
Abstract
The onset of erythropoiesis is under strict developmental control, with direct and indirect inputs influencing its derivation from the hematopoietic stem cell. A major regulator of this transition is KLF1/EKLF, a zinc finger transcription factor that plays a global role in all aspects of erythropoiesis. Here, we have identified a short, conserved enhancer element in KLF1 intron 1 that is important for establishing optimal levels of KLF1 in mouse and human cells. Chromatin accessibility of this site exhibits cell-type specificity and is under developmental control during the differentiation of human CD34+ cells towards the erythroid lineage. This site binds GATA1, SMAD1, TAL1, and ETV6. In vivo editing of this region in cell lines and primary cells reduces KLF1 expression quantitatively. However, we find that, similar to observations seen in pedigrees of families with KLF1 mutations, downstream effects are variable, suggesting that the global architecture of the site is buffered towards keeping the KLF1 genetic region in an active state. We propose that modification of intron 1 in both alleles is not equivalent to complete loss of function of one allele.
Similar content being viewed by others


Introduction
Hematopoietic stem cells are the source of all the mammalian blood lineages, each arising following a process of gradual restriction of downstream cellular potencies1. One of these lineages is erythropoiesis, which gives rise to the red blood cells that make up > 80% of cells in the human body. Critical for this progression are controlling transcription factors, of which Erythroid Krüppel-like Factor (EKLF/KLF1)2 plays a global role in all aspects of red blood cell formation3,4,5,6. KLF1 contains three C2H2 zinc fingers that precisely recognize its cognate DNA site (5′CCMCRCCCN3′) at target genes, enabling a high level of discrimination among similar sites7. It performs this function by binding to its cognate DNA element, interacting with the basal transcription machinery, and recruiting chromatin remodeling proteins and histone modifiers8,9,10. KLF1 integration of these signals leads to establishment of the correct 3-dimensional structure necessary for optimal target gene transcription11. Regulated expression of KLF1 target genes are coordinated within ~ 40–60 nuclear transcription factories12.
Ablation of KLF1 leads to embryonic lethality in the mouse precisely at the time of the switch to adult β-globin13,14. This is only one of many changes that occur in the red cell in its absence, such that KLF1 is now recognized as a global regulator of all aspects of erythropoiesis3,15,16. The range of phenotypic/clinical parameters that follow from its genetic modulation in the human has become extensively catalogued3,4. KLF1 haploinsufficiency leads to benign outcomes (e.g., lower expression of CD44 and Lu/BCAM antigen), but compound heterozygosity can lead to more serious problems such as microcytic hypochromic anemia17 and pyruvate kinase deficiency/chronic non-spherocytic hemolytic anemia18,19,20 or even to hydrops fetalis21. Some mutations lead to a dominant phenotype such as seen in congenital dyserythropoietic anemia type IV22,23,24.
Clinical parameters are affected when one KLF1 allele is inactive, as KLF1 gene variants are associated with altered red cell indices25,26,27,28. Of particular note is the loss of β-like globin switching mechanisms, leading to HPFH (originally mapped in a Maltese family29) and also to persistence of embryonic globin18. There are also benign but significant changes in mean cell volume (MCV) and mean corpuscular hemoglobin (MCH) (both lower), or in zinc protoporphyrin (ZnPP) and reticulocyte count (RTC) (higher). These changes enable identification of KLF1 mutation carriers3,4,30, and it is of considerable interest that allelic distribution of KLF1 mutations are dramatically higher in regions of endemic β-thalassemia31, where the dysregulation of γ-globin expression is of clear clinical benefit. A case report of βo/βo thalassemic twins highlights the remarkable clinical difference in anemia and transfusion dependence that follows from loss of one KLF1 allele32. Collectively these data strongly suggest that a fine-tuned understanding of KLF1 promoter regulatory mechanisms could enable directed modulation of KLF1 levels and provide a significant benefit to thalassemic and sickle cell patients.
KLF1 control elements have been clearly established in vitro33 and in vivo34,35,36,37. These studies have delineated a relatively simple layout of a short (~ 1 kB) region adjacent to the transcriptional start site containing erythroid-specific hypersensitive (EHS1 and 2) upstream enhancer elements that bind hematopoietic general and tissue-restricted regulators38,39,40,41 and respond to BMP4 signaling. Coupled to this, an enhancer element in intron 1 plays a critical role in establishing its maximal expression level37. Epigenetic analyses have determined that the DNA methylation and hydroxymethylation modification status of these controlling regions are important for KLF1 expression control42,43,44.
We recently analyzed the KLF1 transcription unit for variability across of a range of leukemic and malignant erythroid subsets and found only common SNPs27. In the present study, evaluation of the KLF1 transcription unit in Juvenile Myelomonocytic Leukemia (JMML) samples enabled us to more precisely investigate the role of a conserved sequence in intron 1 in modulating expression of KLF1.
Results
Identification of a mutation at the intronic enhancer region of KLF1 in a JMML patient
We sought to investigate KLF1 gene status in cells from patients with JMML, an early childhood myeloproliferative/myelodysplastic disease that is a clonal disorder of pluripotent stem cells45,46,47,48. They are also thrombocytopenic and exhibit hepatosplenomegaly45. We sequenced the complete KLF1 transcription unit27 in genomic DNA from 20 JMML patient samples to search for mutations48. We identified three changes within the samples, two of which are in an upstream region just beyond the critical EHS1 enhancer39,40 (Table S1, Fig. S1a). Of particular interest, the third sample (HM3554) contains a single heterozygous base mutation within a highly conserved region of intron 1 that is not a SNP (Table S1, Fig. S1b). This change did not appear in our previous analysis of over 5000 genomes from direct sequencing and from public databases27. This mutation is located near the intronic enhancer region we had previously discovered in the mouse37. Inclusion of this intron leads to a threefold increase in linked reporter activity beyond that seen with the 5′-promoter alone, as assayed in differentiating embryoid bodies derived from mouse embryonic stem cells (Fig. 1a).

Analysis of the KLF1 intronic enhancer. (a) Reanalysis of data from a study37 on the mouse KLF1 promoter. Constructs containing the KLF1 promoter or one that additionally includes intron 1 were stably integrated into mouse ES cells adjacent to a GFP reporter, yielding two stable ES lines (“P-Klf1-GFP” and “P-Klf1-intron-GFP”). Top: Locations of the mapped upstream enhancer, proximal promoter, and intronic enhancer are as indicated. Bottom: ES cells from each line were differentiated to EBs for the indicated number of days, and samples were quantitatively analyzed for expression of exogenous reporter (green) to monitor the effect of intron 1 inclusion. Data is from analysis of biological triplicates. Endogenous KLF1 expression was also monitored in the same samples to show that its onset/expression is similar in both sets (Fig. S5). (b) Genome browser data aligned at the human KLF1 genomic region (https://main.genome-browser.bx.psu.edu/index.html). Shaded bars show the locations of the enhancer upstream of the gene and of the intron 1 site within the gene (also marked with an asterisk). In vivo GATA1, SMAD1, TAL1, and P300 binding identified from ChIP-seq analyses of erythroid cells are shown. (c) Genome browser data showing onset of chromatin accessibility as monitored by ATAC-seq analyses of primary human cells: top, during erythroid differentiation from sorted adult human CD34+ cells49; middle, from erythroid, megakaryocyte, and hepatic fetal liver cells52; bottom, from adult human hematopoietic subpopulations50. Shaded bars are aligned with and are as in (b). (d) Reporter assay of various renilla reporter constructs after transfection into human JK1 cells. Top: Schematic of constructs containing the KLF1 promoter (as in (a)) are shown. The location of the intron 1 point mutant introduced into P-Klf1-intron-Ren is shown (“M”). Bottom: Results of the assay, showing high renilla levels from the promoter alone that are further stimulated by inclusion of the intron (data from two separate DNA preparations, each performed in triplicate); however, these levels are decreased when the point mutant variant is used (data from three separate DNA preparations, each performed in triplicate). Normalization is to a co-transfected luciferase plasmid, and data is an average of triplicate samples from each DNA preparation.
