
Abstract
The xenotopic expression of the alternative oxidase AOX from the tunicate Ciona intestinalis in diverse models of human disease partially alleviates the phenotypic effects of mitochondrial respiratory chain defects. AOX is a non-proton pumping, mitochondrial inner membrane-bound, single-subunit enzyme that can bypass electron transport through the cytochrome segment, providing an additional site for ubiquinone reoxidation and oxygen reduction upon respiratory chain overload. We set out to investigate whether AOX expression in Drosophila could counteract the effects of mitochondrial DNA (mtDNA) replication defects caused by disturbances in the mtDNA helicase or DNA polymerase γ. We observed that the developmental arrest imposed by either the expression of mutant forms of these enzymes or their knockdown was not rescued by AOX. Considering also the inability of AOX to ameliorate the phenotype of tko25t, a fly mutant with mitochondrial translation deficiency, we infer that this alternative enzyme may not be applicable to cases of mitochondrial gene expression defects. Finding the limitations of AOX applicability will help establish the parameters for the future putative use of this enzyme in gene therapies for human mitochondrial diseases.
Similar content being viewed by others
Introduction
Mutations in the human genes TWNK and POLG, respectively encoding the mitochondrial replicative DNA helicase Twinkle and the catalytic subunit of the mitochondrial replicase pol γ, are associated with diverse human diseases, as well as with aging. In particular, they underlie cases of Progressive External Ophthalmoplegia (PEO) and Alper’s syndrome, for which mitochondrial DNA (mtDNA) depletion, deletions and/or point mutations are frequently observed1,2,3,4. Twinkle and pol γ are essential components of the mtDNA replication machinery. Twinkle hydrolyzes nucleotide tri-phosphate to unwind the parental double-stranded DNA, providing the single-stranded DNA template for pol γ. In concert with the mitochondrial single-stranded DNA-binding protein (mtSSB), Twinkle and pol γ, and possibly replisome proteins that remain to be identified, can promote mtDNA replication and guarantee proper levels of this genome for correct organellar gene expression5,6. mtDNA carries genes that are essential for ATP production via the process of oxidative phosphorylation (OXPHOS).
Mitochondrial diseases exhibit a broad clinical spectrum, often requiring treatments that are specific to each type of disorder, with only a few such treatments shown thus far to be convincingly effective7. However, the fact that OXPHOS is often affected in mitochondrial diseases does provide a possible common target for therapeutic interventions: the respiratory chain (RC). Four of the RC complexes transfer electrons to molecular oxygen, a process coupled to energy transduction forming a proton gradient across the mitochondrial inner membrane, whereas a fifth complex uses this gradient to phosphorylate ADP to ATP. Most organisms possess branches of the electron transfer system known as alternative pathways, which are absent in humans, other vertebrates, and insects8,9. These pathways may represent “safety valves” to alleviate stress arising from RC overload, as is the case in mitochondrial diseases, preventing excess formation of reactive oxygen species (ROS) and allowing reoxidation of primary electron carriers such as NADH. The alternative oxidase (AOX), for example, is a single-subunit enzyme able to transfer electrons from ubiquinol to oxygen, bypassing RC complexes III and IV, without proton pumping. It has been suggested that AOX expression in human mitochondria could be a treatment for patients with mitochondrial diseases, as a bypass therapy7,10.
The transgenic expression of the AOX gene from Ciona intestinalis (Tunicata: Ascidiacea) in cultured human cells, in the mouse, and in the fruitfly Drosophila melanogaster showed several benefits, which are consistent with a role for AOX in buffering OXPHOS stresses, caused by chemical, genetic or physiological damage. It promoted resistance to inhibitors of complexes III and IV11,12,13,14, alleviated the metabolic abnormalities of complex IV-deficient cells derived from patients15 and the deleterious phenotypes of complex IV insufficiency in Drosophila16. AOX function has also been reported to provide some benefit in other models of mitochondrial disorders, such as fly lines with knockdown of pol γ17 or with mutations in dj-1β, the homologue of the human Parkinson’s disease gene DJ112, and in sesB, the homologue of another PEO gene, ANT118. In addition, the rescue by AOX of Drosophila models of Alzheimer’s disease and mid-line closure defects suggests that RC dysfunction and/or excess ROS production are not only involved, but also appear in early stages of these pathogeneses19,20.
We set out to investigate if the beneficial effects of AOX expression in higher metazoans is applicable to fly lines with mtDNA replication defects. Considering the earlier finding that AOX can ameliorate the phenotype of pol γ-depleted flies, which are defective in mtDNA replication, we hypothesized that AOX could have the same effect in flies with defects in the Drosophila homologue of Twinkle. Our data shows that AOX does not rescue developmental lethality, nor alter the biochemical and molecular parameters associated with Twinkle and pol γ mutations or knockdown. This restricts the potential therapeutic use of AOX to specific types of mitochondrial dysfunction.
Results
AOX does not rescue lethality caused by Twinkle mutations
Using standard genetic crosses, we initially created 12 fly lines, in which we combined the AOX variant constructs UAS-empty2nd, UAS-mutAOX2nd, UAS-wtAOX8.1 and UAS-wtAOXF6, with the Twinkle variant constructs UAS-Twinkle WT, UAS-Twinkle K388A, and UAS-Twinkle A442P (see Supplementary Table S1). UAS-empty2nd carries the UAS promoter without any associated transgene, serving as a promoter dilution control, whereas UAS-mutAOX2nd bears the coding sequence for a mutated, inactive form of AOX (mut AOX)21. UAS-wtAOX8.1 and UAS-wtAOXF6, when induced, lead to the expression of AOX at low and high levels, respectively12,21. The Twinkle variant constructs guide the respective overexpression of the wild-type form of the mtDNA helicase (WT), an active-site mutant (K388A), and a PEO mutation-bearing variant (A442P)22. The latter represents a fly mutation homologous to the one found in human PEO families, A475P23.
