
Abstract
The flagellar protein export apparatus switches substrate specificity from hook-type to filament-type upon hook assembly completion, thereby initiating filament assembly at the hook tip. The C-terminal cytoplasmic domain of FlhA (FlhAC) serves as a docking platform for flagellar chaperones in complex with their cognate filament-type substrates. Interactions of the flexible linker of FlhA (FlhAL) with its nearest FlhAC subunit in the FlhAC ring is required for the substrate specificity switching. To address how FlhAL brings the order to flagellar assembly, we analyzed the flhA(E351A/W354A/D356A) ΔflgM mutant and found that this triple mutation in FlhAL increased the secretion level of hook protein by 5-fold, thereby increasing hook length. The crystal structure of FlhAC(E351A/D356A) showed that FlhAL bound to the chaperone-binding site of its neighboring subunit. We propose that the interaction of FlhAL with the chaperon-binding site of FlhAC suppresses filament-type protein export and facilitates hook-type protein export during hook assembly.
Similar content being viewed by others

Introduction
The flagellum of Salmonella enterica (hereafter referred to as Salmonella) is a supramolecular motility machine consisting of the basal body, which acts as a bi-directional rotary motor, the hook, which functions as a universal joint, and the filament, which works as a helical propeller1. For construction of the flagella on the cell surface, the flagellar type III secretion system (fT3SS) transports flagellar building blocks from the cytoplasm to the distal end of the growing flagellar structure2. The fT3SS is divided into three structural parts: a transmembrane export gate complex made of FlhA, FlhB, FliP, FliQ, and FliR, a substrate-chaperone-docking platform composed of the cytoplasmic domains of FlhA and FlhB (FlhAC and FlhBC), and a cytoplasmic ATPase ring complex consisting of FliH, FliI, and FliJ3. The FlhAC–FlhBC-docking platform provides binding sites for the cytoplasmic ATPase complex, flagellar export chaperones (FlgN, FliS, FliT), and export substrates to mediate hierarchical protein targeting and secretion4.
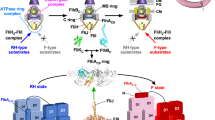
Flagellar assembly begins with the basal body, followed by the hook (FlgE) with the help of the hook cap (FlgD). After completion of hook–basal body (HBB) assembly, the FlgD cap is replaced by FlgK, and then FlgK and FlgL form the hook–filament junction structure at the hook tip, followed by the assembly of the filament cap (FliD). Finally, newly transported flagellin molecules (FliC) assemble into the filament with the help of the filament cap (Fig. 1)5. Flagellar building blocks are classified into two export classes: one is the rod-type (FliE, FlgB, FlgC, FlgF, FlgG, FlgJ) and hook-type class (FlgD, FlgE, and FliK) needed for the assembly of the rod and hook, and the other is the filament-type class (FlgK, FlgL, FlgM, FliC, and FliD) responsible for filament assembly at the hook tip6,7. The FlhAC–FlhBC-docking platform serves as an export switch to induce substrate specificity switching from rod-/hook-type proteins to filament-type ones when the hook reaches its mature length of about 55 nm in Salmonella, thereby terminating hook assembly and initiating filament formation (Fig. 1)8,9,10,11.

The Salmonella flagellum is composed of the basal body, the hook, the hook–filament junction, the filament and the filament cap. Upon completion of basal body assembly, newly exported FlgE molecules polymerize into the hook structure with the help of the hook cap made of FlgD. When the hook reaches its mature length of about 55 nm, the hook cap is replaced by FlgK. FlgK and FlgL self-assemble at the hook tip in this order to form the junction structure. Then, FliD forms the filament cap at the tip of the junction and promotes the assembly of FliC into the filament. A type III protein export apparatus (fT3SS) is located at the flagellar base and transports flagellar building blocks from the cytoplasm to the distal end of the growing flagellar structure. The fT3SS sometimes secretes the FliK ruler to measure the hook length during hook assembly. When the hook reaches its mature length of about 55 nm, the fT3SS switches its substrate specificity, thereby terminating the export of hook-type proteins (FlgD, FlgE, and FliK) and initiating the export of filament-type proteins (FlgK, FlgL, FliD, and FliC). FlgN, FliT, and FliS act as flagellar type III export chaperones specific for FlgK and FlgL, FliD and FliC, respectively. OM outer membrane, PG peptidoglycan layer, CM cytoplasmic membrane.
The fT3SS uses a secreted molecular ruler protein (FliK) to measure the hook length during hook assembly4. FliK is a hook-type protein secreted via the fT3SS during HBB assembly12. FliK not only measures the hook length13,14,15 but also switches substrate specificity of the FlhAC–FlhBC-docking platform (Fig. 1)11,16,17. This has been recently verified by in vitro reconstitution experiments using inverted membrane vesicles18,19. The N-terminal domain of FliK (FliKN) acts as a secreted molecular ruler to measure the hook length13,14,15. When the hook length reaches about 55 nm, a flexible linker region of FliK connecting FliKN and the C-terminal domain (FliKC) promotes a conformational rearrangement of FliKC, allowing FliKC to interact with FlhBC to terminate the export of the rod-type and hook-type proteins20,21.
FlhAC (residues 328–692) consists of four domains, D1, D2, D3, and D4, and a flexible linker (FlhAL) (residues 328–361) connecting FlhAC with the N-terminal transmembrane domain of FlhA (Fig. 2a)22. FlhAC forms a homo-nonamer ring in the fT3SS23 and provides binding sites for flagellar export chaperons (FlgN, FliS, and FliT) in complex with their cognate filament-type proteins (Fig. 2a)24,25,26,27. The flagellar chaperones promote the docking of their cognate filament-type substrates to the FlhAC ring structure to facilitate subsequent unfolding and translocation of the substrates28,29. High-speed atomic force microscopy combined with mutational analysis has shown that FlhAL is required for highly cooperative FlhAC ring formation on mica surface10. Glu-351, Trp-354, and Asp-356 of FlhAL bind to the D1 and D3 domains of its neighboring FlhAC subunit to stabilize FlhAC ring structure (Fig. 2a)10, and the W354A, E351A/D356A, and E351A/W354A/D356A mutations in FlhAL not only inhibit FlhAC ring formation but also reduce the binding affinity of FlhAC for flagellar chaperones in complex with their cognate filament-type substrates, thereby inhibiting the initiation of filament assembly10. Therefore, the FliKC–FlhBC interaction is postulated to modify the binding mode of FlhAL to its nearest subunit in the FlhAC ring structure upon completion of the hook structure, thereby allowing the flagellar chaperones to bind to FlhAC to initiate the export of filament-type proteins10,11,30. However, it remains unknown how FlhAL regulates the interactions of FlhAC with the chaperones during HBB assembly.

