
Abstract
Anticancer peptides (ACPs) perform a promising role in discovering anti-cancer drugs. The growing research on ACPs as therapeutic agent is increasing due to its minimal side effects. However, identifying novel ACPs using wet-lab experiments are generally time-consuming, labor-intensive, and expensive. Leveraging computational methods for fast and accurate prediction of ACPs would harness the drug discovery process. Herein, a machine learning-based predictor, called PLMACPred, is developed for identifying ACPs from peptide sequence only. PLMACPred adopted a set of encoding schemes representing evolutionary-property, composition-property, and protein language model (PLM), i.e., evolutionary scale modeling (ESM-2)- and ProtT5-based embedding to encode peptides. Then, two-dimensional (2D) wavelet denoising (WD) was employed to remove the noise from extracted features. Finally, ensemble-based cascade deep forest (CDF) model was developed to identify ACP. PLMACPred model attained superior performance on all three benchmark datasets, namely, ACPmain, ACPAlter, and ACP740 over tenfold cross validation and independent dataset. PLMACPred outperformed the existing models and improved the prediction accuracy by 18.53%, 2.4%, 7.59% on ACPmain, ACPalter, ACP740 dataset, respectively. We showed that embedding from ProtT5 and ESM-2 was capable of capturing better contextual information from the entire sequence than the other encoding schemes for ACP prediction. For the explainability of proposed model, SHAP (SHapley Additive exPlanations) method was used to analyze the feature effect on the ACP prediction. A list of novel sequence motifs was proposed from the ACP sequence using MEME suites. We believe, PLMACPred will support in accelerating the discovery of novel ACPs as well as other activities of microbial peptides.
Similar content being viewed by others
Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Introduction
Cancer is an alarming health concern and leading cause of deaths worldwide1. Existing conventional clinical therapeutic methods such as chemotherapy, radiotherapy, immunotherapy, surgical interventions and targeted therapy are widely used for cancer treatment2. But, these methods damage the normal cells and sever negative side effects such as infections, bleeding, pronounced immunosuppression on patient body3,4. At present, the scientific research has proven that therapeutic peptides such as anticancer peptide (ACP) and host defense peptides (HDPs) contain high selectivity, specificity that make it more favorable safe drug agent against cancer5. HDPs exist in both amphibians and plants demonstrate the potential to recognize cancer cells in breast cancer, melanoma, and lung cancer with minimum drug resistance in these cancer types6. Similarly, D-K6LP is a polypeptide found on the surface of cancer cells, exhibits anticancer property because of interacting electrostatically with phosphatidylserine7. However, among these agents, ACP is considered competent alternative due to its divers functions likewise low toxicity, high specificity and, minimal innate immune function for developing anticancer vaccines.
ACPs are short length (5-30) polypeptide sequences known for its anticancer activity. Typically, ACP consists of 10–50 amino acids (AAs) and exhibits a complex structure, functioning as a molecular polymer involving AA and proteins8. The polymerization occurs through peptide bonds, connecting several or dozens of AAs. ACP demonstrates the capability to disrupt the structure of tumor cell membranes, consequently impeding the proliferation and migration of cancer cells. Moreover, it can induce apoptosis in cancer cells without causing harm to normal human cells9. ACP’s unique ability to interact specifically with the anionic cell membrane components of cancer cells enables them to selectively eliminate cancer cells with minimal impact on normal cells10. Additionally, certain ACPs, such as cell-penetrating peptides or peptide drugs, have demonstrated the capability to inhibit the cell cycle or other cellular functionalities, thus enhancing their safety profile compared to traditional broad-spectrum drugs11. These attributes have rendered ACPs a highly competitive choice for therapeutics compared to small molecules and antibodies. Recent research has indicated that ACPs exhibit selectivity towards cancer cells while leaving normal physiological functions unaffected12,13. This makes ACP a promising therapeutic approach for cancer treatment. Over the past decade, numerous peptide-based therapies targeting various types of tumors have been assessed and are presently undergoing evaluation across various stages of preclinical and clinical trials14,15. This underscores the significance of developing novel ACPs for the treatment of cancer. Nonetheless, only a limited number of these ACPs may ultimately progress to clinical treatment due to the rigorous selection process16. Moreover, the validation of potential new ACPs through in vitro or in vivo method is both time-consuming and expensive, further compounded by limitations in laboratory resources17. Considering the significant therapeutic potential of ACPs in biomedical applications, there is a pressing demand for high throughput, rapid, and cost-effective discovery of ACPs using computational methods.
Over the past years, the abundance of peptide sequencing accumulating in postgenomic era, has sparked/emerged the researcher’s interest to utilize artificial intelligence knowledge particularly machine learning(ML)18,19,20,21,22,23,24,25, ensemble learning (EL)26,27,28,29,30,31 and deep learning (DL) methods32,33,34,35,36,37,38,39 in ACPs identification. In ML-based ACP predictors, support vector machine (SVM) and random forest (RF) were the widely adopted classifiers. For example, iACP18, AntiCP19, cACP23, and AntiCP_2.024 predictors were developed using traditional ML-classifiers. Similarly, in EL-based methods, majority voting and stacking ensemble strategies were adopted using multi-feature representation to predict the correct ACPs. For example,ACPred-FL26, StackACPred30, and ACPpred-Fuse28 etc. developed EL-based ACP models. Moreover, advanced DL-based methods further improved the performance of predicting ACPs and non-ACPs using sequence information. For example, S Ahmed et al. proposed multi-head multi-headed deep-convolutional neural network(MHCNN)33, Z Lv proposed iACP-DRLF(identify anticancer peptides via deep representation learning features)38, HC Yi proposed deep learning-based long short-term memory model (ACP-DL)40, and W Zhou developed tri-fusion neural network(TriNet)36 for predicting ACP activity. For more details, the readers are referred to comprehensive review articles on existing ACP tools41,42.
