
Abstract
The prevalence and dynamics of circulating tumor DNA (ctDNA) in patients with breast cancer recurrence or de novo metastatic cancer were examined in a retrospective analysis of a prospective observational cohort. Twenty-three recurrent/metastatic breast cancer cases (8 locoregional, 15 distant metastasis) were enrolled, and sequential plasma samples were obtained. Anchor mutations were selected from the target sequencing of each patient’s primary and/or metastatic tumor. An in-house developed assay (UHS assay) was employed for a tumor-informed ctDNA assay during treatment and follow-up. A median of three (range 1–5) anchor mutations per case were applied for ctDNA detection. ctDNA was detected in 14 (63.6%, 14/22) cases at the time of enrollment and 18 (78.5%, 18/23) cases during follow-up. More anchor mutations and higher tumor burden were significantly related to higher ctDNA positive rates (p-value 0.036, 0.043, respectively). The mean enriched variant allele frequency (eVAF) at each time point was significantly higher for stable or progressive disease responses (ANOVA test p-value < 0.001). Eight patients showed an increase in their ctDNA eVAF prior to clinical progression with a mean lead time of 6.2 months (range 1.5–11 months). ctDNA dynamics measured using personalized assay reflected the clinical course of breast cancer recurrence.
Similar content being viewed by others
Introduction
Clinical evaluations and imaging studies are the standard tools for assessing the treatment responses in patients with metastatic breast cancer. While imaging studies enable measurements of tumor size and disease extent, they have the limitation of lacking sensitivity in detecting early changes in tumor burden and differentiating size change and actual progression. Circulating tumor DNA (ctDNA) has the potential to act as a surrogate for treatment responses in these patients1,2. ctDNA is detected in the blood; being a minimally invasive technique, feasible for repeated serial sampling. ctDNA assay results can also reflect real-time tumor burden changes due to their very short half-life of less than 2.5 h3. Previous studies have suggested that changes in ctDNA levels precede radiologic progression in various clinical settings across several solid tumors1,4,5,6. Therapeutically actionable mutations can be also detected in ctDNAs, as can the emergence of resistance mutations, both reflecting spatial and temporal tumor heterogeneity7,8,9.
Circulating free DNA (cfDNA) is found at low levels (approximately 1–10 ng/mL of plasma), which can increase to 5–10 times in patients with cancer10. cfDNA mainly consists of hematopoietic origins and extremely sensitive methods are required to detect the tumor-specific mutations in ctDNA10,11. Digital PCR or tagged amplicon deep sequencing are both highly sensitive methods used for ctDNA detection. Digital PCR is limited in its ability to detect multiple genes in a single test, however. Ultra-deep next-generation sequencing (NGS) can achieve sufficient high sensitivity along with multiplexing, but its high cost limits its use in the clinic.
Our in-house developed ultrahigh sensitive (UHS) assay is a mutant allele-specific enrichment methodology that combines a modified Amplification Refractory Mutation System (ARMS) with conventional iPLEX chemistry which can readily be read by both MassARRAY and real-time quantitative PCR12,13,14,15. In a previous study for non-small cell lung cancer, we demonstrated an average limit of detection of 0.05% for the UHS assay15. Multiplexing is feasible in the UHS assay and only 1 mL of plasma is needed, compared to 20 mL of whole blood needed in other NGS approaches15. The PCR-based methodology and the capacity of the MassARRAY system allow the assay to handle up to 380 samples per pool at once, making the UHS assay a more cost-effective option compared to other assays.
In this present study, we enrolled breast cancer patients with recurrences and/or metastases and performed serial ctDNA tracking on these cases using our bespoke UHS assay. The ctDNA detection rate of this UHS assay in the context of breast cancer recurrence, and the association between genomic tumor burden change and patient clinical course were evaluated.