a Structural model of the FlhAC ring. Only three FlhAC subunits in the FlhAC nonameric ring model are shown. FlhAC (PDB ID: 3A5I) consists of four domains, D1, D2, D3, and D4 and a flexible linker (FlhAL). Glu-351, Trp-354, and Asp-356 of FlhAL binds to the D1 and D3 domains of its neighboring subunit. A well-conserved hydrophobic dimple including Asp-456, Phe-459, and Thr-490 is responsible for the interaction of FlhAC with flagellar export chaperones in complex with filament-type substrates. Phe-459 and Lys-548 are exposed to solvent on the molecular surface when FlhAC adopts the open conformation. FliJ binds not only to FlhAL but also to a large cleft between the D4 domains. b Immunoblotting, using polyclonal anti-FlgD (1st row), anti-FlgE (2nd row), anti-FliK (3rd row), anti-FlgK (4th row), anti-FliC (5th row), anti-FliD (6th row), or anti-FliI (7th row) antibody of whole-cell proteins and culture supernatant fractions prepared from the Salmonella NH001 strain transformed with pTrc99AFF4 (∆flhA), pMM130 (WT), or pYI003 (EWD) and the NH001gM strain transformed with pTrc99FF4A (∆flhA ∆flgM), pMM130 (∆flgM), or pYI003 (EWD ∆flgM). The positions of molecular mass markers are indicated on the left. The regions of interest were cropped from original immunoblots shown in Supplementary Fig. 7. c Relative secretion levels of flagellar proteins. These data are the average of four independent experiments. The average density of each flagellar protein seen in the culture supernatant derived from wild-type cells was set to 1.0, and then relative band density was calculated. Vertical bars indicate standard deviations. Dots indicate individual data points. The source data are shown in Supplementary Data File. Comparisons between datasets were performed using a two-tailed Student’s t-test. A P value of <0.05 was considered to be statistically significant difference. *P < 0.05; *P < 0.01; **P < 0.001; ND no statistical difference.
In the present study, to clarify the role of FlhAL in the export switching mechanism of fT3SS, we analyzed the interaction between FlhAL and FlhAC and provide evidence suggesting that the interaction of FlhAL with the chaperone-binding site of FlhAC brings the order to flagellar protein export in parallel with the assembly order of the flagellar structure.
Results
Isolation of pseudorevertants from the flhA(E351A/W354A/D356A) mutant
Glu-351, Trp-354, and Asp-356 of FlhAL bind to the D1 and D3 domains of its neighboring FlhAC subunit to stabilize FlhAC ring structure (Fig. 2a)10. The flhA(E351A/D356A) (hereafter referred to as flhAED) and flhA(W354A) (hereafter referred to as flhAW) mutants produce the HBBs without the filament attached10. Hook lengths of the flhAED and flhAW mutants are 54.0 ± 22.3 nm [mean ± standard deviation (SD)] and 52.9 ± 19.9 nm, respectively, where their SD values are larger than the wild-type one (51.0 ± 6.9 nm), indicating their hook length is not controlled properly10. Pull-down assays by GST affinity chromatography have revealed that the flhAED and flhAW mutations reduce the binding affinity of FlhAC for flagellar chaperones in complex with their cognate filament-type substrates10. These previous results suggest that the observed interaction between FlhAL and the D1 and D3 domains of its neighboring FlhAC subunit is responsible for making the chaperone-binding site of FlhAC open to allow the chaperones to bind to FlhAC to facilitate the export of filament-type proteins10. However, the flhA(E351A/W354A/D356A) (hereafter referred to as flhAEWD) mutant does not produce the HBBs10, and this raises a question as to why the flhAEWD mutation inhibits HBB assembly.
To address this question, we first carried out quantitative immunoblotting to measure the amount of flagellar building blocks secreted by the fT3SS. The flhAEWD mutation significantly reduced the secretion levels of both hook-type (FlgD, FlgE, FliK) and filament-type substrates (FlgK, FliC, FliD) (Fig. 2b, c), indicating that the flhAEWD mutation significantly reduces the protein transport activity of the fT3SS.
To clarify why and how the flhAEWD mutation inhibits flagellar protein export, we isolated 14 pseudorevertants from the flhAEWD mutant. Motility of the pseudorevertants was somewhat better than that of the flhAEWD mutant but was much poorer than that of wild-type cells (Supplementary Fig. 1a). Export substrates such as FlgD, FlgE, FlgK, and FliD were detected in the culture supernatants of these pseudorevertants (Supplementary Fig. 1b). Consistently, these pseudorevertants produced a couple of flagella on the cell surface (Supplementary Fig. 1c). DNA sequencing revealed that all suppressor mutations are located in the flgMN operon. One was the M1I mutation at the start codon of the flgM gene (isolated twice), presumably inhibiting FlgM translation. Two suppressor mutations produced a stop codon at position of Gln-52 or Ser-85 of FlgM, resulting in truncation of the C-terminal region of FlgM. Nine suppressor mutations were large deletions in flgM. We also found that there was a large deletion in the flgM and flgN genes, thereby disrupting both FlgM and FlgN. A loss-of-function of FlgM results in a considerable increment in the transcription levels of flagellar genes31. Consistently, the cellular levels of flagellar building blocks and the FliI ATPase were higher in the pseudorevertants than those in its parental strain (Fig. 2b).
The interaction of FliJ with FlhAL is required for activation of the fT3SS, and FliH and FliI are required for efficient interaction between FliJ and FlhAL32. It has been reported that overexpression of export substrates and FliJ by FlgM deletion overcomes the loss of both FliH and FliI to a considerable degree33. Because the flhAEWD mutation reduces the binding affinity of FlhAC for FliJ10, this suggests that these flgM mutations increase the cytoplasmic levels of FliH, FliI, FliJ, and export substrates to allow the flhAEWD mutant to export flagellar building blocks for producing a small number of flagella on the cell surface. Therefore, we propose that Glu-351, Trp-354, and Asp-356 of FlhAL also play an important role in the activation mechanism of the fT3SS.