Considering the above backdrop, in this study, we develop PMLACPred, a protein language model for identifying and characterizing ACPs activity (see Fig. 1). First, we used four feature representation schemes, namely (a) component protein sequence representation (CPSR), (b) histogram of oriented gradient-based HOG position-specific scoring matrix (HOG-PSSM), (c) ProtT5, and (d) ESM-2 to extract features from ACP sequence. Then, 2D wavelet denoising (WD) was employed on the extracted features to remove the noise and enhance the prediction of the proposed model. Finally, the processed denoised features are fed into upgraded cascade deep forest (CDF) classifier to build the final ACP-based prediction model. The proposed PMLACPred tool reaches superior performance in term of accuracy up to 94.76%, 96.39% and 98.65% on three independent datasets, i.e., ACPmain, ACPAlter, and ACP740. The contribution of this work can be highlighted as follows:
-
(a)
We extracted local and global features from peptide sequences using transformer-based models. i.e., complementary embedding techniques through ProtT5 and ESM-2, evolutionary-based features through HOG-PSSM, and compositional-based features through CPSR encoding method.
-
(b)
We implemented 2D WD algorithm to effectively de-noise the extracted feature vector and enhance the prediction performance of the proposed ACP prediction model.
-
(c)
We developed PLMACPred, a modified cascade deep forest based model with above mentioned hybrid feature set and obtained the best accuracy outperforming existing models for the same purpose on three benchmark datasets.

The schematic diagram of the PLMACPred method.
Results and discussion
Classifier performance using various feature encoding schemes before and after applying Wavelet Denoising method
In this article, we used four types of single-view feature CPSR, ESM-2, ProtT5 and HOG-PSSM and their combinations (multi-view features). We use the notation F1, F2, F3, F4 to represent CPSR, HOP-PSSM, ESM-2 and ProtT5 features in Tables 1 and 2, respectively. We used SVM and NB as baseline models and cascade deep forest (CDF) as proposed learning model for ACP prediction as depicted in Fig. 1. We used individual features as well as their combination for building ML models. During the training process, we utilized three benchmarking datasets namely ACPmain, ACPAlter and ACP740. After using numerous encoding schemes, we implemented 2D wavelet denoising algorithm to reduce the noise and enhance the prediction performance of classifiers. Table 1 depicts the prediction performance of benchmark datasets ACPmain, ACPAlter and ACP740 using 10-fold CV with and without applying 2D WD algorithm. Similarly, Table 2 shows the prediction performance of three independent datasets with and without using 2D WD method.
We can see from Table 1 that prediction performance of CDF classifier using single features particularly (ProtT5 and ESM-2) and hybrid features (F1+F2+F3) and (F1+F2+F4) has significantly increased by using 2D WD. In case of ACPmain, CDF achieved the highest Acc value of (0.992) with MCC of (0.953) on 10-fold CV method and Acc of (0.948) with a corresponding MCC of (0.896) on independent dataset in Table 2. In case of second dataset ACPalter, again CDF classifier afford the highest performance in term of Acc value 0.988 with MCC of 0.996 using hybrid features after apply 2D WD method. The CDF classifier was also validated on the second independent dataset and attained the Acc of 0.990 and MCC of 0.995(Table 4). In contrast the worst performance is archived by NB classifier using HOG-PSSM feature encoding scheme. We further validated and compare the prediction performance of same set of features using CDF, SVM and NB learning engine on ACP740. We observed, again CDF model beat the other classifiers using hybrid feature combination of F1+F2+F4 and affording Acc of 0.992 and MCC of 0.983 on the training dataset (Table 3) and Acc(0.987) with MCC of(0.973) on validation or independent dataset (Table 2).
Based on the above results, we can observe that better classification performance was achieved by CDF classifier—an ensemble based model. The second observation is, using 2D WD method helped to enhance the overall performance of the model (Fig. 2). Third, after fusing the evolutionary-based, physicochemical-based and deep embedding-based features helped to improve the prediction of ACPs.

Performance of PLMACPpred on ACPmain independent dataset with- and with-out WD. We have only highlighted the number of best performing combination of features with ProtT5 + CPSR + PSSMHOG. After WD, performance improve for all experiment setup.
Comparison with existing methods
To understand the strength and weaknesses of newly designed method, it is important to compare it with state-of-the-art methods. For this purpose, we compared performance of proposed PLMACPred model with existing ML-based and DL-based ACP predictors on three independent datasets namely ACPmain, ACPAlter and ACP740. Table 3 lists the prediction results for ACP identification on ACPmain test dataset from previous models, i.e., iACP18, ACP-MHCNN33, iACP-DRLF38, AntiCP_2.024, AntiCP8, ACPred22, ACPred-FL26, ACPpred-Fuse28, ACP-check39, TriNet,36 and ACPPfel37. Figure 3a–c show the success rates of various computational ACP predictors in term of Acc, Sen, Spe, and MCC for the three datasets. We can observe the prediction outcomes in Table 3 and in Fig. 3a, it is clear that overall efficacy of PLMACPred is superior to existing ACP tools with the best performance in term of Acc(96.60%), Sn(94.80%),Sp(97.10%) and MCC(0.896). The prediction score indicate that our proposed method surpass the recent developed ACP-based predictors such as ACPPfel, TriNet and ACP-check by ACC of 18.53%, Sn of 13.51%, Sp of 18.34% and MCC of 30%.