Results
Patient characteristics
A total of 23 patients with recurrent or de novo metastatic breast cancer were enrolled. Sequential plasma samples were obtained every 3–6 months according to patients’ clinic visits and blood sampling schedules (Supplementary Fig. S1). The clinicopathologic features of these patients are presented in Table 1 and Supplementary Table 1. The mean age at enrollment was 46.8 years (range 34–71 years). The majority of the patients were initially stage 2 and had a hormone receptor (HR)-positive, HER2-negative tumor. Eleven patients recurred within 5 years and seven between 5 and 10 years after their initial diagnosis. Fifteen patients had distant metastasis and eight patients had only locoregional recurrence, including a case that progressed during neoadjuvant chemotherapy. Subtype changes at metastatic sites were noted in five patients among the 13 cases who had immunohistochemical results of biopsied metastatic sites. Three cases were changed from HR-positive HER2-negative to triple-negative breast cancer (TNBC), and two cases were changed from TNBC to HER2-positive and HR-positive HER2-negative, respectively.
Somatic genetic alteration profiling
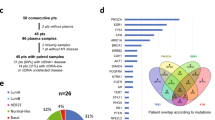
An in-house targeted sequencing panel of 382 genes was performed on the primary and/or metastatic cancer tissues from our present patient series (Fig. 1)16. Primary cancer tissues were obtained from surgical specimens, whereas metastatic cancer tissues were obtained from surgical or core needle biopsy specimens. Six cases underwent targeted sequencing of both their primary and metastatic tumors, 14 patients underwent sequencing of only primary tumors, and three patients of only metastatic tumors. Most metastatic tumors were sampled from regional nodes except for one case which was obtained from a skin metastasis.

(A) Targeted sequencing data from the 23 breast cancer tissue samples classified by subtype and annotated with variation type and mutation frequency. The asterisks are cases that utilized metastatic tumor tissue only for targeted sequencing. (B) Mutational profile of the six pairs of both primary and metastatic tumor tissues. The top panel shows the number of mutations per patient and the side panel represents the number of patients who harbored each mutation. Somatic mutations with a variant allele fraction (VAF) of 5% or higher are shown in the figure.
Somatic genetic alterations were identified in all primary tumors with a median of four (range 2–21) alterations detected per case (Fig. 1A). The most frequently altered gene was TP53 (identified in 43% of patients), followed by PIK3CA (39%), PTEN (17%), MAP3K1 (9%), and SF3B1 (9%), congruent to the mutation profiles in cBioPortal (Supplementary Fig. S2). In the six cases that had paired samples, tumor profiling was similar between primary and metastatic tumors (Fig. 1B).
Semi-quantitative analysis of the UHS assay
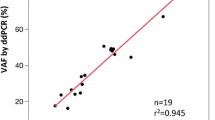
The feasibility of semi-quantitative analysis needed to be verified to apply the UHS assay in treatment response monitoring and MRD detection. A UHS assay was conducted using PIK3CA E545D plasmid that was serially diluted to 5%, 1%, 0.5%, 0.25%, 0.1%, 0.05%, and 0%, along with duplicate tests for each dilution. The UHS data was evaluated by visualizing scatterplots for the relationship between the enriched variant allele frequency (eVAF) obtained from the UHS assay and the initial variant allele frequency (VAF; Supplementary Fig. S3). An eVAF is the ratio of the mutant peak and wild-type peak produced by the MassARRAY system. The scatterplot indicates that the eVAF reached saturation in samples with a VAF > 1% (Supplementary Fig. S3A). In contrast, a linear correlation was identified in positive samples with a 0.05 – 1% VAF, indicating the reliability of data from samples with low concentrations (Supplementary Fig. S3B).
Detection of ctDNA using the UHS platform
Personalized UHS assays were used to analyze serial samples from each patient. A total of 143 blood samples were collected with a median of 6 (range 1–12) consecutive samples per patient. The time interval between each consecutive sample ranged from 1 to 17 months (Fig. 2A,B). Patient-specific anchor mutations were selected from the primary and/or metastatic tumor target sequencing results. A median of nine (range 2–14) anchor mutations per case were initially selected. After excluding nonspecific assays or assays without enrichment, a median of three (range 1–5) anchor mutations per case were applied for ctDNA detection (Supplementary Table S2).