A closer analysis reveals that the mutation is located within a sequence that is highly conserved across mammalian species, and directly adjacent to previously identified GATA and SMAD recognition elements in the mouse37 (Fig. S1b). This region binds GATA1, SMAD1, and TAL1 transcription factors also in human erythroid cells (Fig. 1b), and the increase in chromatin accessibility during human hematopoiesis, development, and erythroid differentiation as judged by ATAC seq analysis is regulated and centered at that site (along with that of the previously-described upstream promoter/enhancer) (Fig. 1c)49,50,51,52. These data suggest that the intron 1 site mutated in the JMML patient is within a functionally important and restricted genetic element that is critical for optimal mammalian KLF1 expression.
Reporter assays demonstrate the relevance of the intronic enhancer mutation on KLF1 expression
We performed reporter assays in transfected JK1 human erythroleukemia cell line to address the importance of the intron 1 mutation. This was assessed by transfection of renilla reporters driven by wild type or site-directed mutated KLF1 promoter and intronic regions (design based on37). Figure 1d shows that, as expected from the murine data, inclusion of intron 1 increases the activity of the promoter ~ 4-fold. However, this increase is reduced by 50% when performing the same assay using a reporter with the intron 1 single nucleotide mutation, verifying its importance. Performing the same assay with the two upstream promoter changes had little effect (Fig. S2).
Genome editing at the mutated intronic region in the KU812 human erythroleukemia cell line leads to reduced KLF1 expression
We next addressed whether the sequence surrounding the mutation is critical for hKLF1 transcription by using TALEN directed-nuclease technology53,54 to delete the site within the KLF1 genome in human cells (Fig. 2a, Table S2), and monitored KLF1 levels. We were aided in the selection process by the use of a cotransfected RFP/GFP reporter55 (Fig. 2a). Using this system we transfected human KU812 cells which, although of low transfection efficiency via lipofection, enabled us to select for double positive RFP+/GFP+ cells (Fig. 2b).

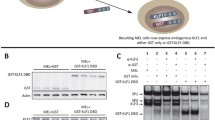
TALEN-mediated indel generation and analysis of KU812 cells. (a) Top, schematic of the location of the two TALEN arms (blue) in the context of the KLF1 gene and the intron 1 mutation site (red). Exact sequence is shown. Bottom, schematic of parental55 and modified constructs containing RFP (in frame) and GFP (out of frame) reporters separated by a target site into which the intron 1 sequence (with the left and right TALEN sites) is incorporated. Successful transfection and activity of the dual TALENs is predicted to yield an indel leading to a frameshift and resultant in-frame expression of GFP (1 out of 3 chance). (b) Data is shown from the transfection of KU812 cells. Selection of RFP+/GFP+ positive cells after cotransfection with left and right TALEN arms greatly increases the chance of finding a clone with the desired indel, even at low frequency. Clones are generated by single-cell sorting and expansion. (c) Genomic sequence analysis of parental and two KU812 subclones (“clone 1” and “clone 2”) containing directed indels at KLF1 intron 1; blue are the TALEN sequences, red is the target JMML site as a reference point. Homo- or heterozygous deletions are as indicated. Analyses of the two indel KU812 cell clones compared to parental cells are shown after a check of KLF1 RNA expression and western blot assessment (uncropped blot is in Fig. S3) of KLF1 and HSP90 protein (insert). (d) ChIP analysis of specific TEL/ETV6 binding in WT or mutant (clone 2, “Mut”) KU812 cells. ETV6 occupancy at the KLF1 intron 1 site, monitored by two different primer pairs, show a positive signal only in WT cells. (e) Positive control targets known to bind ETV6 show signals in all cases (HHEX, GATA2, LMO2 based on114) in WT or mutant cells. For (d) and (e), multiple samples were analyzed, each in triplicate.
The clones encompassed homo- and heterozygous deletions of 3–15 nt centered at the intron 1 mutation site (e.g., Fig. 2c), indicating successful attainment of genomic editing. Clones of non talen-treated parental cells were also obtained to serve as a control to avoid any variables from clonal selection. Western and qRT-PCR analysis was performed for clone 1 (heterozygous deletion), clone 2 (homozygous deletion), and one of the parental clones (Fig. 2c). Western analysis shows that KLF1 protein levels are quantitatively reduced in clone 1 and more reduced in clone 2 compared to a parental clone, effects that mirror the KLF1 transcript expression (Fig. 2c).
These results enable us to sublocalize the functionally critical sequence from within the original large 0.9 kB intron 1 to a small ~ 15 bp region surrounding the intron 1 site mutation, and suggest that a circumscribed deletion centered on this mutation site within normal cells will quantitatively decrease, but not ablate, KLF1 expression.
Identification of TEL/ETV6 protein interaction with the KLF1 intron 1 region
Given the high level of sequence conservation at this site (Fig. S1b), transcription factor interactions may be predicted to be negatively affected by the mutation. We used the UniPROBE factor interaction prediction analysis to identify binding sites that are directly disrupted by the intronic mutation56,57 coupled to analysis via the NIH Roadmap Epigenomics Consortium (Fig. S4). We find that the intron region surrounding the mutated site is enriched for binding sites for the ETS family of proteins58,59 that interact with a consensus motif known to be important for the transition from stem to erythroid cells60. The single base intron 1 mutation is predicted to disrupt the binding for these proteins (the “T” in the sequence GGAAGT, mutated to a “C” in the present case, is particularly critical in all cases)57. Exclusion of those transcription factors not expressed in erythroid cells (analysis via ErythronDB61 and BioGPS62) leaves only a small subset as possible contenders for positive binding such as ERG, ETV6, and ELF2. Of particular interest is TEL/ETV6, a protein that, similar to KLF1, has known roles in enhancing erythroid differentiation. An in vitro binding site selection strategy for ETV6 verifies the importance of a “T” at the position of interest58. In vivo ChIP analyses of WT and intron 1-edited KU812 cells show that TEL/ETV6 binds to this site, and that its binding is abrogated when the target site is mutated (Fig. 2d). TEL/ETV6 binding to three control target sites are not affected in cells containing the mutation at the KLF1 site (Fig. 2e). We conclude that the TEL/ETV6 protein joins GATA1/SMAD/TAL1 in forming an optimal protein/DNA complex at this critical site within intron 1 of the KLF1 gene.