Performance comparison between PLMACPpred and other models in (a) ACPmain, (b) ACPAlter, and (c) ACP740 datasets.
For further validating the robustness of the proposed method, PLMACPred was compared with five deep learning models and six ensemble-based ML ACP models on ACPAlter independent dataset. The detailed comparison outcomes are shown in Table 4 and Fig. 3b. We can observe that that PLMACPred model achieved significantly better performance than ACP-MHCNN, iACP-DRLF, AntiCP_2.0, ACP-DL, ACP-check, ME-ACP, TriNet and ACPPfel. For example, the optimal performance of PLMACPred on this dataset in term of ACC is 99.00%, and MCC of 0.989. The second best performance attained by TriNet which is Acc of 96.60% and MCC of 0.871. The ACPPfel method obtained comparable results with other developed tool i.e., ME-ACP and ACP-check with respect to ACC and MCC measure. Overall, PLMACPred produced dominant performance and enhanced the Acc, Sen, Spe and MCC by 2.4–6%, 1.2–8%, 4.8–6.5%, 5.9–12.90% respectively.
For making more intuitive comparison, we considered another verified Independent dataset ACP740. The validation results of multiple DL-based predictors on ACP740 are depicted in Table 5 and Fig. 3c. Again the proposed PLMACPred model outperforms ACPPfel, TriNet, ME-ACP and ACP-check with increased Acc of 7.59%, Sen of 6.11%, Spe of 5.88%, and MCC of 13.47%. Thus, the empirical outcomes demonstrates that the PLMACPred could produce promising performance for all three datasets in term of all performance indicators namely Acc, MCC, AUC, Spe and Sen.
Motif-based analysis and feature contribution in ML model
Sequence motifs exhibits conserved region over sequence collection often linking towards the function of certain gene or peptides. We discover sequence motifs from the collection of ACPs compared to non-ACPs using MEME tool43. Figure 4 shows the top ranked motifs from ACP in the main training dataset. It is observed that certain enrich motifs such as FLPY- LAGVAAKVLPKIFCKIT, IPCGESCVFIPCITP, GCSCKSKVCYR were found exclusively in ACPs and FAKKLAKLAKK, RKAFRWAWRMLKKAA, DTPLDLAIQHLQRLTIQELPDPPTDLPE, in non-ACPs. From the top motifs, we can observe that in motifs from ACP are mainly C,F, L, Y enriched, and the non-ACP motifs are dominantly A, D, K, P,Q, R, W enriched. Supplementary Table S1 shows the list of exclusive motifs in ACPs and non ACPs.

The identified top motifs from ACP (a–c) vs. non-ACP (d–f).
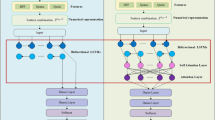
In machine learning, model interpretation plays a significant role to quantify the prediction reliability44. Before developing the model for ACP prediction, the sequence properties in training samples were analyzed using SHAP algorithms to visualize the contribution of important attributes. In Fig. 5, we demonstrated the impact of top 25 ranked engineered features namely CPSR, HOG-PSSM, ESM-2 and ProtT5 in predicting ACPs.

The SHAP diagram for the important featues for PLMACPred from (a) ESM , (b) CPSR, (c) ProtT5, (d) HOG-PSSM.
Material and methods
Benchmark dataset
In designing computational model, the construction of valid benchmark dataset is crucial step to train and test the prediction system45,46,47,48. In ACP prediction, we collected three valid datasets namely ACPmain and ACPAlter datasets from AntiCP2.049 and ACP740 from ACP-DL40 for fair comparison. The origin of all these derived datasets is CancerPPD database50. A series of preprocessing steps such as (labeling, removal of redundant and ambiguous sequences), were undertaken to ensure the quality of the data. By following these steps, ACPmain dataset includes 861 experimentally verified positive peptide samples as ACPs and an equal number of negative peptide samples as non-ACPs. ACPAlter datasets comprises 970 ACPs as positive samples and same number of non-ACPs as negative samples. Similarly, ACP740 dataset contains 740 peptides (including 374 ACPs and 364 non-ACPs). We split the collected benchmark datasets into training and independent testing subset at an 80:20 ratio. Table 6 shows the statistics of ACP and non-ACPs in all three datasets.
Feature encoding methods
Formulating a biological protein/peptide to statistical values using feature encoding methods is a crucial step51,52,53. In this research, to encode ACPs and non-ACPs, we considered compositional-based (CPSR), evolutionary-based (HOG-PSSM) and exploiting the power of large-language-based models to encode microbial peptide sequences into fixed-length feature vector. Each feature encoding method are explained in subsequent subsections.
Composition-based feature extraction method
ACPs are polymers composed of twenty natural AAs residues, while chemically different but structurally identical because of the side chain or functional groups of AAs. These peptides have unique physiochemical properties, frequency of occurrence and sequence length that play an effective role in characterizing the functions of proteins or peptides5. In this study, we use composition-based features called composite protein sequence representation (CPSR) method, previously used for the prediction of membrane proteins54 and anti-MRSA peptides55. CPSR descriptor, extract the set of seven different properties of AAs such as conventional frequency of AAs, sum of hydrophobicity, sequence length, bi-gram exchange group, R-group, electronic and, hydrophobic group given in Supplementary Table S2. The resultant feature vector of CPSR is 71-dimensions. The readers are referenced for more details to our previous study56.