(A) Flowchart of the 23 patients included in this study. Time was measured from the initial breast cancer diagnosis indicated by red squares (primary cancer tissue used for sequencing) or red empty squares (diagnosis of breast cancer). The metastatic cancer tissues used for sequencing are indicated with a blue square. The black circles indicate each point when plasma samples were obtained. Local recurrence is shaded in yellow (1st local recurrence in dark yellow, 2nd in bright yellow), regional recurrence in green and distant metastasis in pink. A case that showed progressive disease during neoadjuvant chemotherapy (NAC) is shaded in turquoise blue. The number of anchor mutations is written in parentheses next to the sample name on the y axis. In cases M032 and N2014126, primary tissues were obtained from palliative surgery specimens, performed after palliative systemic therapy. (B) Timeline plot of all 23 patients during study enrollment. Each time point at which a plasma sample was obtained is indicated with a circle or asterisk. ctDNA positive (one or more anchor mutations detected) is a black dot, ctDNA negative (no anchor mutations detected) is a blank circle, and molecular progression is shown as a blue asterisk. Clinical progression is indicated with a red line. The lead times between molecular progression and clinical progression are illustrated using a green shaded box.
One patient (M022) baseline plasma sample was not available for evaluation due to problems during PCR amplification. Fourteen (63.6%, 14/22) patients were ctDNA-positive at baseline, at the time of enrollment. 78.5% (18/23) of the total study cohort cases were ctDNA-positive at least once during serial follow-up. ctDNA was not detected in any isolated local recurrence cases, whereas all patients with distant metastasis were ctDNA-positive at least once during serial follow-up, showing that tumor burden was significantly related to ctDNA-positive rate (Fig. 3). The baseline ctDNA-positive rate was also lower in patients with only 1–2 anchor mutations compared to patients with 3–5 anchor mutations (40.0% vs. 83.3%, respectively, Chi-square test P = 0.074). Among the nine patients who were ctDNA-negative at baseline, seven had only one or two anchor mutations. Two patients (M003, M024) were tested for ctDNA using four anchor mutations but were both ctDNA-negative at baseline. However, both cases were detected during serial tracking as tumor burden changed.

Bar graph of the ctDNA-positive rate at baseline (upper row) and during serial follow-up (lower row) according to clinicopathologic and ctDNA assay-related factors. The recur site (1st column, ‘Recur Site’) had significant association with ctDNA-positive rate. The tumor subtype of the primary tumor (2nd column, ‘Initial Subtype’) and disease-free interval (3rd column, ‘Disease-Free Interval’), the interval between primary diagnosis and recurrence, were not related to ctDNA-positive rate. Assays that applied more anchor mutations (4th column, ‘Number of Anchor Mutations’) presented with higher ctDNA-positive rate. Using only primary tumor tissue vs both primary and metastatic tissue to select anchor mutations (5th column, ‘Primary vs Paired Tissue’) was also not related to ctDNA-positive rate. HR + HER2− hormone receptor-positive, HER2-negative, HER2+ HER2-positive, DFI disease-free interval, F/U follow-up, De novo de novo metastatic cancer.
The ctDNA-positive rate did not differ according to either the primary tumor subtype or disease-free interval (DFI), the time interval between primary diagnosis and cancer recurrence (Fig. 3). Among patients for whom only primary cancer tissue was available for sequencing (N = 9), the mean DFI was 58.3 months in ctDNA-positive patients at baseline and 38.6 months in ctDNA-negative patients, showing no significant difference (P = 0.458). The ctDNA-positive rate during serial follow-up also did not differ when anchor mutations were selected from paired target sequencing compared to those selected from only primary tumor sequencing (100% vs 75%, P = 0.515). This shows that a long DFI does not negatively affect ctDNA detection rate and that anchor mutations selected from primary tumor tissue could sufficiently detect subsequent recurrence and overcome temporal heterogeneity.
ctDNA dynamics according to the clinical course
The time courses of ctDNA detection in each case in the current study series are plotted in Supplementary Fig. S4 with illustrative examples provided in Fig. 4.

