y. S. Glucocorticoid induces apoptosis of osteoblast cells through the activation of glycogen synthase kinase 3beta. J. Bone Miner. Metab. 27, 140–148 (2009).
Chen, C. et al. Impairment of two types of circulating endothelial progenitor cells in patients with glucocorticoid-induced avascular osteonecrosis of the femoral head. Joint Bone Spine 80, 70–76 (2012).
Varoga, D., Drescher, W., Pufe, M., Groth, G. & Pufe, T. Differential expression of vascular endothelial growth factor in glucocorticoid-related osteonecrosis of the femoral head. Clin. Orthop. Relat. Res 467, 3273–3282 (2009).
Sun, Z. et al. Aberrant CpG islands hypermethylation of ABCB1 in mesenchymal stem cells of patients with steroid-associated osteonecrosis. J. Rheumatol. 40, 1913–1920 (2013).
Sun, Z. et al. Icariin may benefit the mesenchymal stem cells of patients with steroid-associated osteonecrosis by ABCB1-promoter demethylation: a preliminary study. Osteoporos. Int 26, 187–197 (2015).
Hao, C. et al. MiR-708 promotes steroid-induced osteonecrosis of femoral head, suppresses osteogenic differentiation by targeting SMAD3. Sci. Rep. 6, 22599 (2016).
Kai, D., Prabhakaran, M. P., Jin, G., Tian, L. & Ramakrishna, S. Potential of VEGF-encapsulated electrospun nanofibers for in vitro cardiomyogenic differentiation of human mesenchymal stem cells. J. Tissue Eng. Regen. Med. 11, 1002–1010 (2017).
Sun, Z. et al. Growth differentiation factor 5 modulation of chondrogenesis of self-assembled constructs involves gap junction-mediated intercellular communication. Dev. Growth Differ. 54, 809–817 (2012).
Madrigal, A., Tan, L. & Zhao, Y. Expression regulation and functional analysis of RGS2 and RGS4 in adipogenic and osteogenic differentiation of human mesenchymal stem cells. Biol. Res. 50, 43 (2017).
Bullock, W. A. & Robling, A. G. WNT-mediated Modulation of Bone Metabolism: Implications for WNT Targeting to Treat Extra skeletal Disorders. Toxicol. Pathol. 45, 864–868 (2017).
Li, Y. et al. PPAR-γ and Wnt Regulate the Differentiation of MSCs into Adipocytes and Osteoblasts Respectively. Curr. Stem Cell Res Ther. 13, 185–192 (2018).
Li, R. et al. Self-assembled N-cadherin mimetic peptide hydrogels promote the chondrogenesis of mesenchymal stem cells through inhibition of canonical Wnt/β-catenin signaling. Biomaterials 145, 33–43 (2017).
Li, J. P., Zhuang, H. T., Xin, M. Y. & Zhou, Y. L. MiR-214 inhibits human mesenchymal stem cells differentiating into osteoblasts through targeting β-catenin. Eur. Rev. Med. Pharmacol. Sci. 21, 4777–4783 (2017).
Xu, M. L., Bi, C. W. C., Liu, E. Y. L., Dong, T. T. X. & Tsim, K. W. K. Wnt3a induces the expression of acetylcholinesterase during osteoblast differentiation via the Runx2 transcription factor. J. Biol. Chem. 292, 12667–12678 (2017).
Yu, S., Yerges-Armstrong, L. M., Chu, Y., Zmuda, J. M. & Zhang, Y. Transcriptional Regulation of Frizzled-1 in Human Osteoblasts by Sp1. PLoS ONE 11, e0163277 (2016).
Yu, S., Yerges-Armstrong, L. M., Chu, Y., Zmuda, J. M. & Zhang, Y. E2F1 effects on osteoblast differentiation and mineralization are mediated through up-regulation of frizzled-1. Bone 56, 234–241 (2013).
Yu, S., Yerges-Armstrong, L. M., Chu, Y., Zmuda, J. M. & Zhang, Y. AP2 suppresses osteoblast differentiation and mineralization through down-regulation of Frizzled-1. Biochem J. 465, 395–404 (2015).