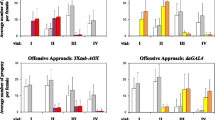
We then crossed the UAS-AOX; UAS-Twinkle lines with the ubiquitous GAL4 driver daGAL4 (see Supplementary Fig. S1 for crossing schemes, and Supplementary Fig. S2 for immunoblot detection of the overexpressed enzymes). Consistent with previously published data22, overexpression of Twinkle WT in the offspring did not affect development, whereas almost all flies expressing the K388A and A442P variants died at the larva L3 or pupal stages (Fig. 1). The lethality of K388A was more severe, as very few larvae were able to enter the pre-pupa stage, dying immediately (Fig. 1b); most L3 larvae were apparently fully developed and mobile at day 5 after egg laying, but remained in this state for an additional 9 days before perishing. Although a very low percentage of adult individuals expressing Twinkle A442P did eclose, indicating that most flies died at the pupa stage, we also observed lethality at the larval stage, judged by the ratio between wild-type and Tubby pupae resulting from the cross UAS-empty2nd; UAS-Twinkle A442P/TM6B-Tubby X daGAL4 (Fig. 1c and Supplementary Fig. S1), which was lower than the expected Mendelian proportion.

AOX does not rescue the lethality caused by the expression of mutant Twinkle proteins. (a) Pupal viability was measured as the mean ratio between the number of adults eclosed and the number of pupae per vial (+/− standard deviation), upon expression (using the daGAL4 driver, left panel) or not (using the w1118 control, right panel) of the indicated AOX and Twinkle variants. (b) A representative developmental assay showing the inability of larvae overexpressing Twinkle K388A to enter the pupal stage: whereas the control adult flies at day 7–9 after egg laying (right panel) were starting to eclose, Twinkle K388A flies were still alive as active larvae on the food, with very few individuals reaching pre-pupa and dying. (c) Larval viability of the UAS-Twinkle A442P lines was measured as the mean ratio between the number of wild-type pupae and the total number of pupae per vial (+/− standard deviation), originated from crosses using UAS-Twinkle A442P / TM6B-Tubby and daGAL4 (induced) or w1118 (uninduced). The expected Mendelian ratio of wild-type pupae (carrying UAS-Twinkle A442P) for these crosses is 0.5 (dashed line). See Supplementary Fig. S1 for details of the genetic crosses. no AOX, mut AOX, low AOX and high AOX indicate respectively the constructs UAS-empty2nd, UAS-mutAOX2nd, UAS-wtAOX8.1 21 and UAS-wtAOXF6 12. Letters a-d indicate significantly different statistical classes (P < 0.05) according to one-way ANOVA, followed by the Tukey post-hoc test, applied separately for the data in (a) left panel, and (c). No significant differences were found (P > 0.05) for the data shown in (a) right panel.
Coexpression of AOX with Twinkle variants in different combinations had only minor effects on adult eclosion rates, and AOX did not rescue the larval lethality caused by Twinkle K388A (Fig. 1a,b). Surprisingly, high levels of functional AOX appeared to intensify the larval lethality caused by Twinkle A442P, as no UAS-wtAOXF6/2; UAS-Twinkle A442P/daGAL4 larva (see Supplementary Fig. S1) ever developed beyond the pre-pupa stage, resembling the more severe K388A phenotype (Fig. 1c). Our data shows that AOX cannot alleviate the developmental disturbances caused by overexpression of Twinkle mutations, and suggests it could in fact aggravate them.
AOX does not alter mtDNA copy number in mutant Twinkle flies
In addition to the expected mtDNA depletion caused by the overexpression of the Twinkle mutants22, we hypothesized that mtDNA copy number might be affected by AOX, since mtDNA maintenance has been linked to mitochondrial membrane potential24 and AOX could potentially lower this parameter by diverting respiratory electron flow. We therefore measured relative mtDNA copy number in our fly lines via quantitative PCR, but found that AOX does not significantly change mtDNA levels in any Twinkle combination (Fig. 2). Overexpressing Twinkle WT did not alter mtDNA copy number in L3 larvae, although a 30–40% increase was observed in adult flies (Fig. 2). Copy number increase upon Twinkle WT overexpression has been reported previously for Drosophila adults22 and cells in culture25, for human cells in culture26 and for mice27,28. As expected, overexpression of Twinkle K388A and A442P depleted mtDNA in L3 larvae to 20–40% of control levels, independently of AOX expression.

AOX does not interfere with mtDNA copy number in mutant Twinkle lines. Total DNA from larva and adult samples and quantitative PCR were performed as described in the Methods. Uninduced and induced indicate samples originated from the crosses UAS-AOX; UAS-Twinkle X w1118 and UAS-AOX; UAS-Twinkle X daGAL4, respectively. Relative mtDNA copy number (mtDNA/nDNA) was normalized by the mean 2−ΔΔCT of control samples from the crosses UAS-empty2nd; UAS-Twinkle X w1118 in every panel. no AOX, mut AOX, low AOX and high AOX indicate respectively the constructs UAS-empty2nd, UAS-mutAOX2nd, UAS-wtAOX8.1 21 and UAS-wtAOXF6 12. Letters a-d indicate significantly different statistical classes (P < 0.05) according to one-way ANOVA, followed by the Tukey post-hoc test, applied separately for the data in each panel.