Examples of the longitudinal dynamics of ctDNA. Time is measured from study enrollment or start of treatment if the patient was enrolled during treatment. Inverted triangles indicate the radiologic response, a partial response as blue, stable disease as green and progressive disease as orange. Each treatment is shaded in different colors. Measurable lesions are plotted in dotted lines to the right y-axis. (A) Patient M019 was diagnosed with bone metastasis and regional lymph node metastasis after a disease-free interval of 61 months. The ctDNA eVAF decreased in this case after a partial response to a 1st line palliative therapy. An abrupt rise and additional ctDNA detection were noted at the 6th serial follow-up, showing a lead time of 8 months before a liver metastasis was radiologically detected. (B) Patient M015 had a locoregional recurrence along with bone metastasis at baseline. PIK3CA mutation eVAF increased as peritoneal seeding progressed. This metastasis was not detected by primary or metastatic tissue targeted sequencing. (C) Patient M005 initially had a bone metastasis but was ctDNA-negative at enrollment. However, as a liver metastasis progressed two PIK3CA mutations were detected by ctDNA. (D) Patient M039 was identified as having mediastinal lymph nodes and pleural metastasis at baseline. During a 1st line treatment, a liver metastasis was identified and progressed along with PIK3CA ctDNA eVAF changes.
The eVAF of each variant at each time point was classified according to clinical response during therapy. The mean eVAF percentage was significantly higher for stable disease (10.32) or progressive disease (23.02) responses compared to complete response (1.39) or partial response (3.17) (ANOVA test P < 0.001; Supplementary Fig. S5).
During serial ctDNA tracking, 11 patients presented with at least one molecular progression event. Among these cases, 8 patients had a molecular progression prior to clinical progression with a lead time of 1.5–11 months (mean 6.2 months; Figs. 2B, 4A). One patient (M027) showed molecular progression simultaneously with clinical disease progression. By contrast, two patients (M018, M024) persisted with a stable disease despite harboring new variants detected at a low level.
A PIK3CA hotspot panel was applied to HR-positive breast cancer patients with serial plasma samples (n = 16). Among these 16 cases, 8 displayed PIK3CA hotspot mutations in the plasma, among which 7 were found to have these mutations in the tumor. A PIK3CA p.H1047R variant was identified only in the plasma ctDNA in one patient (M015, Fig. 4B). This variant was not detected by NGS in the primary or metastatic tissue, but the UHS assay identified it in the metastatic tissue assay (eVAF 28.3%). Whereas, in case M005, PIK3CA mutations were found in the primary lesions, but only detected in the plasma when metastatic disease progressed to the liver (Fig. 4C). Another example of PIK3CA mutation detection is M039, showing that while SF3B1 p.S705C was steadily detected in the serial ctDNA assays, a ctDNA PIK3CA p.E452K variant was detected in accordance with changes in the metastatic site and tumor burden (Fig. 4D).
Patients who had no detectable ctDNA at baseline had a tendency for a longer progression-free survival outcome (log-rank test, P = 0.139; Supplementary Fig. S6). Patients with more than one ctDNA mutation detected at baseline had a tendency towards a poorer prognosis compared to patients with no mutations or only one detected (long-rank test, P = 0.113). However, among patients with a distant metastasis at enrollment, no survival difference was evident between ctDNA-positive and negative cases.
Discussion
In our present prospective, longitudinal monitoring study, we evaluated the association between ctDNA dynamics and the treatment response in breast cancer patients with a recurrence or metastasis. Anchor mutations were selected from tumor targeted sequencing and were analyzed using our in-house developed UHS assay to detect ctDNA in serially obtained plasma samples. We found that the ctDNA-positive rate was higher in the patients with a greater number of anchor mutations and a higher tumor burden. Our longitudinal monitoring of ctDNA by UHS assay in our present series demonstrated a correlation between ctDNA changes and the clinical course. In several cases, molecular progression was identified prior to progression on imaging with a lead time of up to 11 months.
The strong relationship between tumor burden and ctDNA detection presented within this study is indicated in previous studies, showing a higher ctDNA detection rate as breast cancer stage increases17,18. ctDNA was not detected at any timepoints in local recurrence cases in this study which is also consistent with previous reports19,20,21. Another strong predictor of ctDNA detection within the study was the number of anchor mutations. Increasing the number of tracking mutations is expected to increase the ctDNA detection power within the limited amount of plasma21. An exploratory analysis of the cTRAK-TN clinical trial also demonstrated improvements in clinical performance when applying multimutation sequencing compared to digital PCR with only 1–2 mutations22.