Effect of deletion of FlhAL on the interaction between FlhAC and FliJ
The crystal structure of a FliJ homolog, CdsO, in complex with CdsVC, which is a FlhAC homolog, has shown that CdsO binds to a large cleft between domains D4 of neighboring CdsVC subunits in the CdsVC ring structure but not to the linker region of CdsVC34 (Fig. 2a). To confirm the importance of FlhAL in the interaction between FlhAC and FliJ, we analyzed the binding of FlhAC to immobilized GST-FliJ by Bio-layer interferometry (BLI) measurements35. The FliJ–FlhAC interaction showed a complex binding profile (Fig. 3, 1st row) and did not fit the global one-state association-then-dissociation model. Assuming that FlhAC binds to GST-FliJ to form a GST-FliJ/FlhAC complex, followed by a conformational change of this complex, the BLI data fitted well with a two-state reaction model and provided a KD value of 1.36 ± 0.03 μM (mean ± SD, n = 3). Unlike wild-type FlhAC, the association and dissociation processes of FlhAC with the flhAEWD mutation (FlhAC-EWD) or FlhAC lacking FlhAL (FlhAC-ΔL) were observed only at protein concentrations above 10 μM (Fig. 3, 2nd and 3rd rows). Their association and dissociation processes were also different from those of wild-type FlhAC. The association profile of these mutant proteins was composed of two distinct (fast-on and slow-on) processes, and the dissociation profile was also composed of two distinct (fast-off and slow-off) processes. It has been shown that wild-type FlhAC forms dimer in a protein concentration-dependent manner and that FlhAL is required for efficient dimerization of FlhAC27. So, their BLI data were fitted well with curves predicted by the Hill equation, with KD values of 60.7 ± 1.2 μM (n = 3) and 49.0 ± 1.0 μM (n = 3) for the FliJ–FlhAC-EWD and FliJ–FlhAC-ΔL interactions, respectively. Thus, both flhAEWD mutation and deletion of FlhAL reduced the binding affinity of FlhAC for FliJ. Therefore, we conclude that FlhAL is required for the stable interaction between FliJ and FlhAC.

BLI profiles were obtained from the FlhAC–FliJ interaction (1st row), the FlhAC-EWD–FliJ interaction (2nd row), and the FlhAC-ΔL–FliJ interaction (3rd row). GST-FliJ was immobilized to an anti-GST sensor tip. The sensor tip was then dipped into FlhAC, FlhAC-EWD, or FlhAC-ΔL of various concentrations to measure association before being dipped into the kinetic buffer to measure dissociation. Three independent measurements were carried out. All experiments were performed at 25 °C.
Effect of the flhA EWD mutation on flagellar protein export by fT3SS in the absence of FlgM
To quantify the amount of flagellar building blocks secreted by the flhAEWD ΔflgM strain, we introduced the ΔflgM::km allele to the Salmonella NH001 (ΔflhA) strain to produce the ΔflgM and flhAEWD ΔflgM cells. The ΔflgM::km allele restored motility of the flhAEWD mutant in a way similar to other flgM suppressor mutations (Supplementary Fig. 2a, b). The amount of FlgE secreted by the flhAEWD ΔflgM strain was about fivefold higher than that by the ΔflgM strain (Fig. 2b, c), suggesting that this triple mutation significantly increases the binding affinity of the fT3SS for FlgE. However, the flhAEWD mutation did not affect the levels of FlgD and FliK secretion (Fig. 2b, c). These observations suggest that FlhAL may regulate substrate recognition of the fT3SS for hierarchical protein targeting and secretion among the hook-type substrates. The amount of FlgK, FliC, and FliD secreted by the flhAEWD ΔflgM strain was significantly lower than that by the ΔflgM strain (Fig. 2b, c), indicating that the flhAEWD mutation also affects export switching of the fT3SS from hook-type substrates to filament-type ones.
We found that the flhAEWD ΔflgM strain secreted a much larger amount of FlgE into the culture media than the ΔflgM strain, raising the possibility that the length of the hook produced by this mutant may be longer than the wild-type length. To clarify this, we isolated flagella from the ΔflgM and flhAEWD ΔflgM cells and measured their hook length. The hook length of the ΔflgM strain was 52.0 ± 5.1 nm (mean ± SD, n = 157) (Fig. 4, left panels), which is nearly the same as that of the wild-type strain (51.0 ± 6.9 nm)10. This indicates that the loss-of-function mutation of FlgM does not affect the hook length control. In contrast, the average hook length of the flhAEWD ΔflgM strain was 68.8 ± 30.9 nm (n = 122) (Fig. 4, right panels), indicating that the hook length control becomes worse in the presence of the flhAEWD mutation. These suggest that this mutation affects not only the initiation of filament-type protein export but also the termination of hook-type protein export. Because high-speed atomic force microscopy has shown that the flhAEWD mutation also inhibits highly cooperative FlhAC ring formation10, we propose that FlhAL regulates the conformational rearrangement of FlhAC in the ring, which is required for efficient termination of hook assembly and efficient initiation of filament formation at the hook tip.

Electron micrographs of HBBs and histograms of hook length distribution of NH001gM carrying pMM130 (∆flgM) or pYI003 (EWD ∆flgM).
Effect of FlhA linker mutations on the hydrodynamic properties of FlhAC in solution
A well-conserved hydrophobic dimple of FlhAC containing Asp-456, Phe-459, and Thr-490 residues is located at the interface between domains D1 and D2 and is involved in the interactions with the FlgN, FliS, and FliT chaperones in complex with their cognate filament-type substrates (Fig. 2a)25,26,27. The flhAW, flhAED, and flhAEWD mutations reduce the binding affinity of FlhAC for these chaperone/substrate complexes10. Interestingly, the flhA(D456V), flhA(F459A), and flhA(T490M) mutations increase the secretion levels of FlgE and FliK by the ΔfliH-fliI flhB(P28T) mutant36. We found that the flhAEWD mutation increases the secretion level of FlgE by about fivefold, raising the possibility that FlhAL carrying either of flhA linker mutations binds to the hydrophobic dimple of FlhAC not only to facilitate the export of FlgE but also to block the FlhAC–chaperone interaction. If this is the case, FlhAC with these mutations would show distinct hydrodynamic properties compared with wild-type FlhAC. To clarify this possibility, we performed size exclusion chromatography (SEC) with a Superdex 75 column HR 10/30 column. Wild-type His-FlhAC appeared as a single peak at an elution volume of 10.2 ml, which corresponds to the deduced molecular mass of His-FlhAC (about 43 kDa) (Fig. 5a). His-FlhAC with the flhAW (FlhAC-W), flhAED (FlhAC-ED) or flhAEWD mutation (FlhAC-EWD) and FlhAC-ΔL lacking FlhAL appeared as a single peak at an elution volume of 10.3, 10.5, 10.4, and 11.0 ml, respectively (Fig. 5a), indicating that these mutant variants exist as a monomer in solution. FlhAC-ED exhibited a delayed elution behavior compared with the wild type. Furthermore, FlhAC-ED showed a slightly faster mobility in both sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and native PAGE gels (Fig. 5b, c). Far-UV CD measurements revealed that the flhAED mutation did not affect the secondary structures of FlhAC (Supplementary Fig. 3). These suggest that FlhAC-ED adopts a more compact conformation than wild-type FlhAC. The elution peak position of FlhAC-EWD was between those of the wild type and FlhAC-ED (Fig. 5a). Because FlhAC-EWD showed two different bands on SDS-PAGE gels, with a slower mobility band corresponding to wild-type FlhAC and a faster one corresponding to FlhAC-ED (Fig. 5b), we suggest that FlhAC-EWD exists in an equilibrium between the wild-type conformation and the compact conformation. Therefore, we suggest that the flhAED mutation is required to make FlhAC more compact.