Evolutionary-based feature extraction method
Over the recent years, evolutionary-based features have been successfully used for improving the performance of various bioinformatics predictors such as bacteriophage virion protein prediction57, DNA-binding proteins58, missense mutation59, and phosphorylated proteins60. PSSM descriptor are capable of sufficiently/ effectively explore/demonstrate the hidden evolutionary patterns using the alignment of protein sequences61. The PSSM matrix generate L *20 Dimension feature vector by executing PSI-BLAST program62, in which L denotes the peptide’s length and 20 denotes the twenty kinds of AA residues. The retrieved values in PSSM matrix are either positive or negative. The positive values means high correlated and negative values means less correlated features. Since, in our datasets the peptides sequences are variable length, so we cannot directly use PSSM. Conventional, PSSM composition method is often used for this purpose, however one of the major concern of this is approach is the loss of local sequence information63. To tackle this challenge, we introduce histogram of oriented gradient-based(HOG)64 position-specific scoring matrix (HOG-PSSM)65 method, to capture the fixed length feature vector. In, pattern recognition and computer vision histogram of oriented gradient (HOG)64 algorithm has widely been used as feature extractor for human detection66. Motivated by this, we modified HOG encoding method to transform the PSSM matrix by extracting the biological features. The working principle of HOG-PSSM method is explained in the subsequent steps: First, it is important to compute the horizontal Hx(a, b) and vertical gradients Hy(a, b) of the PSSM matrix by the specified formulation:
Subsequently, the gradient’s direction and magnitude can be calculated by the below mathematical expression:
where denotes the gradient magnitude H(a, b) and Θ(a, b) gradient direction of the PSSM matrix. For the 3rd step, the image is segmented into 16 by 16 size connected areas known as cells. Each cell encompasses the feature set compressing gradient magnitude and direction within the sub-matrix.
here i, j represents the sub-matrix subscripts (0≤i≤2, 0 ≤j ≤2) and the subscripts inside the sub-matrix locations (0 ≤ v≤ 9, 0 ≤ v ≤ L/2 -1) are denoted by s, t. Each sub-matrix produces sixteen different histogram channels on the basis of gradient direction. As a result, for each peptide sample HOG-PSSM generates 16*16=256-D (dimensions) feature vector.
Protein language models
In the realm of natural language processing, the evolution of large language models (LLMs)67,68 has become a cornerstone, showcasing remarkable potentials across wide range of problems69. Recently, protein learning representation has become topic of debate to understand protein function and structure70. PLMs71 have been made an extraordinary advancement in the field of bioinformatics and computational biology for the tasks in predicting molecular property72, antihypertensive peptide73, antimicrobial peptides74, etc. PLM typically employ self-supervised learning strategy by using large-scale available protein sequences75. The main advantages of PLM-based feature representation of peptide sequence compared to traditional feature engineering are: embedding vectors are initialized randomly before training and the model could learn the effective feature representation of the peptide sequences automatically rather than consuming much more effort to design the hand-crafted features. Second, the representation vector of the peptide sequence is much denser than lots of statistical features and therefore it can represent more hidden semantics form the sequence. Several PLMs version have been released to explore the protein related texture data. However, we deployed ESM-2 and ProtT5 as explained below as a mainstream methods for representing crucial peptide features.
ESM embedding features
The ESM-2 is the recently developed based on pre-trained language model. ESM framework76, which underwent training on a vast collection of protein sequences trained over 15 billion parameters. This model, leverages the transformer language model architecture. Throughout the initial training phase of ESM, it constructs a contextual sequence feature matrix. This process establishes a dimensional space that captures a variety of dimensions including sequence similarity, site-specific functional attributes, and three-dimensional configurations related to biochemical traits.
ProtT5 embedding features
Recently, Elnaggar et al. proposed ProtTrans71 framework leverages an extensive collection of over 39.3 billion AAs, drawn from the UniRef and BFD data repositories. This model, through its sophisticated pretraining phase, deliver a wide range of biophysical characteristics from proteins without labels. It offers insights into aspects such as the cellular compartmentalization of proteins and their solubility in lipid membranes compared to aqueous environments. Echoing the capabilities of the ESM framework, ProtTrans is similarly equipped to delineate features at the individual residue level within protein chains. It assigns to each protein attribute a 1024-dimensional profile, enriching our understanding of the molecular vicinity of mutations in the sequence. Herein, we named this feature as ProtT5 for easy writing. To our best knowledge, we are the first to introduce ProtT5 and ESM-2 as PLM in peptide sequence encoding to predict bioactive peptide with strong ACP inhibitory activity.
Wavelet denoising
In bioinformatics and machine learning, preprocessing is considered a challenging step to remove the data redundancy and outliers. Over the past years, 2D wavelet denoising (WD) method has been extensively employed in proteomics research77,78. The WD algorithm is also called threshold wavelet, capable of removing irregular non-stationary data and eliminate the noise from 2D data79. The AA residue in peptide sequences are expressed as signal in time and frequency domain. The decomposition of AA signals in peptide sequence through wavelet analysis can efficiently improve the prediction performance of the model by extracting the characteristic signals of each peptide80. In WD process the influencing factors that can significantly reduce the noise effect are comprised of three phases: wavelet transformation or function, the execution of wavelet coefficients and reconstruction of 2D signal81. The entire operation of this algorithm is deduced in Algorithm 1. A 2D data with noise effect can be formulated as:

The pseudocode for 2D WD algorithm is derived from reference79.