Genome editing of KLF1 intron 1 in human CD34+ cells
To test whether quantitative knock down of KLF1 expression could be attained by editing of intron 1 in primary cells, we expanded human CD34+ cells and transfected them with the verified TALEN arms (along with the RFP/GFP reporter), followed by differentiation towards the erythroid lineage using a three-step protocol63,64,65,66. Positively-transfected cells were selected (as before) and cultured as a pool of cells. Sorted cells from transfections without the TALEN arms served as our unaltered control. Deep sequencing of the positive pool indicates that over 60% of the cells are edited at the expected deletion site that overlaps the intron 1 mutation (Fig. 3a). Although encouraging, we found the resultant small deletions had little effect on KLF1 expression, possibly due to the fact that our efficiency was not near 100%, viability was not good after the DNA transfection, and at most we should have detected a 30% drop in expression (50% effect x ~ 60% cells with deletion).

Gene editing and analysis of primary human cells. Human CD34+ cells were transfected, differentiated towards the erythroid lineage with a three-phase protocol, and analyzed by deep sequencing of the KLF1 intron 1 region. Indels were identified, and the top 30 are shown in each case along with the total indel percentage in the population (one representative example from each test is shown). The location of the JMML point mutation (marked in red at the top) serves as a reference point for each design. KLF1 mRNA expression was monitored by RT-qPCR and shown on the right. (a) Cells were transfected with the left and right TALEN arms (indicated at the top in yellow) along with the co-transfected reporter. The RFP+/GFP+ pool was sorted, expanded, and differentiated prior to NGS genomic DNA analysis. KLF1 expression levels are not significantly different (n = 4–6 each). (b) Cells were transfected with an RNP consisting of Cpf1 protein and gRNA (indicated at the top in yellow, with the PAM sequence in blue). The negative control was cells transfected with an RNP containing scrambled gRNA. The cells were expanded and differentiated prior to NGS genomic DNA analysis. NGS of the scrambled control cells showed no changes (not shown). Although there is a range of KLF1 expression after editing, levels are not significantly different from the scrambled control (n = 18 each). (c) Cells were transfected with a 50/50 mix of RNPs consisting of Cpf1 protein and two gRNAs (indicated at the top in yellow, with their PAM sequences in blue). The negative control was cells transfected with an RNP containing scrambled gRNA. The cells were expanded and differentiated prior to NGS genomic DNA analysis. NGS of the scrambled control cells showed no changes (not shown). KLF1 expression is down twofold in the samples dual-edited samples (n = 9 each).
During the initial design of these studies, we had not been able to use a CRISPR/Cas approach due to the inherent requirements of Cas9 gRNA, whose PAM sequence (NGG) limited the target sequences to regions not near the site of interest in intron 1. However, in the interim Cpf1 (aka Cas12a) was discovered and developed as a viable alternate67,68. Its merits in the present situation are that its PAM sequence (TTTV) is significantly different from that of Cas9, and that the Acidaminococcus sp. variant has been engineered for increased activity and fidelity69. This novel recognition sequence enabled us to design a Cpf1 gRNA that directly overlaps the site of interest and exhibits no predicted off-targets even with up to 2 mismatches (Table S3) (CRISPOR program70). We also opted to use an RNP-based approach to enable entry of the Cpf1/gRNA complex into CD34+ cells at high efficiency while retaining high cell viability66,71,72,73. We tested this new design/approach, and indeed find that, even in the absence of selection, > 95% of the cells are edited (Fig. 3b). Use of a scrambled Cpf1/gRNA control routinely gives close to zero editing. The deletions at the region of interest are larger than had been observed with TALENs, but surprisingly we still do not see a significant effect on KLF1 levels on average even though the range of effect was wide (i.e., a subset of the data points were decreased compared to the control).
We next considered whether the Cpf1-directed deletion at the KLF1 intron 1 site might be too limited. Although one might predict that disruption of transcription factor binding should have a major effect on activity of enhancer function, it has been observed that removal of a single site in vivo can have a surprisingly minor effect (concepts discussed in74), as any associated ‘enhanceosome’ components may still form if those protein–protein interactions are strong, and if there are other adjacent DNA binding sites that retain recruitment and cooperativity of other components in the full complex. Indeed, the KLF1 gene forms an extended 3D complex with multiple enhancers in its vicinity75. We inspected the sequence surrounding the original Cpf1 position and found an additional Cpf1 PAM-sequence target site ~ 35 bp away that also exhibits no predicted off-targets even with up to 2 mismatches (Table S3) (CRISPOR program70). We postulated that introduction of both Cpf1 gRNA sequences might enable a larger deletion region to be generated. This dual Cpf1 approach was tested. Again we attain high efficiency of editing in the absence of selection (> 99%), but we now create a larger deletion and produce the desired 50% drop in KLF1 levels (Fig. 3c). This suggests that focused disruption of the ETV6 site is not sufficient, but rather that deletion of a slightly larger region, likely containing other important binding sites [e.g., MYB, SMAD, and GATA proteins (Figs. S1B ad S4)], is needed to more reliably disrupt the enhancing activity of intron 1. Importantly, this verifies the importance of the intron 1 region for optimal KLF1 expression in vivo in primary cells and set the stage for analysis of its downstream effects.
Gene expression analyses of KLF1-edited CD34+ cell-derived erythroid cells
To understand the genetic effects of KLF1 intron 1 gene editing, we undertook two sets of analyses. First, we compared expansion/differentiation properties of control vs intron 1-edited cells and find no significant changes during expansion, and perhaps a minor but still not significant effect on the extent of differentiation (Fig. S6), properties consistent with that from haploinsufficient individuals76. Second, we performed RNA seq on triplicate samples derived from the larger dual guide-edited or control CD34+ cells isolated at the end of the differentiation protocol. A comparative analysis of these two sets of data shows that the editing has a circumscribed/limited effect, with a relatively small number of genes (~ 50 either up/down, p < 0.05, |log2| ≥ 1) altered in their expression (Fig. 4a). Some of the downregulated genes bind KLF1 in vivo via its cognate binding sequence and occasionally overlap with GATA1 binding77,78. The limited effect may be due to the unexpected low outcome of the deletion on KLF1 expression (i.e., 20% rather than 50%) (Fig. 4b). The variation in expression of KLF1 after deletion within intron 1 using the same dual guide approach but with different CD34+ cell sources (comparison of Figs. 3c and 4c, but also foreshadowed by the large range of effect in Fig. 3b) led us to examine whether upstream regulators of KLF1 might variably be increased to compensate10,39. However, there is no coherent increase in expression levels of proteins known to coordinately interact with the critical KLF1 upstream enhancer element (EHS1) within the edited cells (Fig. S7). Given that adults that have one mutated allele actually express 50% of the normal KLF1 level29,76, our results imply that haploinsufficiency by virtue of deletion of intron 1 across both alleles is not equivalent to haploinsufficiency from total functional loss of one allele. This suggests the KLF1 locus is buffered against large changes in expression (more fully discussed below).