Yerges, L. M. et al. Functional characterization of genetic variation in the Frizzled 1 (FZD1) promoter and association with bone phenotypes: more to the LRP5 story? J. Bone Miner. Res 24, 87–96 (2009).
Zhang, Y. et al. Functional and association analysis of frizzled 1 (FZD1) promoter haplotypes with femoral neck geometry. Bone 46, 1131–1137 (2010).
Koo, K. H. et al. Risk period for developing osteonecrosis of the femoral head in patients on steroid treatment. Clin. Rheumatol. 21, 299–303 (2002).
Vivas, D., Caminal, M., Oliver-Vila, I. & Vives, J. Derivation of multipotent mesenchymal stromal cells from ovine bone marrow. Curr. Protoc. Stem Cell Biol. 44, 2B.9.1–2B.9.22 (2018).
Sun, Z., Nair, L. S. & Laurencin, C. T. The paracrine effect of adipose-derived stem cells inhibits IL-1β-induced inflammation in chondrogenic cells through the Wnt/β-catenin signaling pathway. Regen. Eng. Transl. Med 4, 35–41 (2018).
Yu, W. H. et al. PPARγ suppression inhibits adipogenesis but does not promote osteogenesis of human mesenchymal stem cells. Int J. Biochem Cell Biol. 44, 377–384 (2012).
Irizarry, R. A. et al. The human colon cancer methylome shows similar hypo- and hypermethylation at conserved tissue-specific CpG island shores. Nat. Genet 41, 178–186 (2009).
Zhang, C., Zou, Y. L., Ma, J., Dang, X. Q. & Wang, K. Z. Apoptosis associated with Wnt/ β-catenin pathway leads to steroid-induced avascular necrosis of femoral head. BMC Musculoskelet. Disord. 16, 132 (2015).
Baron, R. & Kneissel, M. WNT signaling in bone homeostasis and disease: from human mutations to treatments. Nat. Med 19, 179–192 (2013).
Ko, J. Y. et al. Increased Dickkopf-1 expression accelerates bone cell apoptosis in femoral head osteonecrosis. Bone 46, 584–591 (2010).
Abbey, D. & Seshagiri, P. B. Aza-induced cardiomyocyte differentiation of P19 EC-cells by epigenetic co-regulation and ERK signaling. Gene 526, 364–373 (2013).
Diomede, F. et al. 5-aza exposure improves reprogramming process through embryoid body formation in human gingival stem cells. Front Genet 9, 419 (2018).
Cho, Y. D. et al. Epigenetic priming confers direct cell trans-differentiation from adipocyte to osteoblast in a transgene-free state. J. Cell Physiol. 231, 1484–1494 (2016).
Kornicka, K., Marycz, K., Marędziak, M., Tomaszewski, K. A. & Nicpoń, J. The effects of the DNA methyltranfserases inhibitor 5-Azacitidine on ageing, oxidative stress and DNA methylation of adipose derived stem cells. J. Cell Mol. Med. 21, 387–401 (2017).
Acknowledgements
This work was supported by grants from the National Natural Science Foundation of China (No. 81501894 and No. 81301052), the Health and Family Planning Commission of Wuhan (No.WX16C28), the Fundamental Research and Advanced Technology Program of Henan Province (162300410093), and the Medical Science and Technology Program of Henan Province (201602196).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wu, F., Jiao, J., Liu, F. et al. Hypermethylation of Frizzled1 is associated with Wnt/β-catenin signaling inactivation in mesenchymal stem cells of patients with steroid-associated osteonecrosis. Exp Mol Med 51, 1–9 (2019). https://doi.org/10.1038/s12276-019-0220-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s12276-019-0220-8
- Springer Nature Limited
This article is cited by
-
17 variants interaction of Wnt/β-catenin pathway associated with development of osteonecrosis of femoral head in Chinese Han population
Scientific Reports (2024)
-
DNA methylation in the OPG/RANK/RANKL pathway is associated with steroid-induced osteonecrosis of the femoral head
BMC Musculoskeletal Disorders (2021)