Model development, evaluation and performance metrics
We used off-the-shelf algorithms support vector machine, and Naïve Base classifiers as well as proposed novel model CDF, for training and validating the model.
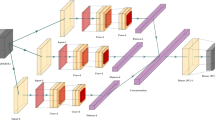
CDF is an ensemble-based framework, proposed by Zhou et al.82, which can serve as a substitute for deep neural networks (DNNs)65. In recent times, CDF model became a has become a dominant learning algorithm in wide range of domains likewise pattern recognition83,84, and bioinformatics85. CDF model structure is an ensemble of trees hierarchically sequenced in multiple layers86. The top-down architecture of CDF enables the classifier ideal for training even limited number of samples. Furthermore, Zhou and Feng pinpointed in their pioneering work that CDF is much easier in tuning the hyper-parameter compare to DNN82. Considering this, an improved version of CDF were developed containing an ensemble of RF87, XGBoost88, and Extremely Randomized Trees (ERT) classifiers89 to build the model. Each layer of this classifiers is composed of four learners of XGBoost, RF and ERT machine learning classifiers that take the feature-vector of the previous layer. The previous level’s class probability is then passed on to the next layer. In order to produce the augmented attributes, the related heterogeneous feature vectors are merged, averaged and the maximum probability values is generated as output.. We set up K=500 decision trees for RF, XGBoost, and ERT. The node split attributes were selected by randomly selecting d features, where d is the total number of features. This training process was terminated when there was no substantial performance improvement. Figure 6 shows the layer-by-layer architecture of the CDF classifier. Hyperparameters of the models were tuned using GridSearch CV of Python. We utilized four evaluation measures i.e., accuracy (Acc), sensitivity (Sn), specificity (Sp), Matthew’s correlation coefficient (MCC) for comprehensive examination of our proposed ACP predictor. The formulation of the evaluation metrics are shown below:

Architecture of the CDF model.
In the above-mentioned notation tp denotes the peptides with ACP activity, and tn denotes the peptides with non-ACP activity. Similarly, fp denotes the number of false peptides that have no ACP activity and fn means the number of false peptides having ACP activity. The aforementioned assessment metrics are threshold dependent. Furthermore, we used the receiver operating characteristic (ROC) curve, along with the area under the ROC curve(AUC) as threshold-independent indexes to evaluate the overall effectiveness of the proposed method90,91. The closer the prediction value is to 1, the better the predictive performance of the classification algorithm and vice versa. We adopted 10-fold CV method to construct an intelligent predictive model for targeting accurate ACPs.
Conclusion
ACPs are short rang therapeutic peptides that play significant role in designing effective anticancer drugs. The pressing demand for predicting novel ACPs via in silico methods remains urgent to understand their functions and potential role in cancer treatment. In this study, we introduced a novel ensemble-based model, PLMACPred for ACP prediction which leveraged the power of PLM, sequence embedding, and biologically relevant features from peptide sequences. The superior performance of PMLACPred on multiple challenging benchmark datasets, solidify its efficiency as valuable prediction tool for the discovery of new ACPs in particular and other therapeutic peptides in general. In future, we will make an effort to develop publicly accessible web server for the proposed method and further extend our research to identifying other activities such as antiviral, antimicrobial, antifungal and anticorona virus etc., in large-scale therapeutics peptides. We believe, PLMACPred will be used heavily as a useful tool for aiding discovery and design of novel ACP in a rapid, high-throughput and cost-effective fashion.
Data availability
Dataset used in this study is shared in Github: https://github.com/Muhammad-Arif-NUST/PLMACPred/.
References
Alsina, M., Arrazubi, V., Diez, M. & Tabernero, J. Current developments in gastric cancer: From molecular profiling to treatment strategy. Nat. Rev. Gastroenterol. Hepatol. 20, 155–170 (2023).
Azad, H. et al. G-acp: A machine learning approach to the prediction of therapeutic peptides for gastric cancer. J. Biomol. Struct. Dyn. https://doi.org/10.1080/07391102.2024.2323141 (2024).
Berger, L. et al. Major complications after intraoperative radiotherapy with low-energy x-rays in early breast cancer. Strahlentherapie und Onkologie 1–11 (2023).
Timmons, P. B. & Hewage, C. M. Ennavia is a novel method which employs neural networks for antiviral and anti- coronavirus activity prediction for therapeutic peptides. Brief. Bioinform. 22, bbab258 (2021).
Kabir, M. et al. Intelligent computational method for discrimination of anticancer peptides by incorporating sequential and evolutionary profiles information. Chemom. Intell. Lab. Syst. 182, 158–165 (2018).
Silva, O. N., Porto, W. F., Ribeiro, S. M., Batista, I. & Franco, O. L. Host-defense peptides and their potential use as biomarkers in human diseases. Drug Discov. Today 23, 1666–1671 (2018).
Huang, Y.-B., Wang, X.-F., Wang, H.-Y., Liu, Y. & Chen, Y. Studies on mechanism of action of anticancer peptides by modulation of hydrophobicity within a defined structural framework. Mol. Cancer Ther. 10, 416–426 (2011).
Chiangjong, W., Chutipongtanate, S. & Hongeng, S. Anticancer peptide: Physicochemical property, functional aspect and trend in clinical application. Int. J. Oncol. 57, 678–696 (2020).