RNA seq analysis of edited CD34+ cell-derived erythroid cells. (a) Volcano plot of RNA expression data from control compared to intron 1-edited CD34+ cells is shown based on analysis of biological triplicate samples harvested at d18 of the three-phase differentiation protocol. Black dots represent genes not changed in expression, green are changed but not significantly, blue are significantly changed but less than |log2| = 1.0, and red are significantly changed and |log2| ≥ 1.0. Some genes in this last category have been identified in the figure. These genes are listed in Tables S5 and S6. (b–d) RNA seq expression data from control (transfected with Cpf1 RNP containing scrambled gRNA; ‘scr’) compared to that from intron 1-edited (transfected with a Cpf1 RNP containing a dual guide mix of intron 1-directed gRNA as in Fig. 3c; ‘dg’). Statistical analysis is from the DESeq2 files (as in (a)). The source file for these genes is Table S7. (b) Expression of KLF1. (c) Expression of genes anticipated to be affected by drop in KLF1 levels. (d) Expression of genes related to β- and α-globin gene regulation, including the BCL11A repressor.
In spite of this, a number of cellular/structural genes anticipated to be altered in expression based on analyses of KLF1 haploinsufficient individuals79 are affected and include DMTN, BSG, and EPB42 (Fig. 4c). However, other targets such as BCAM and GYPA are not affected (Fig. 4c).
With respect to globin regulation, unsurprisingly, expression of the adult β- and α-globin genes are decreased in the edited cells (Fig. 4d). Even though the upstream γ-globin repressor BCL11A (that is directly regulated by KLF129,36) is also decreased in expression (Fig. 4d), levels of other repressors are not significantly affected, leading to minimal effects on γ- and ε-globin expression (Fig. S8). As a control, we edited the + 58 region of the BCL11A gene66 with our methodology and find significant increases in γ-globin expression and the γ/(γ + β) ratio (Fig. S9).
Collectively, these studies show that the dual guide design directed at the intron 1 enhancer element leads to quantitative disruption of KLF1 expression in differentiating human CD34+ cells, and that even less than a two-fold drop leads to measurable changes in expression of its target genes. However, unlike the initial results in cell lines, reproducible knock-down of expression in primary cells requires additional genetic manipulation.
Discussion
Genetic studies in mice and humans have been critical for discovering that two-fold changes in KLF1 expression can lead to clinically useful changes in gene expression. KLF1 as a therapeutic target has largely been abandoned because its absence creates lethal problems for the red cell6,13,14,21. However, the present design stresses the importance of precise quantitative expression control and is based on the observation that a haploinsufficient level of KLF1 is benign, and that manipulations that mimic this effect could have therapeutic benefit because only select targets are dramatically affected by the 50% drop in expression8,79,80,81. Cultured erythroblasts from haploinsufficient individuals proceed through renewal and differentiation stages equivalent to wild type, and overall chromatin accessibility is unchanged76. An alternate gene modification would be to disrupt the coding sequence, but a recent study that depleted KLF1 in this way in human CD34+ cells led to many genes dysregulated in their expression82.
The overall effect on KLF1 expression of deleting the intron 1 site via the dual Cpf1/gRNA approach remained relatively minor when tested in primary cells; this was unexpected, given that the evidence from the reporter transfections and from the Talen cell line editing had shown that even a directed mutation/small deletion exerts an effect on reporter or endogenous KLF1 transcription. However, transfected DNA begins in a non-chromatinized conformation when introduced into the cell, and leukemic cell lines are generally dysregulated; hence the primary cell approach is the most stringent and informative way to mimic the in vivo cellular and epigenetic environments. The quantitative effect of the indel on KLF1 transcription in primary cells, even at a level of 20%, was precisely tracked by a remarkably similar quantitative reduction in expression of a subset of its erythroid targets. This illuminates the extreme level of precision in transcriptional control that, based on KLF1 regulatory properties83, could include precisely attenuated transcriptional pause/release mechanisms for which these target genes may be extremely sensitive.
The inclusion of ETV6 into a positive regulatory role is perhaps unusual, as it has been typecast as a transcriptional repressor. However its molecular role is complex, and in addition a recent study shows that KLF1 levels are decreased in homozygous mutant ETV6 iPS cells that were established as a model of inherited thrombocytopenia84. These cells also exhibited decreased erythroid expansion capability and erythroid colony counts.
With respect to future applications of our approach, KLF1 expression is highly restricted to erythropoietic organs, as evidenced during development85,86, in the adult2, and during hematopoiesis87,88,89. The highly restricted nature of its expression is a crucial property for its potential use as a gene therapy target, as quantitative alteration of its levels is predicted to limit its effect to erythroid-related cells of interest, a unique situation that avoids the complexities that arise in other potential targets that are more generally expressed and required90,91,92,93,94.
A perplexing observation in the field is the extensive variation in HbF levels seen in individuals containing the same monoallelic KLF1 mutation, even within the same family pedigree16,29,95. This was also seen in our own studies (comparison of the dual gRNA data between Figs. 3 and 4). This is thought to be due to variable penetrance and microvariation of KLF1 expression as contributed by additional mutations on the remaining WT allele76,95. Our data suggest that the overall architecture of the KLF1 genomic region is buffered against localized changes having a substantive effect on its expression. This idea is in tune with recent observations demonstrating exactly this phenomenon and explained by the establishment of nested, two-layer networks of enhancers, some of which are distantly located and in non-coding regions, that achieve the net effect of protection against chromosomal perturbations96,97. Consistent with this idea, the KLF1 transcription unit is known to be located in the midst of multiple interacting domains75, and the KLF1 promoter is one of a small subset of tissue-restricted genes whose expression is dependent on multiple regulatory elements98. As a result, reproducible and precise management of KLF1 levels may require additional manipulation(s) in coordination with directed changes at intron 1.
Methods
KU812 and JK1 cells lines have been described99. We initially considered mutating the intron 1 region in HUDEP2 cells100, as they have been widely utilized for human erythroid studies. However, we obtained aberrant results with all our directed mutagenesis attempts at the KLF1 genomic region. Karyotype analysis revealed genomic instability along with trisomy 19 (not shown), which is the genomic location for KLF1. As a result, we switched to using JK1 and/or KU812 human leukemia cell lines for our initial set of gene editing analyses, as both of these appear more stable and, unlike K562, also express KLF1101,102 (discussed in27) and thus more likely to exhibit positive control of an exogenous KLF1 promoter/enhancer construct. Patient DNA samples are not publicly available but were as previously published48 and obtained from Dr M Loh (UCSF), whose studies were approved by the UCSF Committee on Human Research. All participants or their guardians provided informed consent in accordance with the Declaration of Helsinki. As a result, samples and data were analyzed anonymously. Genomic sequencing using primers spanning the complete KLF1 transcription unit, including the upstream promoter, was performed in both directions as previously described27.
Reporter assays were performed after cotransfection of renilla reporter constructs into JK1 cells using X-tremegene HP transfection reagent (Sigma) along with a luciferase expressing plasmid for normalization103. Assays were performed using the dual luciferase kit (Promega) according to manufacturer’s instructions. Mutagenesis was performed with the Quick-change kit (Stratagene).