a Hydrodynamic properties of FlhAC and its FlhA linker mutant variants. Purified protein samples (10 μM) were run on a Superdex 75HR 10/30 column equilibrated with 50 mM Tri-HCl, pH 8.0, 150 mM NaCl. The elution peaks of His-FlhAC (WT, black), His-FlhAC-W (red), His-FlhAC-ED (blue), His-FlhAC-EWD (orange), and His-FlhAC-ΔL (green) are 10.2, 10.3, 10.5, 10.4, and 11.0 ml, respectively. Arrow indicates the elution peaks of γ-globulin (158 kDa), bovine serum albumin (66.4 kDa), and ovalbumin (43 kDa), which are 8.7, 9.7, and 10.7 ml, respectively. b CBB-stained SDS-PAGE gel of purified wild-type FlhAC and its mutant variants. The regions of interest were cropped from an original CBB-stained gel shown in Supplementary Fig. 8a. c Blue Native PAGE gel of purified wild-type FlhAC and its mutant variants. The regions of interest were cropped from an original Blue Native PAGE gel shown in Supplementary Fig. 8b.
Effect of FlhA linker mutations on methoxypolyethylene glycol 5000 maleimide (mPEG-maleimide) modifications of Cys-459 and Cys-548
FlhAC structures adopt open, semi-closed, and closed confromations22,23,24,27,30,37. A large open cleft between domains D2 and D4 is seen in the open form, but not in the closed form. As a result, Phe-459 and Lys-548, which are both located in the cleft between domains D2 and D4, are fully exposed to solvent on the molecular surface of the open conformation of FlhAC but are in close proximity to each other in the closed conformation22,30,37. To test whether mutations in FlhAL bias FlhAC towards the closed structure, we performed Cys modification experiments with mPEG-maleimide. FlhAC with the F459C/K548C substitutions modified by mPEG-maleimide showed much slower mobility shift (Supplementary Fig. 4, left panel), in agreement with a previous report30. The flhAW, flhAED, and flhAEWD mutations did not inhibit Cys modifications with mPEG-maleimide (Supplementary Fig. 4, right panel), indicating that FlhAC with these mutations does not adopt the closed conformation.
Crystal structure of FlhAC-ED
To investigate whether FlhAL binds to the hydrophobic dimple of FlhAC to make FlhAC-ED more compact, we explored crystallization conditions of FlhAC-ED for a molecular packing distinct from the open (PDB code: 3A5I)22 and semi-closed (PDB code: 6AI0)30 forms of wild-type FlhAC. We found a new orthorhombic crystal that diffracted up to 2.8 Å resolution, with unit cell dimensions a = 71.7 Å, b = 96.2 Å, c = 114.1 Å (Table 1) and the asymmetric unit containing two FlhAC molecules (A and B). Mol-A adopts an open conformation similar to the 3A5I structure (Supplementary Fig. 5a, c) whereas Mol-B shows a semi-closed conformation similar to the 6AI0 structure (Supplementary Fig. 5b, d). The residues from Val-349 to Val-357 in FlhAL of Mol-A form an α-helix, which interacts with the hydrophobic dimple of a neighboring Mol-A molecule related by a crystallographic symmetry (Fig. 6a). Trp-354 fits into the hydrophobic dimple, and Ala-351 hydrophobically contacts with Pro-442 on the periphery of the dimple (Fig. 6b and Supplementary Fig. 6a). These interactions resemble the interaction between the N-terminal α-helix of FliS and the hydrophobic dimple of FlhAC (PDB ID: 6CH3)27 (Fig. 6c). Ile-7 and Tyr-10 of the N-terminal α-helix of FliS is in the corresponding position of Ala-351 and Trp-354 of FlhAL, respectively. Tyr-10 fits into the hydrophobic dimple of FlhAC, and Ile-7 interacts with Pro-442 of FlhAC (Fig. 6d). These observations suggest that FlhAL and flagellar chaperones bind competitively to a common binding site on FlhAC and that the dissociation of FlhAL from this binding site is required for the binding of the flagellar chaperones to FlhAC. When Ala-351 and Ala-356 of FlhAC-ED in the crystal structure were replaced back to the original Glu-351 and Asp-356 residues, respectively, the side chain arm of Glu-351 can form a hydrophobic contact with Pro-442 (Supplementary Fig. 6b), suggesting that FlhAL can bind to the hydrophobic dimple of FlhAC even in the wild type without the flhAED mutation. Because the introduced Ala residues would increase the helical propensity of residues 349–357 of FlhAL as seen in the crystal, we suggest that the flhAED mutation allowed residues 349–357 of FlhAL to efficiently form an α-helix to stabilize the binding of FlhAL to the hydrophobic dimple of FlhAC.

a FlhAL of Mol-A (magenta) interacts with neighboring Mol-A (cyan) related by a crystallographic symmetry. b Close-up view of the interaction between FlhAL and the hydrophobic dimple shown by a red box in a. Residues that form the hydrophobic dimple are indicated by balls. The side chains of Ala-351, Trp-354, and Ala-356 in FlhAL are shown in stick models. c Interaction between FlhAC (green) and FliS (orange) fused with the C-terminal region of FliC (yellow) (PDB code: 6CH3). d Close-up view of the interaction between the extreme N-terminal region of FliS (FliSEN) and the hydrophobic dimple shown by a red box in c. The residues that form the hydrophobic dimple are indicated by ball. The side chains of Ile-7 and Tyr-10 of FliSEN are shown in stick models.