We demonstrated a correlation between ctDNA eVAF value dynamics and patients’ clinical course in this study and a significantly higher mean eVAF was identified at disease progression. The feasibility of using ctDNA dynamics to monitor treatment response was demonstrated in many previous studies2,23,24,25. Previous studies have also demonstrated a correlation between the presence of detectable ctDNA or early change in ctDNA to the progression-free survival in metastatic breast cancer cases1,5, 24, 26, 27. These studies applied results from panel-based ctDNA assays or detection of specific gene alterations (e.g., TP53, PIK3CA, ESR1) for survival analysis. Our study used a tumor-informed assay with a limited number of gene alterations per assay, limiting the association of ctDNA detection and patient outcome. The small number and heterogeneous patient cohort also limited statistical power for survival analysis.
The detection of minimal residual disease using ctDNA mutation tracking has been demonstrated in several studies with a lead time of up to 2 years over clinical relapse19,28. In our current study, molecular progression was identified with a lead time of up to 11 months prior to clinical progression during serial ctDNA monitoring. Identification of earlier molecular progression can guide an early intervention using targeted therapy, thereby promoting longer survival in advanced breast cancer cases. The clinical benefits of early therapeutic targeting by ctDNA analysis were shown in the PADA-1 trial, and additional, minimal residual disease-guided interventional studies in adjuvant settings are ongoing to confirm the clinical utility of ctDNA monitoring29,30,31,32,33. The UHS assay is an effective and affordable ctDNA analysis method with high sensitivity and is only needed of a small amount of plasma. In this study, targeted panel sequencing was used to select anchor mutations which resulted in a limited number of mutations to track for each patient. The UHS assay would need amendments, and whole exome or whole genome tumor sequencing needs to be considered to select a sufficient number of anchor mutations for higher sensitivity of ctDNA detection. Parsons et al. have demonstrated higher sensitivity when whole genome sequencing was applied to select patient-specific mutations for tracking compared to first-generation assays that use whole exome sequencing34.
There were some limitations of our present study of note. The first of these involved the heterogeneity of our cohort regarding recurrence sites and treatments. Also, the intervals between each blood sample were uneven, thus limiting any assessment of the correlation between ctDNA dynamics and the clinical course of the patient. In addition, because these plasma sampling intervals were irregular, lead time assessments could be less accurate compared to more frequent, regular sampling settings. A correlation between ctDNA dynamics and the clinical course was observed from our present investigation, but a clear standard by which to assess an increase or decrease in ctDNA is needed in future studies.
ctDNA detection is highly associated with the tumor burden and number of anchor mutations in breast cancer, and ctDNA dynamics are correlated with the clinical course of these patients. Our in-house UHS assay has proven to be a feasible and cost-effective multigene mutation profiling method for ctDNA detection in breast cancer. Early detection of recurrence and disease progression can inform future studies of personalized treatments and may enhance survival outcomes via the treatment of molecular disease.
Methods
Patients
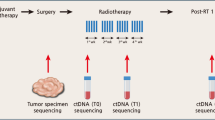
This is a retrospective ctDNA study from an observational prospective cohort comprised of patients who had been diagnosed with breast cancer recurrence or de novo metastatic breast cancer between February 2016 and January 2019 at Asan Medical Center (AMC). These patients were enrolled at the time of recurrence/metastasis and their baseline plasma was collected (Supplementary Fig. 2). They were followed in accordance with standard-of-care management protocols at AMC, and clinical data were collected as per standard practice. Serial blood samples were collected every 3–6 months according to patients’ clinic visits and clinically indicated blood draws. Study protocols were compliant to the Declaration of Helsinki and were approved by the Institutional Review Board of AMC (IRB no. 2019-1480). Informed consent was obtained from all the study participants before sampling.
Selection of anchor mutations
To select anchor mutations for tracking, we conducted targeted sequencing using the primary or metastatic tumor. Details of the targeted sequencing process are provided in the Supplementary Methods. Briefly, target capture using the Agilent SureSelectXT custom kit (OP_AMCv3, RNA bait; Agilent Technologies) targeting a total of 382 genes, including the entire exons of 199 genes, 184 hot spots, and the partial introns of 8 genes that are often rearranged in cancer16. This in-house targeted panel was designed to include genes related to cancer and therapeutic targets and undergo regular updates to align with the latest treatment trends.