Mouse embryonic stem cell culture and differentiation into embryoid bodies was performed as described37 using cell lines generated by targeting the P-Klf1-GFP or P-Klf1-intron-GFP constructs into the site-specific Ainv18 homing site37. These contain a single copy, unidirectionally inserted sequence into the same site and avoids random integration.
Browser data showing ATAC-seq analyses of primary human cells were derived from49,50,52. Data for ChIP transcription factor binding was selected from the Penn State University VISION hematopoiesis human GRCh37/hg19 genome browser (https://main.genome-browser.bx.psu.edu/index.html), focusing on results using CD34+ or bone marrow derived erythroid cells.
Human CD34+ cells were purchased from AllCells or obtained from the Yale Cooperative Center of Excellence in Hematology. These were differentiated ex vivo under three-phase protocols, initially based on63,104 but then later based on64,65,66. Briefly, CD34+ cells were thawed on day 0 into X-VIVO 15 (Lonza, 04–418Q) supplemented with 100 ng/ml human stem cell factor (SCF), 100 ng/ml human thrombopoietin, and 100 ng/ml recombinant human Flt3-ligand. Transfection was performed before the expansion phase I, and cells were allowed to recover for 24–48 h in X-VIVO media prior to switching to phase I. The base erythroid differentiation medium (EDM) consists of IMDM supplemented with 330 μg/ml holo-human transferrin, 10 μg/ml recombinant human insulin, 2 IU/ml heparin, 5% human Octoplas AB plasma (OctaPharma), 3 IU/ml erythropoietin, 1% l-glutamine, and 1% penicillin/streptomycin. During phase I (days 0–7), EDM was further supplemented with 10E–6 M hydrocortisone, 100 ng/ml human SCF, and 5 ng/ml human IL-3. During phase II (days 7–11), EDM was supplemented with 100 ng/ml human SCF alone. During phase III (days 11–18), EDM had no additional supplements. All analyses were performed after harvesting the cells on day 11 or 18. FACS was performed on an Attune NxT Flow Cytometer using PE-conjugated mouse anti-human CD71 (Cat. 555537) and APC-conjugated mouse anti-human CD235a (Cat. 551336) antibodies from BD Biosciences. FCS Express version 7 software was used for subsequent analysis. Brightfield photography was performed with a Nikon Micro-phot-FX microscope equipped with a Q-Imaging camera.
Cloning of left and right TALEN arms was performed by insertion into a CMV promoter-containing plasmid, downstream of T7 polymerase promoter, HA tag, and NLS sequences, but upstream (in frame) of the Fok1 nuclease and bGH poly(A) signals (PNA Bio Inc). In addition, an RFP/GFP surrogate reporter was synthesized that contained the intron 1 sequence between them as a linker, but out of frame for GFP. Proper nuclease activity after transfection/expression leads to frameshift mutations and expression of GFP, enabling enrichment of a low number of positively transfected cells by sorting for RFP+/GFP+ double positivity (Fig. 2a,b)55.
Transfection of TALEN DNAs into KU812 cells was performed using the Neon Transfection System (1450v, 10 ms, 3 pulses), sorted for RFP+/GFP+, and the positive pools were diluted to single cells into 96-well plates. After expansion, DNA was isolated and analyzed by PCR at the region of interest (using sequencing primer pairs27) to determine whether any of these clones contained a deletion. Individual clones that were positive were expanded and used for subsequent analyses. Genomic PCR of cell line material used 25 ng purified DNA (Qiagen), followed by direct sequence analysis of the product (Macrogen).
CD34+ cells were transfected with the Amaxa Nucleofector II using program U-008 for TALEN DNA. For RNP we used the Amaxa 4D Nucleofector with program EO-100. Biological replicates (typically three) were each ultimately analyzed in triplicate.
AsCpf1 protein (enhanced69) was purchased from IDT. RNPs were freshly formed by mixing 105 pmol of Cpf1 protein with 120 pmol of gRNA (IDT) for 15 at room temperature, and kept on ice until use.
Positive pools of CD34+ cells were analyzed by next generation sequencing (MGH DNA Core Facility) following genomic DNA isolation and PCR. Alternatively, TIDE105 or ICE106 analysis was performed on these or on KU812 cell DNA samples using sequencing primer pairs27.
Predicted off-target effects of the TALEN pair was tested using the PROGNOS analysis program107. We found no predicted off-targets when 0, or even 3, mismatches are allowed per arm (Table S2)107. Predicted off-targets for the two gRNA designs used the CRISPOR program70, which showed no predicted off-targets even allowing for up to 2 mismatches (Table S3).
Sequences of all primers and oligos used for analyses are listed in Table S4. Indels directed at the ‘+58’ region of the BCL11A gene were generated by using gRNA #1617 and analyzed using its associated genomic primer pairs108.
RNA isolation and RT-qPCR analysis was as previously described37,109, and normalized to catalase108. RNA seq analysis was performed as described23 using NEBNext polyA selection (Cat. # E7490S) and RNA Library Prep Kit (Cat. # E7770L). Sequencing was performed on the Illumina NovaSeq 6000 System at the Icahn School of Medicine Genomics Core Facility. Raw data have been submitted to the Gene Expression Omnibus (GSE223212). The derived data used for the analysis of Fig. 4a (filtered by p < 0.05, |log2| ≥ 1) are listed in Tables S5 (downregulated) and S6 (upregulated); the derived data for the analyses of Fig. 4c–e, Figs. S7, S8 is listed in Table S7 (filtered by removal of padj = NA110).
Bioinformatic analysis was performed as described86. Statistical analysis and representations were performed in R with package DESeq2110. Figures were made with packages ggplot2 and EnhancedVolcano along with other packages111,112 (see Table S8 for additional links).
Chromatin immunoprecipitation (based on113) of TEL/ETV6 in KU812 cells after cross-linking with 1.0% formaldehyde was performed with a mix of antibodies (containing anti-TEL from Santa Cruz sc-1668335 and sc-8547 along with home-made rabbit polyclonal; generous gifts from Graves and Clark59). Chromatin was sonicated using a Sonics Vibra Cell 500 for ten 40-s pulses at 21% amplitude with 1-min intervals. Positive control targets (HHEX, GATA2, LMO2) were chosen based on114.
References
Laurenti, E. & Gottgens, B. From haematopoietic stem cells to complex differentiation landscapes. Nature (London) 553, 418–426 (2018).
Miller, I. J. & Bieker, J. J. A novel, erythroid cell-specific murine transcription factor that binds to the CACCC element and is related to the Krüppel family of nuclear proteins. Mol. Cell. Biol. 13, 2776–2786 (1993).
Perkins, A. et al. Kruppeling erythropoiesis: An unexpected broad spectrum of human red blood cell disorders due to KLF1 variants. Blood 127, 1856–1862 (2016).
Waye, J. S. & Eng, B. Kruppel-like factor 1: Hematologic phenotypes associated with KLF1 gene mutations. Int. J. Lab. Hematol. 37(Suppl 1), 78–84 (2015).
Gnanapragasam, M. N. & Bieker, J. J. Orchestration of late events in erythropoiesis by KLF1/EKLF. Curr. Opin. Hematol. 24, 183–190 (2017).