Developmental effects of Twinkle mutants are tissue-specific
Next, we checked if AOX could improve mitochondrial function upon expression of the Twinkle mutants, although it cannot rescue the flies from lethality. Surprisingly, none of the Twinkle variants altered mitochondrial oxygen consumption driven by complex I or IV substrate oxidation in the body wall tissues of L3 larvae (Fig. 3 and Supplementary Fig. S3). Even though there was an increase in respiration in all Twinkle overexpressors in the presence of functional AOX, this was not statistically significant, except in the presence of the complex III inhibitor antimycin A, as expected (Fig. 3, middle panel). Because the larval body wall consists mostly of muscular and nervous tissues, we tested if tissue-specific expression of the Twinkle mutations using the mhcGAL4 and elavGAL4 drivers, respectively, could reproduce the lethal phenotype observed in the presence or absence of AOX. Consistent with the lack of detectable OXPHOS dysfunction in the larval body wall, flies overexpressing Twinkle K388A or A442P in muscles or neurons completed normal development (Supplementary Fig. S4). Again, coexpression of AOX had no effect on the developmental parameters analyzed. In addition to the observation that the L3 larvae expressing K388A and A442P via daGAL4 induction appear actively “healthy” for several days after the expected time of pupation, our mitochondrial respiration data and developmental assays using mhcGAL4 and elavGAL4 suggest that the lethality caused by the Twinkle variants are independent of the action of these mutants in tissues traditionally affected by mitochondrial disorders.

Mitochondrial respiration is not affected by overexpression of Twinkle nor AOX. Oxygen consumption was measured as described in the Methods in samples expressing the indicated AOX and Twinkle variants (UAS-AOX; UAS-Twinkle X daGAL4). Respiration linked to complex I substrate oxidation (upper panel) was calculated as the mean oxygen consumption after addition of pyruvate, proline, malate and ADP, followed by inhibition with antimycin A and rotenone. AOX activity (antimycin A-resistant respiration) was calculated as the mean oxygen consumption after addition of pyruvate, proline, malate, ADP and antimycin A, followed by inhibition with propyl-gallate and rotenone. Complex IV activity was calculated as the mean oxygen consumption after addition of TMPD and ascorbate, followed by inhibition with potassium cyanide. no AOX, mut AOX, low AOX and high AOX indicate respectively the constructs UAS-empty2nd, UAS-mutAOX2nd, UAS-wtAOX8.1 21 and UAS-wtAOXF6 12. No significant differences were found (P > 0.05) according to one-way ANOVA, followed by the Tukey post-hoc test.
The knockdown of Twinkle is pupal lethal and cannot be rescued by AOX
To eliminate the possibility that the inability of AOX to rescue our Twinkle models was because of the overexpression system, we also created and tested four UAS-AOX lines combined with UAS-Twinkle RNAi (see Supplementary Table S1). To our knowledge, this is the first report describing the use of this RNAi line from the Vienna Drosophila Research Center (VDRC), so we first verified its effect on the levels of Twinkle mRNA in controls (UAS-Twinkle RNAi; UAS-AOX X w1118) and induced individuals (UAS-Twinkle RNAi; UAS-AOX X daGAL4) to show the efficacy of the construct (see Supplementary Fig. S1 for crossing schemes). As expected, Twinkle mRNA was knocked down to 40–50% of the control levels in all lines (Supplementary Fig. S5a). Flies with low levels of Twinkle presented mtDNA depletion (Supplementary Fig. S5c) and reached the pupal stage, unlike most mutant overexpressors, but ~75% of them failed to eclose. AOX did not significantly alter viability (Supplementary Fig. S5b), except when expressed at a high level, which once again appeared to exacerbate the lethality caused by Twinkle disturbances. This data demonstrates that the AOX bypass does not mitigate the effects of Twinkle defects caused either by overexpression of mutants or by depletion of the endogenous enzyme.
AOX is unable to rescue other mtDNA replication defects
Considering that Twinkle and pol γ are part of the same replication machinery, our findings contrast those of Humphrey et al.17, in which AOX expression in Drosophila was shown to counteract deleterious effects of pol γ (tamas) knockdown in development (using the ubiquitous Act5CGAL4 driver) and in neurodegeneration models (using specific neural drivers). We therefore created and analyzed eight additional UAS-AOX lines combined with UAS-tamas RNAi or with a polymerase-active-site mutant of the catalytic subunit of pol γ, tamas Q1009A (see Supplementary Table S1). This RNAi line is the same as was used by Humphrey et al.17, whereas the tamas Q1009A mutation has been engineered in its natural genomic location, presenting the same expression pattern for the gene as in a wild-type line29. Both models promoted distinct developmental defects (see Supplementary Figs S1 and S6 for crossing schemes): UAS-tamas RNAi, when induced, led to mtDNA depletion (Supplementary Fig. S5d) and ~100% lethality at the pupal stage (Fig. 4a), whereas tamas Q1009A individuals died as L3 larvae (Fig. 4b). Coexpression of AOX, at low or high levels, had no effect on the phenotypes of these pol γ-deficient flies (Fig. 4 and Supplementary Fig. S5d), indicating that this alternative enzyme is in general unable to ameliorate mtDNA replication defects caused by disturbances in different components of the mtDNA replisome.