Tracking of anchor mutations using cfDNA
Approximately 8–12 mL of venous blood was collected from each study subject at each time-point in three EDTA tubes. Each tube was immediately centrifuged at 1600g for 10 min with further centrifugation of the separated plasma at 3000g for 10 min at room temperature. Plasma aliquots were stored at − 80 °C until use.
One milliliter of plasma was used to extract cfDNAs using an STB cell-free DNA kit (Syntekabio, Seoul, Republic of Korea), following the manufacturer’s instructions. All extracted cfDNAs were quantified using a Bioanalyzer High Sensitivity DNA Analysis kit (Agilent, Santa Clara, USA), and kept at − 80 °C until use.
All cfDNAs extracted in sufficient quantity (from 2.7 to 24.2 ng) were used to construct DNA libraries and applied to the UHS experiment to preserve the limited quantities of cfDNA available. Approximately 10 ng of input was used for all DNA samples in the initial multiplex PCR. Serial sequential steps including multiplexing PCR followed the protocol as described in a previous study14.
The designed UHS primers for each anchor mutations were tested in both tumor and normal samples to confirm sufficient enrichment. Mutations with nonspecific assays or assays without enrichment at the UHS assay were excluded as an anchor mutation. Hotspot mutations of PIK3CA (p.H1047R, p.H1047L, p.E542K, p.E545K) gene were designed and tested in all HR positive cases.
Assessment of semi-quantitation using the UHS assay
Synthetic plasmids were used as positive and negative controls to evaluate the analytical performance of the UHS assay. The sequence of PIK3CA E545D was synthesized as a 150-bp single-stranded DNA fragment, including 74 bp upstream and 74 bp downstream of the E545D-altered position. The wild-type sequence was synthesized without a nucleotide substitution at the E545D position, maintaining its original sequence. The synthesized DNA fragments were then amplified using PCR for TA cloning. Double-stranded DNA was cloned into the TA vector using the pMD20-T vector from the Mighty TA-cloning kit (TAKARA). Plasmids from all picked clones were subjected to Sanger sequencing to verify their sequences.
A plasmid vector containing the partial sequence of PIK3CA E545D was diluted with a plasmid vector containing the wild-type sequence. A mutant-to-wild-type allele fractions of 5%, 1%, 0.5%, 0.25%, 0.1%, 0.05%, and 0% were made through dilution. A UHS assay was performed to compare the eVAF obtained through the UHS assay to the initial VAF, along with duplicate tests for each dilution.
Response evaluation by imaging analysis
CT, ultrasound, PET/CT and bone scintigraphy scanning, depending on the site of metastasis, were conducted as per standard practices. RECIST 1.1 was used to evaluate the tumor responses to treatment35.
Statistical analysis
Raw data produced by the MassARRAY system were analyzed by assessing the height value for extension analytes. To clearly distinguish mutant samples, an eVAF was devised, using the following equation:
A UHS assay on tumor tissue was initially performed to confirm each mutation assay. For each mutation, ctDNA positivity was defined based on the eVAF value of the wild type samples. If the wild type eVAF value was lower than 5%, an eVAF of 5% and over was considered positive. If the wild type eVAF value exceeded 5%, ctDNA positivity was defined according to the wild type eVAF value. At each time point, ctDNA positive was defined as the detection of one or more mutation. When no mutations were detected at a specific time point, this was determined to be a ctDNA-negative result.
A molecular progression was defined as a newly detected mutation with an eVAF of more than twofold higher than the baseline value or a mutation that eVAF increased more than twofold from the previous time point. A lead time to progression was calculated as the difference between the time point of a progressive disease and the first time point of molecular progression that was sustained as ctDNA-positive at the next visit. A molecular progression identified within one month of progressive disease was considered a concurrent event.
Statistical analyses were conducted using both Prism 9 and R software. Differences between groups were compared by Student’s t-test or ANOVA test. The Kaplan–Meier method with the log-rank test was employed for survival analysis. P-values < 0.05 were considered statistically significant.
Data availability
The datasets generated and analyzed during the current study can be made available as a limited set of de-identified data by the corresponding author upon reasonable request.