Effect of FlhA linker mutations on the interaction of FlhAC with the FlgN chaperone
We found that FlhAL with the flhAED mutation binds to the chaperone-binding site in its neighboring subunit in the crystal. If this interaction reflects the functional state of FlhAC, the flhAED mutation would affect the docking process of FlgN to FlhAC. To clarify this hypothesis, we performed BLI measurements. When GST-FlgN was tethered to a sensor chip and then allowed FlhAC of various concentrations to bind to immobilized GST-FlgN, the interaction between FlgN and FlhAC showed a typical BLI profile (Fig. 7). The association and dissociation rate constants were measured to be about 8.23 ± 0.20 × 103 M−1 S−1 and 7.81 ± 0.03 × 10−4 S−1, respectively, giving a KD value of 95.0 ± 2.0 nM (mean ± SD, n = 3). This KD value is in agreement with previous data obtained by surface plasmon resonance24. Unlike wild-type FlhAC, FlhAC-ED and FlhAC-EWD did not bind to immobilized GST-FlgN at protein concentrations less than 10 μM, indicating that FlhAL with either of these two mutations inhibits the binding of FlhAC to FlgN. The association and dissociation processes of FlhAC-ED and FlhAC-EWD were observed with an increase in the protein concentration (Fig. 7). However, these mutations caused fast-on and fast-off binding profiles (Fig. 7). Assuming that GST-FlgN binds to FlhAC-ED or FlhAC-EWD by inducing the dissociation of FlhAL with either of these flhA mutations from the chaperone-binding site so that GST-FlgN forms a complex with FlhAC-ED or FlhAC-EWD on the sensor chip, their BLI data fitted well with a two-state reaction model, giving KD values of 37.8 ± 1.9 μM (n = 3) and 31.2 ± 1.1 μM (n = 3) for the FlgN–FlhAC-ED and FlgN–FlhAC-EWD interactions, respectively.

BLI profiles were obtained from the FlhAC–FlgN interaction (upper, left panel), the FlhAC-ED–FlgN interaction (upper, right panel), the FlhAC-EWD–FlgN interaction (lower, left panel) and the FlhAC-ΔL–FlgN interaction (lower, right panel). GST-FlgN was immobilized to an anti-GST sensor tip. The sensor tip was then dipped into FlhAC, FlhAC-ED, FlhAC-EWD, or FlhAC-ΔL of various concentrations to measure association before being dipped into the kinetic buffer to measure dissociation. Three independent measurements were carried out. All experiments were performed at 25 °C.
We next investigated whether deletion of FlhAL affect the binding process of FlgN to FlhAC. The association and dissociation processes of FlhAC-ΔL were clearly observed at protein concentrations above 5 μM, and the BLI signals for the FlgN–FlhAC-ΔL interaction were much stronger at the same protein concentrations compared to the FlgN–FlhAC-ED and FlgN–FlhAC-EWD interactions (Fig. 7). Furthermore, the association and dissociation profiles of FlhAC-ΔL were different from those of FlhAC-ED and FlhAC-EWD. Its BLI data did not fit the global one-state association-then-dissociation model, but fitted with a heterogeneous reaction model, showing a KD value of 3.1 ± 0.1 μM (n = 3). Thus, the binding affinity of FlhAC-ΔL for FlgN was higher than those of FlhAC-ED and FlhAC-EWD. This suggests that FlhAL with either flhAED or flhAEWD mutation inhibits the binding of FlgN to FlhAC. Because the binding affinity of FlhAC-ΔL for FlgN was much lower than that of wild-type FlhAC, we suggest that FlhAL is required to keep FlhAC in the open form so that FlgN can efficiently and stably bind to the well-conserved hydrophobic dimple of FlhAC.
Discussion
The FlhAC ring serves as the docking platform for flagellar export chaperones in complex with their cognate substrates and facilitates the export of filament-type proteins to form the filament at the hook tip after completion of hook assembly24,25,26,27. The FlhAC ring also ensures the strict order of flagellar protein export, thereby allowing the huge and complex flagellar structure to be built efficiently on the cell surface10,11,30,36. An interaction of FlhAL with its neighboring FlhAC subunit in the nonamer ring is required for the initiation of filament-type protein export upon completion of hook assembly10. However, it remained unclear how the FlhAC ring mediates such hierarchical protein export during flagellar assembly.
In this study, we first performed genetic analyses of the flhAEWD mutant and found that this mutation reduces the protein transport activity of the fT3SS significantly (Fig. 2b, c). We also found that both the flhAEWD mutation and deletion of FlhAL reduce the binding affinity of FlhAC for FliJ (Fig. 3). Because the interaction between FliJ and FlhAL is required for activation of the fT3SS32, we propose that Glu-351, Trp-354, and Asp-356 of FlhAL is required for stable interaction of FlhAL with FliJ to fully activate the fT3SS to facilitate flagellar protein export.
It has been reported that either flhA(D456V), flhA(F459A), or flhA(T490M) mutation in the flagellar chaperone-binding site in FlhAC increases the levels of FlgE and FliK secretion by the ΔfliH-fliI flhB(P28T) mutant36, suggesting that this chaperone-binding site is also involved in the export of hook-type substrates. Here, we found that the flhAEWD mutation significantly increased the secretion level of FlgE by a ΔflgM mutant (Fig. 2b, c), thereby producing longer hooks (Fig. 4). This indicates that the flhAEWD mutation affects the termination of hook-type protein export, suggesting that an interaction between FlhAL and the chaperone-binding site of FlhAC coordinates the export of hook-type proteins with hook assembly in a highly organized and well-controlled manner. Furthermore, we also found that this triple mutation also reduced the secretion levels of filament-type substrates significantly (Fig. 2b, c), thereby reducing the number of flagellar filaments per cell (Supplementary Figs. 1 and 2). Taken all together, we propose that FlhAL serves as a structural switch for substrate specificity switching of the fT3SS from hook type to filament type and that Glu-351, Trp-354, and Asp-356 of FlhAL are directly involved in this export switching mechanism.