AOX does not rescue the developmental defects caused by genetic disturbances of pol γ. (a) Transcript levels of tamas, the coding gene for the catalytic subunit of pol γ (left panel), were measured as described in the Methods. Pupal viability (right panel) was measured as the mean ratio between the number of adults eclosed and the number of pupae per vial (+/− standard deviation). Uninduced and induced indicate samples originated from the crosses UAS-tamas RNAi; UAS-AOX X w1118 and UAS-tamas RNAi; UAS-AOX X daGAL4, respectively. (b) Viability of the tamas Q1009A lines was measured as the mean ratio between the number of wild-type adults and the total number of individuals per vial (+/− standard deviation). Left panel, progeny originated from crosses using tamas Q1009A/CyO; UAS-AOX and w1118 (uninduced) or daGAL4 (induced) to generate a heterozygous tamas Q1009A progeny, serving as controls. The expected Mendelian ratio of wild-type adults (carrying one tamas Q1009A allele) for these crosses is 0.5 (dashed line). Right panel, progeny originated from crosses using tamas Q1009A/CyO; UAS-AOX and tamas Q1009A/CyO (uninduced) or tamas Q1009A/CyO; daGAL4 (induced) to generate a homozygous tamas Q1009A progeny. The expected Mendelian ratio of wild-type adults (carrying two tamas Q1009A alleles) for these crosses is zero, as tamas Q1009A in homozygozity is lethal29. See Supplementary Fig. S6 for details of the genetic crosses. no AOX, mut AOX, low AOX and high AOX indicate respectively the constructs UAS-empty3rd, UAS-mutAOX3rd, UAS-wtAOX7.1 21 and UAS-wtAOXF24 12. *indicates statistical differences (P < 0.05) according to Student’s t-tests. Letters a-b indicate significantly different statistical classes (P < 0.05) according to one-way ANOVA, followed by the Tukey post-hoc test, applied separately for the data in (a) right panel, and (b) right and left panels. No significant differences were found (P > 0.05) for the data shown in (b).
Discussion
It has been shown that the xenotopic expression of C. intestinalis AOX in mammalian and insect mitochondria is efficient in rescuing diverse models of mitochondrial dysfunction, especially those with OXPHOS complex IV deficiencies30,31. AOX appears to be safely expressed and to function only when the cytochrome segment of the RC is deficient11,12, which makes it an attractive enzyme to be used in future therapies for mitochondrial diseases10. Nevertheless, there are aspects of AOX expression in heterologous systems that warrant further evaluation, such as the effects of its thermogenic and antioxidant properties. Heat production by mitochondrial uncoupling is to date only detectable in AOX-expressing cells in the presence of antimycin A32, but the lowering of ROS apparently occurs even under normal physiological conditions when OXPHOS is properly functioning, at least in adult flies33. In addition, the fact that AOX can rescue developmental defects not related, a priori, to mitochondrial dysfunction, such as cleft thorax, notched wings, leg malformations, and bristle abnormalities in Drosophila20, indicates that yet unknown cellular mechanism(s) is(are) operating when AOX is present in the mitochondria of higher animals.
At the other end of the spectrum of AOX applicability, there is a reported case of a true mitochondrial dysfunction, whose associated phenotypes were not ameliorated by the transgenic expression of this alternative enzyme, namely flies bearing the tko25t mutation34. This mutant has a defective mitochondrial ribosomal protein S12, impairing organellar protein synthesis and causing developmental delay and neurological problems35. Because another alternative enzyme, the alternative NADH dehydrogenase Ndi1, was also unable to rescue tko25t, the authors concluded that this mutant has a more complex metabolic defect, e.g. affecting multiple RC components, or some other process34. Here, we describe another case of AOX inability to rescue mitochondrial dysfunction in Drosophila. Overexpression of Twinkle mutants or knockdown of the endogenous enzyme caused developmental lethality at different stages with varying levels of mtDNA depletion, which was not alleviated by AOX. AOX was also unable to rescue the lethality caused by expression of a catalytically inactive pol γ or knockdown of its catalytic subunit, nor any of the molecular and biochemical parameters associated with mutant Twinkle expression. At high levels, AOX was in fact more detrimental for flies with Twinkle defects. One possible explanation is that the mutant Twinkle, or the depletion of the wild-type enzyme, in some way affects the organization of the inner mitochondrial membrane, and the further expression of a membrane-targeted protein (active or inactive AOX) synergistically impairs even more larval/pupal viability. Twinkle has been associated with a specific lipid domain of the inner mitochondrial membrane36,37, consistent with this hypothesis.
However, similar aggravations of developmental problems were also observed for tko25t in the presence of Ndi1, or Ndi1 plus AOX34. We speculate that in both cases, the metabolic consequences of severely defective mtDNA replication/mitochondrial translation may result in the activation of AOX, leading to an additional decrease in ATP production due to partial diversion of electron flow. Both the multi-subunit mitochondrial ribosomal machinery, of which the tko gene product is an essential part, and the modularly-architectured, multimeric Twinkle enzyme are dependent on the energy of nucleotide tri-phosphate hydrolysis for their proper functions in organellar protein synthesis and genome maintenance, respectively (reviewed in3,38). For example, the recombinant human wild-type Twinkle not only requires Mg2+ and ATP for dsDNA unwinding and ssDNA translocation, but the enzyme also presents significantly higher stability in vitro in the presence of such compounds39. Therefore, fluctuations in the ATP level in a sensitive mitochondrial genetic condition, such as that found in the tko25t and Twinkle models, may worsen the defects caused by such mutant proteins, which may in turn require even higher than normal nucleotide levels to maintain their fragile stability and/or function.
One of the primary effects of disturbing Twinkle or pol γ is the decrease of intact copies of mtDNA (via rearrangements, nucleotide substitutions or degradation)22,29, which in turn impairs mitochondrial RNA and protein synthesis40. tko25t produces a defect directly in the mitochondrial translation apparatus, downstream of Twinkle and pol γ, although its phenotypic consequences appear less severe than those of our mtDNA replication models. Nonetheless, the end result is similar: incorrect or insufficient subunits of all four OXPHOS complexes partly encoded in mtDNA, which AOX cannot circumvent. The fact that the mitochondrial genome encodes seven subunits of OXPHOS complex I and two subunits of complex V (~70% of all mtDNA genes) may explain the inability of AOX to rescue mitochondrial gene expression defects, as this enzyme is predicted to counteract complexes III and/or IV deficiencies.