Zhang, Q.-Y. et al. Antimicrobial peptides: Mechanism of action, activity and clinical potential. Mil. Med. Res. 8, 1–25 (2021).
Pan, F. et al. Anticancer effect of rationally designed α-helical amphiphilic peptides. Colloids Surf. B Biointerfaces 220, 112841 (2022).
Tornesello, A. L., Borrelli, A., Buonaguro, L., Buonaguro, F. M. & Tornesello, M. L. Antimicrobial peptides as anticancer agents: Functional properties and biological activities. Molecules 25, 2850 (2020).
Zafar, S. et al. Novel therapeutic interventions in cancer treatment using protein and peptide-based targeted smart systems. Semin. Cancer Biol. 69, 249–267 (2021).
Herrera-León, C. et al. The influence of short motifs on the anticancer activity of hb43 peptide. Pharmaceutics 14, 1089 (2022).
Nhàn, N. T. T., Yamada, T. & Yamada, K. H. Peptide-based agents for cancer treatment: Current applications and future directions. Int. J. Mol. Sci. 24, 12931 (2023).
Araste, F. et al. Peptide-based targeted therapeutics: Focus on cancer treatment. J. Controll. Release 292, 141–162 (2018).
Hilchie, A., Hoskin, D. & Power Coombs, M. Anticancer activities of natural and synthetic peptides. Antimicrob. Pept. Basics Clin. Appl. https://doi.org/10.1007/978-981-13-3588-4_9 (2019).
Ramazi, S., Mohammadi, N., Allahverdi, A., Khalili, E. & Abdolmaleki, P. A review on antimicrobial peptides databases and the computational tools. Database 2022, baac011 (2022).
Chen, W., Ding, H., Feng, P., Lin, H. & Chou, K.-C. iacp: A sequence-based tool for identifying anticancer peptides. Oncotarget 7, 16895 (2016).
Li, F.-M. & Wang, X.-Q. Identifying anticancer peptides by using improved hybrid compositions. Sci. Rep. 6, 33910 (2016).
Boopathi, V. et al. macppred: A support vector machine-based meta-predictor for identification of anticancer peptides. Int. J. Mol. Sci. 20, 1964 (2019).
Manavalan, B. et al. Mlacp: Machine-learning-based prediction of anticancer peptides. Oncotarget 8, 77121 (2017).
Schaduangrat, N., Nantasenamat, C., Prachayasittikul, V. & Shoombuatong, W. Acpred: A computational tool for the prediction and analysis of anticancer peptides. Molecules 24, 1973 (2019).
Akbar, S., Rahman, A. U., Hayat, M. & Sohail, M. cacp: Classifying anticancer peptides using discriminative intelligent model via chou’s 5-step rules and general pseudo components. Chemom. Intell. Lab. Syst. 196, 103912 (2020).
Agrawal, P., Bhagat, D., Mahalwal, M., Sharma, N. & Raghava, G. P. Anticp 2.0: An updated model for predicting anticancer peptides. Brief. Bioinform. 22, 1153 (2021).
Charoenkwan, P. et al. Improved prediction and characterization of anticancer activities of peptides using a novel flexible scoring card method. Sci. Rep. 11, 3017 (2021).
Wei, L., Zhou, C., Chen, H., Song, J. & Su, R. Acpred-fl: A sequence-based predictor using effective feature representation to improve the prediction of anti-cancer peptides. Bioinformatics 34, 4007–4016 (2018).
Wei, L., Zhou, C., Su, R. & Zou, Q. Pepred-suite: Improved and robust prediction of therapeutic peptides using adaptive feature representation learning. Bioinformatics 35, 4272–4280 (2019).
Rao, B., Zhou, C., Zhang, G., Su, R. & Wei, L. Acpred-fuse: Fusing multi-view information improves the prediction of anticancer peptides. Brief. Bioinform. 21, 1846–1855 (2020).
Liang, X. et al. Large-scale comparative review and assessment of computational methods for anti-cancer peptide identification. Brief. Bioinform. 22, bbaa12 (2021).
Arif, M. et al. Stackacpred: Prediction of anticancer peptides by integrating optimized multiple feature descriptors with stacked ensemble approach. Chemom. Intell. Lab. Syst. 220, 104458 (2022).
Akbar, S., Hayat, M., Iqbal, M. & Jan, M. A. iacp-gaensc: Evolutionary genetic algorithm based ensemble classification of anticancer peptides by utilizing hybrid feature space. Artif. Intell. Med. 79, 62–70 (2017).
Grisoni, F. et al. Designing anticancer peptides by constructive machine learning. ChemMedChem 13, 1300–1302 (2018).
Ahmed, S. et al. Acp-mhcnn: An accurate multi-headed deep-convolutional neural network to predict anticancer peptides. Sci. Rep. 11, 23676 (2021).
Cao, R., Wang, M., Bin, Y. & Zheng, C. Dlff-acp: Prediction of acps based on deep learning and multi-view features fusion. PeerJ 9, e11906 (2021).
Lane, N. & Kahanda, I. Deepacppred: A novel hybrid cnn-rnn architecture for predicting anti-cancer peptides. In Practical Applications of Computational Biology & Bioinformatics, 14th International Conference (PACBB 2020) Vol. 14 (eds Panuccio, G. et al.) 60–69 (Springer, 2021).
Zhou, W. et al. Trinet: A tri-fusion neural network for the prediction of anticancer and antimicrobial peptides. Patterns 4, 100702 (2023).