Caria, C. A., Faa, V. & Ristaldi, M. S. Kruppel-like factor 1: A pivotal gene regulator in erythropoiesis. Cells 11, 3069 (2022).
Donze, D., Townes, T. M. & Bieker, J. J. Role of erythroid Krüppel-like factor (EKLF) in human g- to β-globin switching. J. Biol. Chem. 270, 1955–1959 (1995).
Siatecka, M. & Bieker, J. J. The multifunctional role of EKLF/KLF1 during erythropoiesis. Blood 118, 2044–2054 (2011).
Tallack, M. R. & Perkins, A. C. KLF1 directly coordinates almost all aspects of terminal erythroid differentiation. IUBMB Life 62, 886–890 (2010).
Yien, Y. Y. & Bieker, J. J. EKLF/KLF1, a tissue-restricted integrator of transcriptional control, chromatin remodeling, and lineage determination. Mol. Cell. Biol. 33, 4–13 (2013).
Drissen, R. et al. The active spatial organization of the beta-globin locus requires the transcription factor EKLF. Genes Dev. 18, 2485–2490 (2004).
Schoenfelder, S. et al. Preferential associations between co-regulated genes reveal a transcriptional interactome in erythroid cells. Nat. Genet. 42, 53–61 (2010).
Nuez, B., Michalovich, D., Bygrave, A., Ploemacher, R. & Grosveld, F. Defective haematopoiesis in fetal liver resulting from inactivation of the EKLF gene. Nature (London) 375, 316–318 (1995).
Perkins, A. C., Sharpe, A. H. & Orkin, S. H. Lethal β-thalassemia in mice lacking the erythroid CACCC-transcription factor EKLF. Nature (London) 375, 318–322 (1995).
Tallack, M. R. et al. A global role for KLF1 in erythropoiesis revealed by ChIP-seq in primary erythroid cells. Genome Res. 20, 1052–1063 (2010).
Borg, J., Patrinos, G. P., Felice, A. E. & Philipsen, S. Erythroid phenotypes associated with KLF1 mutations. Haematologica 96, 635–638 (2011).
Huang, J. et al. Compound heterozygosity for KLF1 mutations is associated with microcytic hypochromic anemia and increased fetal hemoglobin. Eur. J. Hum. Genet. 23, 1341–1348 (2015).
Viprakasit, V. et al. Mutations in Kruppel-like factor 1 cause transfusion-dependent hemolytic anemia and persistence of embryonic globin gene expression. Blood 123, 1586–1595 (2014).
Xu, L. et al. Compound heterozygosity for KLF1 mutations causing hemolytic anemia in children: A case report and literature review. Front. Genet. 12, 691461 (2021).
Tangsricharoen, T. et al. Severe neonatal haemolytic anaemia caused by compound heterozygous KLF1 mutations: Report of four families and literature review. Br. J. Haematol. 194, 626–634 (2021).
Magor, G. W. et al. KLF1-null neonates display hydrops fetalis and a deranged erythroid transcriptome. Blood 125, 2405–2417 (2015).
Arnaud, L. et al. A dominant mutation in the gene encoding the erythroid transcription factor KLF1 causes a congenital dyserythropoietic anemia. Am. J. Hum. Genet. 87, 721–727 (2010).
Varricchio, L. et al. Genetic disarray follows mutant KLF1-E325K expression in a congenital dyserythropoietic anemia patient. Haematologica 104, 2372–2380 (2019).
Kulczynska-Figurny, K., Bieker, J. J. & Siatecka, M. Severe anemia caused by dominant mutations in Kruppel-like factor 1 (KLF1). Mutat. Res. 786, 108336 (2020).
Astle, W. J. et al. The allelic landscape of human blood cell trait variation and links to common complex disease. Cell 167, 1415-1429 e1419 (2016).
Ganesh, S. K. et al. Multiple loci influence erythrocyte phenotypes in the CHARGE Consortium. Nat. Genet. 41, 1191–1198 (2009).
Gnanapragasam, M. N. et al. Survey and evaluation of mutations in the human KLF1 transcription unit. Sci. Rep. 8, 6587 (2018).
Wang, Q. et al. Rare variant contribution to human disease in 281,104 UK Biobank exomes. Nature (London) 597, 527–532 (2021).
Borg, J. et al. Haploinsufficiency for the erythroid transcription factor KLF1 causes hereditary persistence of fetal hemoglobin. Nat. Genet. 42, 801–805 (2010).
Natiq, A. et al. Hereditary persistence of fetal hemoglobin in two patients with KLF1 haploinsufficiency due to 19p13.2–p13.12/13 deletion. Am. J. Hematol. 92, E2–E3 (2017).
Liu, D. et al. KLF1 mutations are relatively more common in a thalassemia endemic region and ameliorate the severity of beta-thalassemia. Blood 124, 803–811 (2014).
Xie, X. M., Liu, Y. N., Li, J., Jiang, F. & Li, D. Z. A Kruppel-like factor 1 gene mutation ameliorates the severity of beta-thalassemia: A case report. Hemoglobin 43, 137–139 (2019).
Chen, X., Reitman, M. & Bieker, J. J. Chromatin structure and transcriptional control elements of the erythroid Kruppel-like factor (EKLF) gene. J. Biol. Chem. 273, 25031–25040 (1998).
Adelman, C. A., Chattopadhyay, S. & Bieker, J. J. The BMP/BMPR/Smad pathway directs expression of the erythroid-specific EKLF and GATA1 transcription factors during embryoid body differentiation in serum-free media. Development 129, 539–549 (2002).
Xue, L., Chen, X., Chang, Y. & Bieker, J. J. Regulatory elements of the EKLF gene that direct erythroid cell-specific expression during mammalian development. Blood 103, 4078–4083 (2004).
Zhou, D., Liu, K., Sun, C. W., Pawlik, K. M. & Townes, T. M. KLF1 regulates BCL11A expression and gamma- to beta-globin gene switching. Nat. Genet. 42, 742–744 (2010).
Lohmann, F. & Bieker, J. J. Activation of Eklf expression during hematopoiesis by Gata2 and Smad5 prior to erythroid commitment. Development 135, 2071–2082 (2008).
Crossley, M., Tsang, A. P., Bieker, J. J. & Orkin, S. H. Regulation of the erythroid Kruppel-like factor (EKLF) gene promoter by the erythroid transcription factor GATA-1. J. Biol. Chem. 269, 15440–15444 (1994).
Lohmann, F. et al. The DEK oncoprotein is a critical component of the EKLF/KLF1 enhancer in erythroid cells. Mol. Cell. Biol. 35, 3726–3738 (2015).
Chen, X., Lohmann, F. & Bieker, J. J. Association of Ddx5/p68 protein with the upstream erythroid enhancer element (EHS1) of the Klf1 gene. bioRxiv 2019, 743435 (2019).
Ghosh, S. et al. Identification of RIOK2 as a master regulator of human blood cell development. Nat. Immunol. 23, 109–121 (2022).
Madzo, J. et al. Hydroxymethylation at gene regulatory regions directs stem/early progenitor cell commitment during erythropoiesis. Cell Rep. 6, 231–244 (2014).
Fluhr, S. et al. Epigenetic dysregulation of the erythropoietic transcription factor KLF1 and the beta-like globin locus in juvenile myelomonocytic leukemia. Epigenetics 12, 715–723 (2017).