Although in accord with the above hypothesis, our data appears inconsistent with the previously published findings showing significant improvement in the phenotypes associated with the knockdown of pol γ in diverse cell types of Drosophila17. We speculate that differences in the genetic background of the fly lines, in dietary conditions, and in experimental design might explain these discrepancies. Even though conditions appear to be homogenous within each study, they are difficult to compare side-by-side. Our implementation of two controls (for promoter dilution and for AOX enzymatic activity), plus two expression levels for AOX, and two defective pol γ models (knockdown and mutant expression), does represent a robust experimental design. On the other hand, the use by Humphrey et al.17 of several neural-specific GAL4 drivers does make their report reliable regarding pol γ depletion in the nervous system. Deficiencies of RC complex III and/or IV in specific neuronal classes are thus implied to underlie neurodegeneration in pol γ disease, in contrast to the broader effects of mtDNA replication defects documented in our study. Another important difference is that the drivers used for ubiquitous knockdown are different: daGAL4, used here, and Act5CGAL4 used by Humphrey et al.17 may differ in their exact strength, tissue and stage-specificity, and response to conditions. Notably, in combination with daGAL4, we found UAS-tamas RNAi to be pupal lethal, whereas in combination with Act5CGAL4, Humphrey et al.17 found it to be only semilethal, with AOX impacting the number of progeny. Clearly, the matter needs further investigation.
Our combined biochemical and genetic data also revealed an unexpected phenomenon regarding the overexpression of Twinkle variants in Drosophila: the absence of OXPHOS defects in larval body-wall mitochondria despite severe mtDNA depletion in whole L3 larval homogenates, as well as the absence of developmental lethality in the case of tissue-specific expression of mutant Twinkle in muscle and neurons. Maternal protein, RNA and even mtDNA may supply enough mitochondrial activity in those tissues to complete development. Because the large storage of triglycerides in the L3 larvae could interfere with the respiration measurements, we used a previously validated protocol29 in which the animals were dissected to eliminate all internal organs and the hemolymph, preserving the larval body wall (comprising muscle, nerve and epidermis) for these experiments. The results imply either that the larval phenotype described by us and others22 upon mutant Twinkle overexpression is caused by effects in one or more of these internal organs, or else could be due to an additional but unknown biological role of Twinkle, separate from mtDNA maintenance. The findings suggest a crucial role for mitochondria in the brain, fat body or other tissues that generate and respond to signals governing metamorphosis, over which AOX has no influence. Future studies using other tissue-restricted drivers for the larva, including ring gland, fat body, Malpighian tubule, salivary gland, glia, neuroendocrine cells, among others, will help us understand these roles.
As the human diseases associated with Twinkle and pol γ mutations account for a significant portion of all mitochondrial disorders, AOX may have limited applicability in therapy. Indeed, taking into account the combined data on tko25t, Twinkle and pol γ, we would argue that consideration of the possible use of AOX in therapy should exclude deficiencies in mtDNA maintenance or gene expression, unless clear benefits can be robustly demonstrated in a specific, clinically relevant model.
Methods
Fly stocks and maintenance
The standard line w1118, balancer lines CyO, TM3,Sb and TM6B-Tubby, and driver lines daughterless-GAL4 (daGAL4), myosin heavy chain-GAL4 (mhcGAL4) and embryonic lethal abnormal visionC155-GAL4 (elavGAL4) were obtained from stock centers. The lines UAS-AOXF6, UAS-AOXF24 12, UAS-wtAOX8.1, UAS-wtAOX7.1, UAS-empty2nd, UAS-empty3rd, UAS-mutAOX2nd and UAS-mutAOX3rd 21 were used in genetic crosses with the balancers described above to establish double transgenic lines in combination with UAS-Twinkle WT, UAS-Twinkle K388A, UAS-Twinkle A442P22, UAS-Twinkle RNAi (VDRC ID 108644/KK), UAS-tamas RNAi (VDRC ID 106955/KK) and tamas Q1009A29. Supplementary Table S1 shows the genotypes of all lines created for this work. All fly lines were first backcrossed into the w1118 background for six-to-ten generations, and were maintained in standard diet12 at 18 or 25 °C, with 12 hr light/dark cycles.
Developmental assays
10–15 virgin adult females of the indicated genotypes (Supplementary Table S1) were crossed with 5–7 adult control (w1118) or GAL4-expressing males (daGAL4 or mhcGAL4) per vial, and allowed to lay eggs for 24 hrs at 25 °C in standard diet12, where the offspring were kept until eclosion (see Supplementary Figs S1 and S5 for crossing schemes). For the neuron-specific transgene expression, elavGAL4 females were crossed with UAS construct-bearing males. The offspring of the UAS-Twinkle RNAi lines were kept at 29 °C to enhance the knockdown and its associated phenotype. Development was analyzed visually at the larval stage and quantitatively at the pupal stage, as the mean ratio between the number of eclosed adults and the number of pupae per vial, for a total of 8 vials per experiment, with 3 biological replicates.