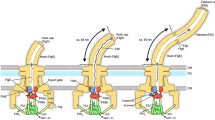
It has been reported that the flhAW, flhAED, and flhAEWD mutations inhibit interactions between FlhAC and flagellar chaperones in complex with their cognate filament-type substrates10, suggesting that FlhAL regulates the binding affinity of FlhAC for flagellar chaperones. The crystal structure of FlhAC-ED we solved in this study showed that FlhAL of a Mol-A molecule bound to the hydrophobic dimple of the flagellar chaperone-binding site of its nearest Mol-A in the crystal (Fig. 6). Although the relative orientations of these Mol-A molecules in the crystal differs from those in the FlhAC nonameric ring, FlhAL should be able to bind to the hydrophobic dimple of FlhAC in the nonamer ring structure as well because of a highly flexible nature of FlhAL (Fig. 8). The C-terminal region of FlhAL is flexible enough to allow such subunit orientations without changing the essential interaction between FlhAL and the chaperone-binding site of FlhAC (Fig. 8), as it has been shown to have various conformations in the known FlhAc structures28. BLI measurements indicated that FlhAL with either flhAED or flhAEWD mutation inhibits the docking process of FlgN to FlhAC (Fig. 7). Because we also found that FlhAL is required for stable interaction between FlgN and FlhAC (Fig. 7), we propose that the interaction between FlhAL and the hydrophobic dimple of its neighboring FlhAC subunit suppresses the docking of flagellar chaperones to the FlhAC ring platform during HBB assembly and that the hook assembly completion induces the detachment of FlhAL from the dimple through an interaction between FliKC and FlhBC and its attachment to the D1 and D3 domains to induce structural remodeling of the entire FlhAC ring, thereby terminating hook assembly and initiating filament formation (Fig. 8). Because FlhAC-ED monomer adopts a more compact conformation compared with the wild-type FlhAC monomer as judged by SEC (Fig. 5a), FlhAL may bind to FlhAC in a cis manner as well. Therefore, it is also possible that FlhAL may block the docking of the flagellar chaperones to FlhAC by covering the binding site of the same FlhAC molecule.

Trp-354 of FlhAL binds to a well-conserved hydrophobic dimple containing Asp-456, Phe-459, and Thr-490 of its neighboring FlhAC subunit in the FlhAC ring not only to inhibit the interaction of FlhAC with flagellar chaperones in complex with their cognate filament-type substrates but also to facilitate the export of the hook protein during hook assembly. When the hook reaches its mature length of about 55 nm, an interaction between FliKC and FlhBC triggers a conformational rearrangement of the FlhAC ring so that FlhAL dissociates from the hydrophobic dimple and binds to the D1 and D3 domains of the neighboring FlhAC subunit, allowing the chaperones to bind to FlhAC to facilitate the export of their cognate substrates for filament assembly.
Methods
Bacterial strains, plasmids, transductional crosses, and DNA manipulations
Bacterial strains and plasmids used in this study are listed in Table 2. P22-mediated transductional crosses were performed with P22HTint. DNA manipulations were performed using standard protocols38. Site-directed mutagenesis were carried out using the QuikChange site-directed mutagenesis method as described in the manufacturer’s instructions (Stratagene). DNA sequencing reactions were carried out using BigDye v3.1 (Applied Biosystems) and then the reaction mixtures were analyzed by a 3130 Genetic Analyzer (Applied Biosystems).
Motility assays
We transformed Salmonella enterica strains NH001 and NH001gM with a pTrc99A-based plasmid encoding wild-type FlhA or its mutant variant. Fresh transformants were inoculated into soft agar plates [1% (w/v) triptone, 0.5% (w/v) NaCl, 0.35% Bacto agar] containing 100 μg ml−1 ampicillin and incubated at 30 °C. At least five independent measurements were performed.
Secretion assays
S. enterica cells were grown in T-broth [1% (w/v) tryptone, 0.5% (w/v) NaCl] containing ampicillin at 30 °C with shaking until the cell density had reached an OD600 of ca. 1.4–1.6. Cultures were centrifuged to obtain cell pellets and culture supernatants. The cell pellets were resuspended in a sample buffer solution [62.5 mM Tris-HCl, pH 6.8, 2% SDS, 10% glycerol, 0.001% bromophenol blue] containing 1 μl of 2-mercaptoethanol. Proteins in the culture supernatants were precipitated by 10% trichloroacetic acid and suspended in a Tris/SDS loading buffer (one volume of 1 M Tris, nine volumes of 1× sample buffer solution)39 containing 1 μl of 2-mercaptoethanol. Both whole cellular proteins and culture supernatants were normalized to a cell density of each culture to give a constant cell number. After boiling proteins in both whole cellular and culture supernatant fractions at 95 °C for 3 min, these protein samples were separated by SDS-PAGE (normally 12.5% acrylamide) and transferred to nitrocellulose membranes (Cytiva) using a transblotting apparatus (Hoefer). Then, immunoblotting with polyclonal anti-FlgD, anti-FlgE, anti-FlgK, anti-FliC, anti-FliD, anti-FliI, or anti-FliK antibody was carried out using iBand Flex Western Device (Thermo Fisher Scientific). Detection was performed with Amersham ECL Prime western blotting detection reagent (Cytiva). Chemiluminescence signals were captured by a Luminoimage analyzer LAS-3000 (GE Healthcare). The band intensity of each blot was analyzed using an image analysis software, CS Analyzer 4 (ATTO, Tokyo, Japan). More than three independent experiments were performed.
Electron microscopy observation of negatively stained Salmonella cells
S. enterica cells were exponentially grown in 5 ml L-broth [1% (w/v) tryptone, 0.5% (w/v) yeast extract, 0.5% (w/v) NaCl] containing ampicillin at 30 °C. Five microliters of the cell culture was applied to carbon-coated copper grids and then negatively stained with 0.5% (w/v) phosphotungstic acid, pH 6.5. Micrographs were recorded at a magnification of ×1200 with a JEM-1010 transmission electron microscope (JEOL) operating at 100 kV.
Observation of flagellar filaments with a fluorescent dye
S. enterica cells were grown in T-broth containing ampicillin. The cells were attached to a coverslip (Matsunami glass, Japan), and unattached cells were washed away with motility buffer (10 mM potassium phosphate pH 7.0, 0.1 mM EDTA, 10 mM l-sodium lactate). Then, the flagellar filaments were labeled using anti-FliC antibody and anti-rabbit IgG conjugated with Alexa Fluor 594 (Invitrogen) as described previously40. After washing twice with the motility buffer, epi-fluorescence of Alexa Fluor 594 was observed by an inverted fluorescence microscope (IX-83, Olympus) with a ×150 oil immersion objective lens (UApo150XOTIRFM, NA 1.45, Olympus) and an Electron-Multiplying Charge-Coupled Device camera (iXonEM + 897-BI, Andor Technology)41. Fluorescence images were analyzed using ImageJ software version 1.52 (National Institutes of Health).