Zhang, X. et al. DNMT3A and TET2 compete and cooperate to repress lineage-specific transcription factors in hematopoietic stem cells. Nat. Genet. 48, 1014–1023 (2016).
Emanuel, P. D. Juvenile myelomonocytic leukemia. Curr. Hematol. Rep. 3, 203–209 (2004).
Tefferi, A. & Gilliland, D. G. Oncogenes in myeloproliferative disorders. Cell Cycle 6, 550–566 (2007).
Van Etten, R. A. & Shannon, K. M. Focus on myeloproliferative diseases and myelodysplastic syndromes. Cancer Cell 6, 547–552 (2004).
Stieglitz, E. et al. The genomic landscape of juvenile myelomonocytic leukemia. Nat. Genet. 47, 1326–1333 (2015).
Schulz, V. P. et al. A unique epigenomic landscape defines human erythropoiesis. Cell Rep. 28, 2996-3009 e2997 (2019).
Ulirsch, J. C. et al. Interrogation of human hematopoiesis at single-cell and single-variant resolution. Nat. Genet. 51, 683–693 (2019).
Ludwig, L. S. et al. Transcriptional states and chromatin accessibility underlying human erythropoiesis. Cell Rep. 27, 3228-3240 e3227 (2019).
Zhang, K. et al. A single-cell atlas of chromatin accessibility in the human genome. Cell 184, 5985-6001 e5919 (2021).
Joung, J. K. & Sander, J. D. TALENs: A widely applicable technology for targeted genome editing. Nat. Rev. Mol. Cell. Biol. 14, 49–55 (2013).
Miller, J. C. et al. A TALE nuclease architecture for efficient genome editing. Nat. Biotechnol. 29, 143–148 (2011).
Kim, H., Um, E., Cho, S. R., Jung, C. & Kim, J. S. Surrogate reporters for enrichment of cells with nuclease-induced mutations. Nat. Methods 8, 941–943 (2011).
Newburger, D. E. & Bulyk, M. L. UniPROBE: An online database of protein binding microarray data on protein-DNA interactions. Nucleic Acids Res. 37, D77–D82 (2009).
Hume, M. A., Barrera, L. A., Gisselbrecht, S. S. & Bulyk, M. L. UniPROBE, update 2015: New tools and content for the online database of protein-binding microarray data on protein-DNA interactions. Nucleic Acids Res. 43, D117–D122 (2015).
Wei, G. H. et al. Genome-wide analysis of ETS-family DNA-binding in vitro and in vivo. EMBO J. 29, 2147–2160 (2010).
Hollenhorst, P. C., McIntosh, L. P. & Graves, B. J. Genomic and biochemical insights into the specificity of ETS transcription factors. Annu. Rev. Biochem. 80, 437–471 (2011).
Thoms, J. A. I. et al. Disruption of a GATA2-TAL1-ERG regulatory circuit promotes erythroid transition in healthy and leukemic stem cells. Blood 138, 1441–1455 (2021).
Kingsley, P. D. et al. Ontogeny of erythroid gene expression. Blood 121, e5–e13 (2013).
Wu, C., Jin, X., Tsueng, G., Afrasiabi, C. & Su, A. I. BioGPS: Building your own mash-up of gene annotations and expression profiles. Nucleic Acids Res. 44, D313–D316 (2016).
Antoniani, C. et al. Induction of fetal hemoglobin synthesis by CRISPR/Cas9-mediated editing of the human beta-globin locus. Blood 131, 1960–1973 (2018).
Grevet, J. D. et al. Domain-focused CRISPR screen identifies HRI as a fetal hemoglobin regulator in human erythroid cells. Science 361, 285–290 (2018).
Breda, L. et al. Forced chromatin looping raises fetal hemoglobin in adult sickle cells to higher levels than pharmacologic inducers. Blood 128, 1139–1143 (2016).
Wu, Y. et al. Highly efficient therapeutic gene editing of human hematopoietic stem cells. Nat. Med. 25, 776–783 (2019).
Hur, J. K. et al. Targeted mutagenesis in mice by electroporation of Cpf1 ribonucleoproteins. Nat. Biotechnol. 34, 807–808 (2016).
Liu, Z. et al. Genome editing of CCR5 by AsCpf1 renders CD4(+)T cells resistance to HIV-1 infection. Cell Biosci. 10, 85 (2020).
Kleinstiver, B. P. et al. Engineered CRISPR-Cas12a variants with increased activities and improved targeting ranges for gene, epigenetic and base editing. Nat. Biotechnol. 37, 276–282 (2019).
Concordet, J. P. & Haeussler, M. CRISPOR: Intuitive guide selection for CRISPR/Cas9 genome editing experiments and screens. Nucleic Acids Res. 46, W242–W245 (2018).
Vakulskas, C. A. et al. A high-fidelity Cas9 mutant delivered as a ribonucleoprotein complex enables efficient gene editing in human hematopoietic stem and progenitor cells. Nat. Med. 24, 1216–1224 (2018).
Gundry, M. C. et al. Highly efficient genome editing of murine and human hematopoietic progenitor cells by CRISPR/Cas9. Cell Rep. 17, 1453–1461 (2016).
Weber, L. et al. Editing a gamma-globin repressor binding site restores fetal hemoglobin synthesis and corrects the sickle cell disease phenotype. Sci. Adv. 6, eaay9392 (2020).
Blobel, G. A., Higgs, D. R., Mitchell, J. A., Notani, D. & Young, R. A. Testing the super-enhancer concept. Nat. Rev. Genet. 22, 749–755 (2021).
Hua, P. et al. Defining genome architecture at base-pair resolution. Nature (London) 595, 125–129 (2021).
Heshusius, S. et al. Epigenomic analysis of KLF1 haploinsufficiency in primary human erythroblasts. Sci. Rep. 12, 336 (2022).
Su, M. Y. et al. Identification of biologically relevant enhancers in human erythroid cells. J. Biol. Chem. 288, 8433–8444 (2013).
Tallack, M. R. et al. Novel roles for KLF1 in erythropoiesis revealed by mRNA-seq. Genome Res. 22, 2385–2398 (2012).
Singleton, B. K., Burton, N. M., Green, C., Brady, R. L. & Anstee, D. J. Mutations in EKLF/KLF1 form the molecular basis of the rare blood group In(Lu) phenotype. Blood 112, 2081–2088 (2008).
Singleton, B. K., Frayne, J. & Anstee, D. J. Blood group phenotypes resulting from mutations in erythroid transcription factors. Curr. Opin. Hematol. 19, 486–493 (2012).
Tallack, M. R. & Perkins, A. C. Three fingers on the switch: Kruppel-like factor 1 regulation of gamma-globin to beta-globin gene switching. Curr. Opin. Hematol. 20, 193–200 (2013).
Lamsfus-Calle, A. et al. Comparative targeting analysis of KLF1, BCL11A, and HBG1/2 in CD34(+) HSPCs by CRISPR/Cas9 for the induction of fetal hemoglobin. Sci. Rep. 10, 10133 (2020).