Immunoblotting
Protein extracts from 10 whole 1-to-3-day-old adult males of the indicated genotype were prepared by gentle grinding of tissues in 200 μl PBS containing 1.5% Triton X-100 on ice. The suspension was centrifuged at 16,000 gmax for 10 min at 4 °C, and protein concentration of the supernatant was measured by the Bradford method. 100 μg of total protein extracts were mixed with 5X Laemmli buffer (10% SDS, 50% glycerol, 25% 2-mercaptoethanol, 0.02% bromophenol blue and 0.3125 M Tris-HCl, pH 6.8), denatured at 95 °C for 5 min and resolved by SDS-PAGE on 12% polyacrylamide gels at 120 V for approximately 1.5 h. Proteins were transferred to nitrocellulose membranes using the semi-dry gel transfer system MSD10 (Major Science, USA) for 15 min at room temperature. Membranes were blocked in PBST (8 mM Na2HPO4, 2 mM KH2PO4, 150 mM NaCl, 30 mM KCl, 0.05% Tween 20, pH 7.4) containing 5% dried nonfat milk for 2 h at room temperature or overnight at 4 °C. Primary antibodies [rabbit polyclonal anti-AOX12 (1:10,000), rabbit polyclonal anti-Dmhelicase25 (1:4,000), and mouse monoclonal anti-PDH E1α (1:5,000, Abcam, UK)] were incubated for at least 1 h at room temperature, followed by three washes (10 min each) with PBST. Secondary antibodies [HRP-conjugated goat anti-rabbit and anti-mouse IgGs (1:10,000, Bio-Rad, USA)] were incubated for at least 2 h at room temperature, and washed as described above. Membranes were incubated in Luminol substrate detection system Immun-Star HRP (Bio-Rad, USA), and chemiluminescence signals were detected on a ChemiDoc Imaging System (Bio-Rad, USA).
Nucleic acid isolation and quantitative PCR
Total DNA was extracted as described previously24, using 5–10 L3 larvae (5 days after egg laying) or adult males (1–3 days after eclosion) per preparation. To estimate relative mtDNA copy number, 15 ng of total DNA were used as template in a 20 μl quantitative PCR reaction, containing 10 μl PowerUp SYBR Green Master MixTM (Thermo Fisher Scientific) and 10 μM of each primer. The primers used were 16S-R (5′-TCGTCCAACCATTCATTCCA-3′) and 16S-L (5′-TGGCCGCAGTATTTTGACTG-3′) to amplify the mitochondrial 16S rRNA gene, and RpL32F (5′-AGGCCCAAGATCGTGAAGAA-3′) and RpL32A (5′-TGTGCACCAGGAACTTCTTGAA-3′) to amplify the single-copy, nuclear RpL32 gene. Reactions were performed in a StepOnePlusTM Real-Time PCR instrument (Applied Biosystems), under the manufacturer’s recommended conditions: 95 °C for 2 min, followed by 40 cycles of 95 °C for 3 s and 60 °C for 30 s. The ΔΔCT values were calculated by comparing the ratios between the mitochondrial and nuclear target genes. The relative mtDNA copy number values were derived by comparing each group of UAS-AOX; UAS-Twinkle lines to the same control sample (UAS-empty2nd; UAS-Twinkle X w1118) that was arbitrarily given the value 1.0.
Total RNA was prepared from 5 larvae using the TRIzol® plus PureLink RNA Mini Kit, and was reverse-transcribed using the SuperScript™ IV VILO™ Master Mix with ezDNase™ Enzyme kit (Thermo Fisher Scientific), according to the manufacturer’s recommendations. The quantitative PCR reactions to estimate the relative amounts of mtDNA-helicase (Twinkle) and tamas transcripts were performed essentially as described above, except that 1 μl of a 1:25 dilution of the reverse transcription reaction was used as template, along with 10 μM of the specific primers: TwinkleF (5′CCAAAGCGGTTCTAGTCAGC3′) and TwinkleR (5′GACTGGCATCGTAGTGCAAC3′) for mtDNA-helicase; tamasF (5′AAAATGTTCTCCATCACAAAGG3′) and tamasR (5′TTGGGACGATGAAATACCTC3′) for tamas; and RpL32F and RpL32A (described above) for RpL32. The relative transcript values were derived from the standard curve method, by comparing the ratio between mtDNA helicase and RpL32, or tamas and RpL32 for each UAS-RNAi; UAS-AOX X daGAL4 line to its control sample (UAS-RNAi; UAS-AOX X w1118) that was arbitrarily given the value 1.0.
Mitochondrial oxygen consumption
Five L3 larvae (5 days after egg laying) of the indicated genotype were dissected as previously29 in ice-cold PBS, eliminating the internal organs and the hemolymph, to obtain the larval body wall, which was then weighed in an analytical balance, gently homogenized in isolation buffer (250 mM sucrose, 5 mM Tris-HCl, 2 mM EGTA, 1% BSA, pH 7.4), and transferred to the oxygraph O2k chambers (Oroboros, Inc.). Oxygen consumption was measured at 25 °C in assay buffer (120 mM KCl, 0.5 mM KH2PO4, 3 mM HEPES, 1 mM EGTA, 1 mM MgCl2, 1% BSA, pH 7.2) after addition of substrates for complex I (5 mM sodium pyruvate, 5 mM L-proline and 1.5 mM malate), and complex IV (1.9 mM N,N,N′,N′-tetramethyl-p-phenylenediamine [TMPD] and 4 mM ascorbate) in the presence of 1 mM ADP. Complex I- and IV-driven respiration was corrected for the signal obtained after addition of 50 nM antimycin A and 5 nM rotenone, and 100 nM potassium cyanide, respectively. The antimycin A-resistant complex I-driven oxygen consumption, corrected for the signal obtained after addition of 200 nM propyl-galate and 5 nM rotenone, was attributed to AOX.
Statistical analyses
Statistical differences among the indicated groups of flies were tested using one-way ANOVA with Tukey post hoc test, as indicated in the figure legends, except for differences in Twinkle and tamas transcript levels, for which Student t-tests were applied. The analyses were performed using the software SAS University Edition v9.4 (SAS Institute Inc.).
Data Availability Statement
Any material or fly line used in this work is available from the corresponding author upon individual requests at any moment.