Bio-layer interferometry
His-FlhAC and its mutant variants were purified by Ni affinity chromatography, followed by SEC as described previously28. GST-FliJ and GST-FlgN were purified by GST affinity chromatography as described previously25,28. Purified protein samples were dialyzed overnight against a kinetic buffer [PBS (8.8 g of NaCl, 0.2 g of KCl, 3.63 g of Na2HPO4·12H2O, 0.24 g of KH2PO4, pH 7.4 per liter), 0.1% bovine serum albumin, 0.002% Tween-20] at 4 °C with three changes of PBS.
BLI measurements were carried out using a BLItz (FortéBio). GST-FliJ or GST-FlgN was immobilized to an anti-GST sensor tip (FortéBio). The sensor tip was then dipped into His-FlhAC or its mutant variants to measure association before being dipped into the kinetic buffer to measure dissociation. Data were reference subtracted and fit to various model using BLItz Pro software (FortéBio) and BIAevaluation software (GE Healthcare).
Hook length measurements
The HBBs were purified from NH004gM carrying pMM130 or pYI003 as described previously36. Salmonella cells were grown in L-broth containing ampicillin at 30 °C with shaking until the cell density had reached an OD600 of ca. 1.0. The cultures were centrifuged (10,000g, 10 min, 4 °C), and the cell pellets were suspended in 20 ml of ice-cold 0.1 M Tris-HCl pH 8.0, 0.5 M sucrose, followed by addition of EDTA and lysozyme at the final concentrations of 10 mM and 0.1 mg ml−1, respectively. The cell suspensions were stirred for 30 min at 4 °C, and then were solubilized on ice for 1 h by adding Triton X-100 and MgSO4 at final concentrations of 1% (w/v) and 10 mM, respectively. The cell lysates were adjusted to pH 10.5 with 5 M NaOH and then centrifuged (10,000g, 20 min, 4 °C) to remove cell debris. After ultracentrifugation (45,000g, 60 min, 4 °C), pellets were resuspended in 10 mM Tris-HCl, pH 8.0, 5 mM EDTA, 1% Triton X-100 and the solution was loaded a 20–50% (w/w) sucrose density gradient in 10 mM Tris-HCl, pH 8.0, 5 mM EDTA, 1% Triton X-100. After ultracentrifugation (49,100g, 13 h, 4 °C), intact flagella were collected and ultracentrifuged (60,000g, 60 min, 4 °C). Pellets were suspended in 50 mM glycine, pH 2.5, 0.1% Triton X-100 to depolymerize the flagellar filaments. After ultracentrifugation (60,000g, 60 min, 4 °C), pellets were resuspended in 50 μl of 10 mM Tris-HCl, pH 8.0, 5 mM EDTA, 0.1% Triton X-100. The HBBs were negatively stained with 2% (w/v) uranyl acetate. Electron micrographs were recorded with a JEM-1011 transmission electron microscope (JEOL, Tokyo, Japan) operated at 100 kV and equipped with a F415 CCD camera (TVIPS, Gauting, Germany). Hook length was measured by ImageJ version 1.52 (National Institutes of Health).
Size exclusion chromatography
SEC was performed with a Superdex 75HR 10/30 column (GE Healthcare). Purified His-FlhAC and its mutant variants (10 μM) were run on the SEC column equilibrated with 50 mM Tri-HCl, pH 8.0, 150 mM NaCl at a flow rate of 0.5 ml min−1. γ-Globulin (158 kDa), bovine serum albumin (66.4 kDa) and ovalbumin (43 kDa) were used as size markers. All fractions were run on SDS-PAGE and then analyzed by Coomassie Brilliant blue (CBB) staining.
Native PAGE
Purified His-FlhAC and its mutant variants (14.4 μM) were run on Native PAGE Novex Bis-Tris gels as described in the manufacturer’s instructions (Invitrogen).
Far-UV CD spectroscopy
Far-UV CD spectroscopy of His-FlhAC or its mutant variants was carried out at room temperature using a Jasco-720 spectropolarimeter (JASCO International Co., Tokyo, Japan) as described previously42. The CD spectra of His-FlhAC and its mutant forms were measured in 20 mM Tris-HCl, pH 8.0 using a cylindrical fused quartz cell with a path length of 0.1 cm in a wavelength range of 200–260 nm. Spectra were obtained by averaging five successive accumulations with a wavelength step of 0.5 nm at a rate of 20 nm min−1, response time of 8 s, and bandwidth of 2.0 nm.
Cystein modification by mPEG-maleimide
His-FlhAC(F459C), His-FlhAC(K548C), His-FlhAC(F459C/K548C), His-FlhAC-W(F459C/K548C), His-FlhAC-ED(F459C/K548C), and His-FlhAC-EWD(F459C/K548C) were dialyzed overnight against PBS (8 g of NaCl, 0.2 g of KCl, 3.63 g of Na2HPO4 12H2O, 0.24 g of KH2PO4, pH 7.4 per liter) at 4 °C. Twenty-five microliters of mPEG-maleimide reaction buffer (PBS containing 4 mM mPEG-maleimide) was added to 25 μl of 10 μM protein solutions. After incubation at 37 °C for 30 min, 5 μl of 2-mercaptoethanol was added to quench the reaction, and then 5 μl of 10% SDS was added. After centrifugation (20,000g, 20 min, 4 °C) to remove any aggregates, 60 μl of each soluble solution was mixed with 60 μl of 2× SDS loading buffer. After boiling at 95 °C for 3 min, each protein solution was run on SDS-PAGE and then analyzed by CBB staining.