Liu, M. et al. Acppfel: Explainable deep ensemble learning for anticancer peptides prediction based on feature optimization. Front. Genet. 15, 1352504 (2024).
Lv, Z., Cui, F., Zou, Q., Zhang, L. & Xu, L. Anticancer peptides prediction with deep representation learning features. Brief. Bioinform. 22, bbab008 (2021).
Zhu, L., Ye, C., Hu, X., Yang, S. & Zhu, C. Acp-check: An anticancer peptide prediction model based on bidirectional long short-term memory and multi-features fusion strategy. Comput. Biol. Med. 148, 105868 (2022).
Yi, H.-C. et al. Acp-dl: A deep learning long short-term memory model to predict anticancer peptides using high-efficiency feature representation. Mol. Ther. Acids 17, 1–9 (2019).
Basith, S., Manavalan, B., Hwan Shin, T. & Lee, G. Machine intelligence in peptide therapeutics: A next-generation tool for rapid disease screening. Med. Res. Rev. 40, 1276–1314 (2020).
Song, X., Zhuang, Y., Lan, Y., Lin, Y. & Min, X. Comprehensive review and comparison of anticancer peptides identification models. Curr. Protein Pept. Sci. 22, 201–210 (2021).
Vens, C., Rosso, M.-N. & Danchin, E. G. Identifying discriminative classification-based motifs in biological sequences. Bioinformatics 27, 1231–1238 (2011).
Jiménez-Luna, J., Grisoni, F. & Schneider, G. Drug discovery with explainable artificial intelligence. Nat. Mach. Intell. 2, 573–584 (2020).
Ahmed, S., Arif, M., Kabir, M., Khan, K. & Khan, Y. D. Predaodp: Accurate identification of antioxidant proteins by fusing different descriptors based on evolutionary information with support vector machine. Chemom. Intell. Lab. Syst. 228, 104623 (2022).
Zulfiqar, H. et al. Deep-stp: A deep learning-based approach to predict snake toxin proteins by using word embeddings. Front. Med. 10, 1291352 (2023).
Zulfiqar, H. et al. Empirical comparison and recent advances of computational prediction of hormone binding proteins using machine learning methods. Comput. Struct. Biotechnol. J. https://doi.org/10.1016/j.csbj.2023.03.024 (2023).
Liu, X.-W. et al. ipadd: A computational tool for predicting potential antidiabetic drugs using machine learning algorithms. J. Chem. Inf. Model. 63, 4960–4969 (2023).
Agrawal, P. et al. Cppsite 2.0: A repository of experimentally validated cell-penetrating peptides. Nucleic Acids Res. 44, D1098–D1103 (2016).
Tyagi, A. et al. Cancerppd: A database of anticancer peptides and proteins. Nucleic Acids Res. 43, D837–D843 (2015).
Ge, F. et al. Vpatho: A deep learning-based two-stage approach for accurate prediction of gain-of-function and loss-of- function variants. Brief. Bioinform. 24, bbac535 (2023).
Ge, F. et al. Review of computational methods and database sources for predicting the effects of coding frameshift small insertion and deletion variations. ACS Omega 9, 2032–2047 (2024).
Musleh, S., Arif, M., Alajez, N. M. & Alam, T. Unified mrna subcellular localization predictor based on machine learning techniques. BMC Genom. 25, 151 (2024).
Arif, M., Hayat, M. & Jan, Z. imem-2lsaac: A two-level model for discrimination of membrane proteins and their types by extending the notion of saac into chou’s pseudo amino acid composition. J. Theor. Biol. 442, 11–21 (2018).
Arif, M. et al. imrsapred: Improved prediction of anti-mrsa peptides using physicochemical and pairwise contact-energy properties of amino acids. ACS Omega 9, 2874–2883 (2024).
Arif, M. et al. Targetcpp: accurate prediction of cell-penetrating peptides from optimized multi-scale features using gradient boost decision tree. J. Comput.-Aided Mol. Des. 34, 841–856 (2020).
Arif, M. et al. Pred-bvp-unb: Fast prediction of bacteriophage virion proteins using un-biased multi-perspective properties with recursive feature elimination. Genomics 112, 1565–1574 (2020).
Hu, J. et al. Improving dna-binding protein prediction using three-part sequence-order feature extraction and a deep neural network algorithm. J. Chem. Inf. Model. 63, 1044–1057 (2023).
Ge, F., Hu, J., Zhu, Y.-H., Arif, M. & Yu, D.-J. Targetmm: Accurate missense mutation prediction by utilizing local and global sequence information with classifier ensemble. Comb. Chem. High Throughput Screen. 25, 38–52 (2022).
Ahmed, S., Kabir, M., Arif, M., Ali, Z. & Swati, Z. N. K. Prediction of human phosphorylated proteins by extracting multi-perspective discriminative features from the evolutionary profile and physicochemical properties through lfda. Chemom. Intell. Lab. Syst. 203, 104066 (2020).
Zhou, S., Zhou, Y., Liu, T., Zheng, J. & Jia, C. Predllps_pssm: A novel predictor for liquid–liquid protein separation identification based on evolutionary information and a deep neural network. Brief. Bioinform. 24, bbad299 (2023).
Sf, A. Gapped blast and psi-blast: A new generation of protein database search programs. Nucleic Acids Res. 25, 3389–3402 (1997).
Fu, X. et al. Improved dna-binding protein identification by incorporating evolutionary information into the chou’s pseaac. IEEE Access 6, 66545–66556 (2018).