References
Euro, L., Farnum, G. A., Palin, E., Suomalainen, A. & Kaguni, L. S. Clustering of Alpers disease mutations and catalytic defects in biochemical variants reveal new features of molecular mechanism of the human mitochondrial replicase, Pol gamma. Nucleic Acids Res 39, 9072–9084, https://doi.org/10.1093/nar/gkr618 (2011).
Fernandez-Millan, P. et al. The hexameric structure of the human mitochondrial replicative helicase Twinkle. Nucleic Acids Res 43, 4284–4295, https://doi.org/10.1093/nar/gkv189 (2015).
Kaguni, L. S. & Oliveira, M. T. Structure, function and evolution of the animal mitochondrial replicative DNA helicase. Crit Rev Biochem Mol Biol 51, 53–64, https://doi.org/10.3109/10409238.2015.1117056 (2016).
Kauppila, T. E. S., Kauppila, J. H. K. & Larsson, N. G. Mammalian mitochondria and aging: an update. Cell Metab 25, 57–71, https://doi.org/10.1016/j.cmet.2016.09.017 (2017).
McKinney, E. A. & Oliveira, M. T. Replicating animal mitochondrial DNA. Genet Mol Biol 36, 308–315, https://doi.org/10.1590/S1415-47572013000300002 (2013).
Ciesielski, G. L., Oliveira, M. T. & Kaguni, L. S. Animal mitochondrial DNA replication. Enzymes 39, 255–292, https://doi.org/10.1016/bs.enz.2016.03.006 (2016).
Schiff, M., Benit, P., Jacobs, H. T., Vockley, J. & Rustin, P. Therapies in inborn errors of oxidative metabolism. Trends Endocrinol Metab 23, 488–495, https://doi.org/10.1016/j.tem.2012.04.006 (2012).
McDonald, A. & Vanlerberghe, G. Branched mitochondrial electron transport in the Animalia: presence of alternative oxidase in several animal phyla. IUBMB Life 56, 333–341, https://doi.org/10.1080/1521-6540400000876 (2004).
McDonald, A. E., Vanlerberghe, G. C. & Staples, J. F. Alternative oxidase in animals: unique characteristics and taxonomic distribution. J Exp Biol 212, 2627–2634, https://doi.org/10.1242/jeb.032151 (2009).
Rustin, P. & Jacobs, H. T. Respiratory chain alternative enzymes as tools to better understand and counteract respiratory chain deficiencies in human cells and animals. Physiol Plant 137, 362–370, https://doi.org/10.1111/j.1399-3054.2009.01249.x (2009).
Hakkaart, G. A., Dassa, E. P., Jacobs, H. T. & Rustin, P. Allotopic expression of a mitochondrial alternative oxidase confers cyanide resistance to human cell respiration. EMBO Rep 7, 341–345, https://doi.org/10.1038/sj.embor.7400601 (2006).
Fernandez-Ayala, D. J. et al. Expression of the Ciona intestinalis alternative oxidase (AOX) in Drosophila complements defects in mitochondrial oxidative phosphorylation. Cell Metab 9, 449–460, https://doi.org/10.1016/j.cmet.2009.03.004 (2009).
El-Khoury, R. et al. Alternative oxidase expression in the mouse enables bypassing cytochrome c oxidase blockade and limits mitochondrial ROS overproduction. PLoS Genet 9, e1003182, https://doi.org/10.1371/journal.pgen.1003182 (2013).
Szibor, M. et al. Broad AOX expression in a genetically tractable mouse model does not disturb normal physiology. Dis Model Mech 10, 163–171, https://doi.org/10.1242/dmm.027839 (2017).
Dassa, E. P. et al. Expression of the alternative oxidase complements cytochrome c oxidase deficiency in human cells. EMBO Mol Med 1, 30–36, https://doi.org/10.1002/emmm.200900001 (2009).
Kemppainen, K. K. et al. Expression of alternative oxidase in Drosophila ameliorates diverse phenotypes due to cytochrome oxidase deficiency. Hum Mol Genet 23, 2078–2093, https://doi.org/10.1093/hmg/ddt601 (2014).
Humphrey, D. M. et al. Alternative oxidase rescues mitochondria-mediated dopaminergic cell loss in Drosophila. Hum Mol Genet 21, 2698–2712, https://doi.org/10.1093/hmg/dds096 (2012).
Vartiainen, S. et al. Phenotypic rescue of a Drosophila model of mitochondrial ANT1 disease. Dis Model Mech 7, 635–648, https://doi.org/10.1242/dmm.016527 (2014).
El-Khoury, R. et al. Expression of the alternative oxidase mitigates beta-amyloid production and toxicity in model systems. Free Radic Biol Med 96, 57–66, https://doi.org/10.1016/j.freeradbiomed.2016.04.006 (2016).
Andjelkovic, A., Kemppainen, K. K. & Jacobs, H. T. Ligand-bound GeneSwitch causes developmental aberrations in Drosophila that are alleviated by the alternative oxidase. G3 (Bethesda) 6, 2839–2846, https://doi.org/10.1534/g3.116.030882 (2016).
Andjelkovic, A. et al. Diiron centre mutations in Ciona intestinalis alternative oxidase abolish enzymatic activity and prevent rescue of cytochrome oxidase deficiency in flies. Sci Rep 5, 18295, https://doi.org/10.1038/srep18295 (2015).
Sanchez-Martinez, A. et al. Modeling pathogenic mutations of human twinkle in Drosophila suggests an apoptosis role in response to mitochondrial defects. PLoS One 7, e43954, https://doi.org/10.1371/journal.pone.0043954 (2012).
Spelbrink, J. N. et al. Human mitochondrial DNA deletions associated with mutations in the gene encoding Twinkle, a phage T7 gene 4-like protein localized in mitochondria. Nat Genet 28, 223–231, https://doi.org/10.1038/90058 (2001).