Mukherjee, K. & Bieker, J. J. EKLF/Klf1 regulates erythroid transcription by its pioneering activity and selective control of RNA Pol II pause-release. Cell Rep. 41, 111830 (2022).
Borst, S. et al. Study of inherited thrombocytopenia resulting from mutations in ETV6 or RUNX1 using a human pluripotent stem cell model. Stem Cell Rep. 16, 1458–1467 (2021).
Southwood, C. M., Downs, K. M. & Bieker, J. J. Erythroid Kruppel-like Factor (EKLF) exhibits an early and sequentially localized pattern of expression during mammalian erythroid ontogeny. Dev. Dyn. 206, 248–259 (1996).
Mukherjee, K. et al. EKLF/KLF1 expression defines a unique macrophage subset during mouse erythropoiesis. Elife 10, e61070 (2021).
Frontelo, P. et al. Novel role for EKLF in megakaryocyte lineage commitment. Blood 110, 3871–3880 (2007).
Cui, K. et al. Chromatin signatures in multipotent human hematopoietic stem cells indicate the fate of bivalent genes during differentiation. Cell Stem Cell 4, 80–93 (2009).
Li, B., Ding, L., Li, W., Story, M. D. & Pace, B. S. Characterization of the transcriptome profiles related to globin gene switching during in vitro erythroid maturation. BMC Genom. 13, 153 (2012).
Bauer, D. E., Kamran, S. C. & Orkin, S. H. Reawakening fetal hemoglobin: Prospects for new therapies for the beta-globin disorders. Blood 120, 2945–2953 (2012).
Yu, Y. et al. Bcl11a is essential for lymphoid development and negatively regulates p53. J. Exp. Med. 209, 2467–2483 (2012).
Brendel, C. et al. Lineage-specific BCL11A knockdown circumvents toxicities and reverses sickle phenotype. J. Clin. Investig. 126, 3868–3878 (2016).
Liu, P. et al. Bcl11a is essential for normal lymphoid development. Nat. Immunol. 4, 525–532 (2003).
Lazarus, K. A. et al. BCL11A interacts with SOX2 to control the expression of epigenetic regulators in lung squamous carcinoma. Nat. Commun. 9, 3327 (2018).
Eernstman, J. et al. Novel variants in Krueppel like factor 1 that cause persistence of fetal hemoglobin in In(Lu) individuals. Sci. Rep. 11, 18557 (2021).
Elkon, R. & Agami, R. Two-layer design protects genes from mutations in their enhancers. Nature (London) 609, 477–478 (2022).
Lin, X. et al. Nested epistasis enhancer networks for robust genome regulation. Science 377, 1077–1085 (2022).
Bergman, D. T. et al. Compatibility rules of human enhancer and promoter sequences. Nature (London) 607, 176–184 (2022).
Drexler, H. G. Guide to Leukemia-Lymphoma Cell Lines 2nd edn. (Elsevier, 2010).
Kurita, R. et al. Establishment of immortalized human erythroid progenitor cell lines able to produce enucleated red blood cells. PLoS One 8, e59890 (2013).
Bieker, J. J. Isolation, genomic structure, and expression of human Erythroid Kruppel-like Factor (EKLF). DNA Cell Biol. 15, 347–352 (1996).
Li, B. et al. Characterization of transcription factor networks involved in umbilical cord blood CD34+ stem cells-derived erythropoiesis. PLoS One 9, e107133 (2014).
Siatecka, M., Xue, L. & Bieker, J. J. Sumoylation of EKLF promotes transcriptional repression and is involved in inhibition of megakaryopoiesis. Mol. Cell. Biol. 27, 8547–8560 (2007).
Yien, Y. Y. & Bieker, J. J. Functional interactions between erythroid Kruppel-like factor (EKLF/KLF1) and protein phosphatase PPM1B/PP2Cbeta. J. Biol. Chem. 287, 15193–15204 (2012).
Brinkman, E. K., Chen, T., Amendola, M. & van Steensel, B. Easy quantitative assessment of genome editing by sequence trace decomposition. Nucleic Acids Res. 42, e168 (2014).
Hsiau, T. et al. Inference of CRISPR edits from Sanger Trace Data. CRISPR J. 5, 123–130 (2019).
Fine, E. J., Cradick, T. J., Zhao, C. L., Lin, Y. & Bao, G. An online bioinformatics tool predicts zinc finger and TALE nuclease off-target cleavage. Nucleic Acids Res. 42, e42 (2014).
Zeng, J. et al. Therapeutic base editing of human hematopoietic stem cells. Nat. Med. 26, 535–541 (2020).
Gnanapragasam, M. N. et al. EKLF/KLF1-regulated cell cycle exit is essential for erythroblast enucleation. Blood 128, 1631–1641 (2016).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014).
Lawrence, M. et al. Software for computing and annotating genomic ranges. PLoS Comput. Biol. 9, e1003118 (2013).
Soneson, C., Love, M. I. & Robinson, M. D. Differential analyses for RNA-seq: Transcript-level estimates improve gene-level inferences. F1000Res 4, 1521 (2015).
Planutis, A. et al. Neomorphic effects of the neonatal anemia (Nan-Eklf) mutation contribute to deficits throughout development. Development 144, 430–440 (2017).
Unnikrishnan, A. et al. A quantitative proteomics approach identifies ETV6 and IKZF1 as new regulators of an ERG-driven transcriptional network. Nucleic Acids Res. 44, 10644–10661 (2016).
Acknowledgements
We thank Dr. Mignon Loh (UCSF) for JMML patient DNA samples. We thank Drs Barbara Graves and Kathleen Clark (University of Utah) for antibodies and discussion related to ETV6, and Dr. John Pimanda (Prince of Wales Hospital) for target binding sequence information. We thank Drs. Dan Bauer and Divya Vinjamur (Bauer lab; Harvard University), Scott Peslak and Aoi Wakabayashi (Blobel lab; University of Pennsylvania), and Laura Breda (Rivella lab; University of Pennsylvania) for cell culture protocol advice. We acknowledge Dr. Kaustav Mukherjee (MSSM) for ongoing discussions. We thank Drs. Vesna Najfeld and Joseph Tripodi (MSSM) for karyotype analysis, and Drs. Vivian Simon and Lubbertus Mulder (MSSM) for Amaxa usage. This work was supported by Doris Duke Charitable Foundation award 2019113 to JJB and JAG, by PHS grant R01 DK46865 to JJB, and by a Cooley’s Anemia Foundation Fellowship to MNG. The Yale Cooperative Center of Excellence in Hematology is supported by DK106829.
Author information
Authors and Affiliations
Contributions
M.N.G. and A.P. performed experiments; A.P. carried out bioinformatics; M.N.G. and J.J.B. conceptualized the study; J.G. and J.J.B. oversaw the study and provided support; M.N.G. and J.J.B. wrote the initial draft, then all authors contributed to the final version.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Gnanapragasam, M.N., Planutis, A., Glassberg, J.A. et al. Identification of a genomic DNA sequence that quantitatively modulates KLF1 transcription factor expression in differentiating human hematopoietic cells. Sci Rep 13, 7589 (2023). https://doi.org/10.1038/s41598-023-34805-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-023-34805-5
- Springer Nature Limited