References
Velimirovic, M. et al. Rising circulating tumor DNA as a molecular biomarker of early disease progression in metastatic breast cancer. JCO Precis. Oncol. 4, 1246–1262 (2020).
Jacob, S. et al. The use of serial circulating tumor DNA to detect resistance alterations in progressive metastatic breast cancer. Clin. Cancer Res. 27, 1361–1370 (2021).
Diehl, F. et al. Circulating mutant DNA to assess tumor dynamics. Nat. Med. 14, 985–990 (2008).
Clatot, F. et al. Risk of early progression according to circulating ESR1 mutation, CA-15.3 and cfDNA increases under first-line anti-aromatase treatment in metastatic breast cancer. Breast Cancer Res. 22, 56 (2020).
Hrebien, S. et al. Early ctDNA dynamics as a surrogate for progression-free survival in advanced breast cancer in the BEECH trial. Ann. Oncol. 30, 945–952 (2019).
Martínez-Sáez, O. et al. Circulating tumor DNA dynamics in advanced breast cancer treated with CDK4/6 inhibition and endocrine therapy. NPJ Breast Cancer. 7, 8 (2021).
Kingston, B. et al. Genomic profile of advanced breast cancer in circulating tumour DNA. Nat. Commun. 12, 2423 (2021).
Turner, N. C. et al. Circulating tumour DNA analysis to direct therapy in advanced breast cancer (plasmaMATCH): A multicentre, multicohort, phase 2a, platform trial. Lancet Oncol. 21, 1296–1308 (2020).
Rolfo, C. et al. Liquid biopsy for advanced NSCLC: A consensus statement from the international association for the study of lung cancer. J. Thorac. Oncol. 16, 1647–1662 (2021).
Corcoran, R. B. & Chabner, B. A. Application of cell-free DNA analysis to cancer treatment. N. Engl. J. Med. 379, 1754–1765 (2018).
Panet, F. et al. Use of ctDNA in early breast cancer: Analytical validity and clinical potential. NPJ Breast Cancer. 10, 50 (2024).
Gabriel, S., Ziaugra, L., Tabbaa, D. SNP genotyping using the Sequenom Massarray iPLEX platform. In Current Protocols in Human Genetics/Editorial Board, Jonathan L Haines [et al], Chapter 2, Unit 2.12 (2009).
Little, S. Amplification-refractory mutation system (ARMS) analysis of point mutations. Curr. Protoc. Hum. Genet. Chapter 9, Unit 9.8 (2001).
Shin, S. J. et al. Feasibility of multiplexed gene mutation detection in plasma samples of colorectal cancer patients by mass spectrometric genotyping. PLoS One. 12, e0176340 (2017).
Lee, J. Y. et al. Revolutionizing non-small cell lung cancer diagnosis: Ultra-high-sensitive ctDNA analysis for detecting hotspot mutations with long-term stored plasma. Cancer Res. Treat. 56, 484–501 (2024).
Kim, J. E. et al. Mutation burden and I index for detection of microsatellite instability in colorectal cancer by targeted next-generation sequencing. J. Mol. Diagn. 21, 241–250 (2019).
Cohen, J. D. et al. Detection and localization of surgically resectable cancers with a multi-analyte blood test. Science. 359, 926–930 (2018).
Zhang, X. et al. Parallel analyses of somatic mutations in plasma circulating tumor DNA (ctDNA) and matched tumor tissues in early-stage breast cancer. Clin. Cancer Res. 25, 6546–6553 (2019).
Coombes, R. C. et al. Personalized detection of circulating tumor DNA antedates breast cancer metastatic recurrence. Clin. Cancer Res. 25, 4255–4263 (2019).
Lipsyc-Sharf, M. et al. Circulating tumor DNA and late recurrence in high-risk hormone receptor-positive, human epidermal growth factor receptor 2-negative breast cancer. J. Clin. Oncol. 40, 2408–2419 (2022).
Parsons, H. A. et al. Sensitive detection of minimal residual disease in patients treated for early-stage breast cancer. Clin. Cancer Res. 26, 2556–2564 (2020).