Wang, X., Han, T. X. & Yan, S. An hog-lbp human detector with partial occlusion handling. In 2009 IEEE 12th International Conference on Computer Vision (eds Wang, X. et al.) 32–39 (IEEE, 2009).
Arif, M. et al. Deepcppred: A deep learning framework for the discrimination of cell-penetrating peptides and their uptake efficiencies. IEEE/ACM Trans. Comput. Biol. Bioinform. 19, 2749–2759 (2021).
Pang, Y., Yuan, Y., Li, X. & Pan, J. Efficient hog human detection. Signal Process. 91, 773–781 (2011).
Achiam, J. et al. Gpt-4 technical report. Preprint at https://arXiv.org/quant-ph/2303.08774 (2023).
Zhang, M., Gong, C., Ge, F. & Yu, D.-J. Fcmstrans: Accurate prediction of disease-associated nssnps by utilizing multiscale convolution and deep feature combination within a transformer framework. J. Chem. Inf. Model. 64(4), 1394–406 (2024).
Chowdhery, A. et al. Palm: Scaling language modeling with pathways. J. Mach. Learn. Res. 24, 1–113 (2023).
Zhuo, L. et al. Protllm: An interleaved protein-language llm with protein-as-word pre-training. Preprint at https://arXiv.org/quant-ph/2403.07920 (2024).
Elnaggar, A. et al. Prottrans: Toward understanding the language of life through self-supervised learning. IEEE Trans. Pattern Anal. Mach. Intell. 44, 7112–7127 (2021).
Liu, Z. et al. Molca: Molecular graph-language modeling with cross-modal projector and uni-modal adapter. Preprint at https://arXiv.org/quant-ph/2310.12798 (2023).
Du, Z. et al. plm4ace: A protein language model based predictor for antihypertensive peptide screening. Food Chem. 431, 137162 (2024).
Dee, W. Lmpred: Predicting antimicrobial peptides using pre-trained language models and deep learning. Bioinform. Adv. 2, vbac021 (2022).
Rives, A. et al. Biological structure and function emerge from scaling unsupervised learning to 250 million protein sequences. Proc. Natl. Acad. Sci. 118, e2016239118 (2021).
Lin, Z. et al. Evolutionary-scale prediction of atomic-level protein structure with a language model. Science 379, 1123–1130 (2023).
Lio, P. Wavelets in bioinformatics and computational biology: State of art and perspectives. Bioinformatics 19, 2–9 (2003).
Yu, B. & Zhang, Y. A simple method for predicting transmembrane proteins based on wavelet transform. Int. J. Biol. Sci. 9, 22 (2013).
Wang, S. & Wang, X. Prediction of protein structural classes by different feature expressions based on 2-d wavelet denoising and fusion. BMC Bioinform. 20, 1–17 (2019).
Kandaswamy, A., Kumar, C. S., Ramanathan, R. P., Jayaraman, S. & Malmurugan, N. Neural classification of lung sounds using wavelet coefficients. Comput. Biol. Med. 34, 523–537 (2004).
Tian, B. et al. Predicting protein–protein interactions by fusing various chou’s pseudo components and using wavelet denoising approach. J. Theor. Biol. 462, 329–346 (2019).
Zhou, Z.-H. & Feng, J. Deep forest. Natl. Sci. Rev. 6, 74–86 (2019).
Cai, R. & Chen, C. Learning deep forest with multi-scale local binary pattern features for face anti-spoofing. Preprint at https://arXiv.org/quant-ph/1910.03850 (2019).
Wang, Y. et al. Deep forest for radar hrrp recognition. J. Eng. 2019, 8018–8021 (2019).
Chen, Z.-H. et al. An improved deep forest model for predicting self-interacting proteins from protein sequence using wavelet transformation. Front. Genet. 10, 430173 (2019).
Utkin, L. V., Kovalev, M. S. & Meldo, A. A. A deep forest classifier with weights of class probability distribution subsets. Knowl.-Based Syst. 173, 15–27 (2019).
Breiman, L. Random forests. Mach. Learn. 45, 5–32 (2001).
Chen, T. & Guestrin, C. Xgboost: A scalable tree boosting system. In: Proc. 22nd acm sigkdd international conference on knowledge discovery and data mining, 785–794 (2016).
Geurts, P., Ernst, D. & Wehenkel, L. Extremely randomized trees. Mach. Learn. 63, 3–42 (2006).
Bao, W. & Yang, B. Protein acetylation sites with complex-valued polynomial model. Front. Comput. Sci. 18, 183904 (2024).
Bao, W., Liu, Y. & Chen, B. Oral_voting_transfer: Classification of oral microorganisms’ function proteins with voting transfer model. Front. Microbiol. 14, 1277121 (2024).
Acknowledgements
This work was supported by the College of Science and Engineering, Hamad Bin Khalifa University (HBKU), Doha 34110, Qatar.
Author information
Authors and Affiliations
Contributions
M.A. and T.A conceived and designed the project, wrote initial draft. M.A, S.M conducted the experiment(s). All authors read and reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
41598_2024_67433_MOESM1_ESM.pdf
Supplementary Table S1: Exclusive motifs found in ACPs and non-ACP peptides in training dataset. Supplementary Table S2: Composition-based ACPs features.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Arif, M., Musleh, S., Fida, H. et al. PLMACPred prediction of anticancer peptides based on protein language model and wavelet denoising transformation. Sci Rep 14, 16992 (2024). https://doi.org/10.1038/s41598-024-67433-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-67433-8
- Springer Nature Limited