Fukuoh, A. et al. Screen for mitochondrial DNA copy number maintenance genes reveals essential role for ATP synthase. Mol Syst Biol 10, 734, https://doi.org/10.15252/msb.20145117 (2014).
Matsushima, Y. & Kaguni, L. S. Differential phenotypes of active site and human autosomal dominant progressive external ophthalmoplegia mutations in Drosophila mitochondrial DNA helicase expressed in Schneider cells. J Biol Chem 282, 9436–9444, https://doi.org/10.1074/jbc.M610550200 (2007).
Goffart, S. et al. Twinkle mutations associated with autosomal dominant progressive external ophthalmoplegia lead to impaired helicase function and in vivo mtDNA replication stalling. Hum Mol Genet 18, 328–340, https://doi.org/10.1093/hmg/ddn359 (2009).
Tyynismaa, H. et al. Twinkle helicase is essential for mtDNA maintenance and regulates mtDNA copy number. Hum Mol Genet 13, 3219–3227, https://doi.org/10.1093/hmg/ddh342 (2004).
Ikeda, M. et al. Overexpression of TFAM or twinkle increases mtDNA copy number and facilitates cardioprotection associated with limited mitochondrial oxidative stress. PLoS One 10, e0119687, https://doi.org/10.1371/journal.pone.0119687 (2015).
Bratic, A. et al. Complementation between polymerase- and exonuclease-deficient mitochondrial DNA polymerase mutants in genomically engineered flies. Nat Commun 6, 8808, https://doi.org/10.1038/ncomms9808 (2015).
El-Khoury, R. et al. Engineering the alternative oxidase gene to better understand and counteract mitochondrial defects: state of the art and perspectives. Br J Pharmacol 171, 2243–2249, https://doi.org/10.1111/bph.12570 (2014).
Camargo, A. F. et al. Xenotopic expression of alternative electron transport enzymes in animal mitochondria and their impact in health and disease. Cell Biol Int 42, 664–669, https://doi.org/10.1002/cbin.10943 (2018).
Chretien, D. et al. Mitochondria are physiologically maintained at close to 50 degrees C. PLoS Biol 16, e2003992, https://doi.org/10.1371/journal.pbio.2003992 (2018).
Sanz, A., Fernandez-Ayala, D. J., Stefanatos, R. K. & Jacobs, H. T. Mitochondrial ROS production correlates with, but does not directly regulate lifespan in Drosophila. Aging (Albany NY) 2, 200–223, https://doi.org/10.18632/aging.100137 (2010).
Kemppainen, K. K., Kemppainen, E. & Jacobs, H. T. The alternative oxidase AOX does not rescue the phenotype of tko25t mutant flies. G3 (Bethesda) 4, 2013–2021, https://doi.org/10.1534/g3.114.013946 (2014).
Toivonen, J. M. et al. Technical knockout, a Drosophila model of mitochondrial deafness. Genetics 159, 241–254 (2001).
Rajala, N., Gerhold, J. M., Martinsson, P., Klymov, A. & Spelbrink, J. N. Replication factors transiently associate with mtDNA at the mitochondrial inner membrane to facilitate replication. Nucleic Acids Res 42, 952–967, https://doi.org/10.1093/nar/gkt988 (2014).
Gerhold, J. M. et al. Human mitochondrial DNA-protein complexes attach to a cholesterol-rich membrane structure. Sci Rep 5, 15292, https://doi.org/10.1038/srep15292 (2015).
Greber, B. J. & Ban, N. Structure and function of the mitochondrial ribosome. Annu Rev Biochem 85, 103–132, https://doi.org/10.1146/annurev-biochem-060815-014343 (2016).
Ziebarth, T. D. et al. Dynamic effects of cofactors and DNA on the oligomeric state of human mitochondrial DNA helicase. J Biol Chem 285, 14639–14647, https://doi.org/10.1074/jbc.M109.099663 (2010).
Gustafsson, C. M., Falkenberg, M. & Larsson, N. G. Maintenance and expression of mammalian mitochondrial DNA. Annu Rev Biochem 85, 133–160, https://doi.org/10.1146/annurev-biochem-060815-014402 (2016).
Acknowledgements
The authors would like to thank Dr. Alvaro Sanchez-Martinez for providing the mutant Twinkle fly lines, Dr. Nils-Göran Larsson for providing the tamas Q1009 fly line, Dr. Laurie S. Kaguni for providing a sample of the anti-Dmhelicase antibody, Drs. Helen A. Penha and Andressa de Souza for technical assistance, and Dr. Nick Hudson for critical reading of the manuscript. The authors acknowledge the support of the Fundação de Amparo à Pesquisa do Estado de São Paulo (grant numbers 2014/02253-6 to M.T.O. and 2015/14547-7 to A.P.C.R.), the Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (fellowship to A.F.C.), the Finnish Cultural Foundation (grant to A.A.), the European Research Council (advanced grant 232738), the Academy of Finland (decisions 256615 and 272376), the Tampere University Medical Research Fund and the Sigrid Juselius Foundation (to H.T.J.).
Author information
Authors and Affiliations
Contributions
A.P.C.R., A.F.C., A.A. and M.T.O. performed the experiments; A.P.C.R., A.F.C. and M.T.O. designed the experiments; A.P.C.R., A.F.C., H.T.J. and M.T.O. analyzed the data; H.T.J. and M.T.O. wrote the paper.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Rodrigues, A.P.C., Camargo, A.F., Andjelković, A. et al. Developmental arrest in Drosophila melanogaster caused by mitochondrial DNA replication defects cannot be rescued by the alternative oxidase. Sci Rep 8, 10882 (2018). https://doi.org/10.1038/s41598-018-29150-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-29150-x
- Springer Nature Limited