Coakley, M. et al. Comparison of circulating tumor DNA assays for molecular residual disease detection in early-stage triple-negative breast cancer. Clin. Cancer Res. 30, 895–903 (2024).
Gouda, M. A. et al. Longitudinal monitoring of circulating tumor DNA to predict treatment outcomes in advanced cancers. JCO Precis. Oncol. 6, e2100512 (2022).
Gerratana, L. et al. Longitudinal dynamics of circulating tumor cells and circulating tumor DNA for treatment monitoring in metastatic breast cancer. JCO Precis. Oncol. 5, 943–952 (2021).
Dawson, S. J. et al. Analysis of circulating tumor DNA to monitor metastatic breast cancer. N. Engl. J. Med. 368, 1199–1209 (2013).
Cullinane, C. et al. Association of circulating tumor DNA with disease-free survival in breast cancer: A Systematic review and meta-analysis. JAMA Netw. Open. 3, e2026921 (2020).
Chiu, J. et al. Potential value of ctDNA monitoring in metastatic HR+/HER2− breast cancer: Longitudinal ctDNA analysis in the phase Ib MONALEESASIA trial. BMC Med. 21, 306 (2023).
Garcia-Murillas, I. et al. Mutation tracking in circulating tumor DNA predicts relapse in early breast cancer. Sci. Transl. Med. 7, 302ra133 (2015).
Kadcyla And Neratinib for Interception of HER2+ Breast Cancer With Molecular Residual Disease. https://ClinicalTrials.gov/show/NCT05388149.
A PoC Study to Evaluate Treatments' Efficacy by Monitoring MRD Using ctDNA in HR-positive/HER2-negative EBC Population. https://ClinicalTrials.gov/show/NCT05708235.
CDK 4/6 Inhibitor, Ribociclib, With Adjuvant Endocrine Therapy for ER-positive Breast Cancer. https://ClinicalTrials.gov/show/NCT03285412.
DNA-Guided Second Line Adjuvant Therapy For High Residual Risk, Stage II–III, Hormone Receptor Positive, HER2 Negative Breast Cancer. https://ClinicalTrials.gov/show/NCT04567420.
Bidard, F.-C. et al. Switch to fulvestrant and palbociclib versus no switch in advanced breast cancer with rising ESR1 mutation during aromatase inhibitor and palbociclib therapy (PADA-1): A randomised, open-label, multicentre, phase 3 trial. Lancet Oncol. 23, 1367–1377 (2022).
Parsons, H. A. et al. Circulating tumor DNA association with residual cancer burden after neoadjuvant chemotherapy in triple-negative breast cancer in TBCRC 030. Ann. Oncol. 34, 899–906 (2023).
Schwartz, L. H. et al. RECIST 1.1-Update and clarification: From the RECIST committee. Eur. J. Cancer. 62, 132–137 (2016).
Acknowledgements
This study was supported by a grant (2020IP0059) from the Asan Institute for Life Sciences, Asan Medical Center, Seoul, Korea.
Author information
Authors and Affiliations
Contributions
J.K. and S.M.C. contributed to the concept and design of the study. W.K.C., S.B.L., I.Y.C., H.J.K., B.S.K., J.W.L., B.H.S., S.H.A., J.H.J., J.E.K., J.H.A., K.H.K., S.B.K., H.J.L. and G.G. contributed to data collection. J.Y.L. and H.P. performed the genomic sequencing and ctDNA assay. T.K.Y., J.Y.L., H.P., J.K. and S.M.C. analyzed and interpreted the patient data. T.K.Y. and J.Y.L. were major contributors in writing the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing non-financial Interests but the following competing financial Interests: JK and SMC are co-founders and in the advisory board of Genopeaks Inc. but declares no non-financial competing interests. Author TKY holds shares and is in the advisory board of Genopeaks Inc but declares no non-financial competing interest. Author KHJ and SBK hold shares in Genopeaks Inc but declares no non-financial competing interest.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Yoo, TK.R., Lee, JY., Park, H. et al. Longitudinal dynamics of circulating tumor DNA for treatment monitoring in patients with breast cancer recurrence. Sci Rep 14, 20252 (2024). https://doi.org/10.1038/s41598-024-70887-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-70887-5
- Springer Nature Limited