X-ray crystallographic study of FlhAC(E351A/D356A)
Initial crystallization screening was performed at 20 °C by the sitting-drop vapor-diffusion method using Wizard Classic I and II, Wizard Cryo I and II (Rigaku Reagents, Inc.), Crystal Screen, and Crystal Screen 2 (Hampton Research). Crystals suitable for X-ray analysis were obtained from drops prepared by mixing 0.5 μl protein solution with 0.5 μl reservoir solution containing 0.1 M Tris-HCl, pH 8.5, 20% (v/v) PEG 8000, and 200 mM MgCl2. X-ray diffraction data were collected at synchrotron beamline BL41XU in SPring-8 (Harima, Japan) with the approval of the Japan Synchrotron Radiation Research Institute (JASRI) (Proposal No. 2016B2544 and 2018A2568). The FlhAC(E351A/D356A) crystal was soaked in a solution containing 90% (v/v) of the reservoir solution and 10% (v/v) glycerol for a few seconds and was directly transferred into liquid nitrogen for freezing. The X-ray diffraction data were collected at the wavelength of 1.000 Å under nitrogen gas flow at 100 K. The diffraction data were processed with MOSFLM43 and were scaled with Aimless44. The initial phase was determined by molecular replacement using the software package Phenix45 with the wild-type FlhAC structure in the orthorhombic crystal form (PDB code: 6AI0) as a search model. The atomic model was constructed with Coot46 and refined with Phenix45. During the refinement process, iterative manual modification was performed. The Ramachandran statistics indicated that 96.0%, 3.9%, and 0.1% residues were in the most favorable, allowed, and outlier regions, respectively. The diffraction data statistics and refinement statistics are summarized in Table 1.
Statistics and reproducibility
Statistical tests, sample size, and number of biological replicates are reported in the figure legends. Statistical analyses were done using KaleidaGraph software (HULINKS). Comparisons between datasets were performed using a two-tailed Student’s t-test. A P value of <0.05 was considered to be statistically significant difference. *P < 0.05; **P < 0.01; ***P < 0.001.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
The X-ray crystal structure and structure factors of FlhAC(E351A/D356A) have been deposited in Protein Data Bank under the accession code 7CTN. All data generated during this study are included in this published article, Supplementary Information and Supplementary Data file. Strains, plasmids, polyclonal antibodies, and all other data are available from the corresponding author on reasonable request.
References
Nakamura, S. & Minamino, T. Flagella-driven motility of bacteria. Biomolecules 9, 279 (2019).
Minamino, T. Protein export through the bacterial flagellar type III export pathway. Biochim. Biophys. Acta 1843, 1642–1648 (2014).
Minamino, T., Kawamoto, A., Kinoshita, M. & Namba, K. Molecular organization and assembly of the export apparatus of flagellar type III secretion systems. Curr. Top. Microbiol. Immunol. 427, 91–107 (2020).
Minamino, T. Hierarchical protein export mechanism of the bacterial flagellar type III protein export apparatus. FEMS Microbiol. lett. 365, fny117 (2018).
Kubori, T., Shimamoto, N., Yamaguchi, S., Namba, K. & Aizwa, S. Morphological pathway of flagellar assembly in Salmonella typhimurium. J. Mol. Biol. 226, 433–446 (1992).
Minamino, T. & Macnab, R. M. Components of the Salmonella flagellar export apparatus and classification of export substrates. J. Bacteriol. 181, 1388–1394 (1999).
Minamino, T., Doi, H. & Kutsukake, K. Substrate specificity switching of the flagellum-specific export apparatus during flagellar morphogenesis in Salmonella typhimurium. Biosci. Biotechnol. Biochem. 63, 1301–1303 (1999).
Minamino, T. & Macnab, R. M. Domain structure of Salmonella FlhB, a flagellar export component responsible for substrate specificity switching. J. Bacteriol. 182, 4906–4919 (2000).
Fraser, G. M. et al. Substrate specificity of type III flagellar protein export in Salmonella is controlled by subdomain interactions in FlhB. Mol. Microbiol. 48, 1043–1057 (2003).
Terahara, N. et al. Insight into structural remodeling of the FlhA ring responsible for bacterial flagellar type III protein export. Sci. Adv. 4, eaao7054 (2018).
Minamino, T., Inoue, Y., Kinoshita, M. & Namba, K. FliK-driven conformational rearrangements of FlhA and FlhB are required for export switching of the flagellar protein export apparatus. J. Bacteriol. 202, e00637–19 (2020).
Minamino, T., González-Pedrajo, B., Yamaguchi, K., Aizawa, S.-I. & Macnab, R. M. FliK, the protein responsible for flagellar hook length control in Salmonella, is exported during hook assembly. Mol. Microbiol. 34, 295–304 (1999).
Moriya, N., Minamino, T., Hughes, K. T., Macnab, R. M. & Namba, K. The type III flagellar export specificity switch is dependent on FliK ruler and a molecular clock. J. Mol. Biol. 359, 466–477 (2006).
Shibata, S. et al. FliK regulates flagellar hook length as an internal ruler. Mol. Microbiol. 64, 1404–1415 (2007).
Erhardt, M., Singer, H. M., Wee, D. H., Keener, J. P. & Hughes, K. T. An infrequent molecular ruler controls flagellar hook length in Salmonella enterica. EMBO J. 30, 2948–2961 (2011).
Minamino, T., Ferris, H. U., Morioya, N., Kihara, M. & Namba, K. Two parts of the T3S4 domain of the hook-length control protein FliK are essential for the substrate specificity switching of the flagellar type III export apparatus. J. Mol. Biol. 362, 1148–1158 (2006).
Minamino, T., Moriya, N., Hirano, T., Hughes, K. T. & Namba, K. Interaction of FliK with the bacterial flagellar hook is required for efficient export specificity switching. Mol. Microbiol. 74, 239–251 (2009).
Terashima, H. et al. In vitro reconstitution of functional type III protein export and insights into flagellar assembly. mBio 9, e00988–18 (2018).
Terashima, H. et al. In vitro autonomous construction of the flagellar axial structure in the inverted membrane vesicles. Biomolecules 10, 126 (2020).
Kinoshita, M., Aizawa, S. I., Inoue, Y., Namba, K. & Minamino, T. The role of intrinsically disordered C-terminal region of FliK in substrate specificity switching of the bacterial flagellar type III export apparatus. Mol. Microbiol. 105, 572–588 (2017).
Kinoshita, M. et al. The flexible linker of the secreted FliK ruler is required for export switching of the flagellar protein export apparatus. Sci. Rep. 10, 838 (2020).
Saijo-Hamano, Y. et al. Structure of the cytoplasmic domain of FlhA and implication for flagellar type III protein export. Mol. Microbiol. 76, 260–268 (2010).
Abrusci, P. et al. Architecture of the major component of the type III secretion system export apparatus. Nat. Struct. Mol. Biol. 20, 99–104 (2013).
Bange, G. et al. FlhA provides the adaptor for coordinated delivery of late flagella building blocks to the type III secretion system. Proc. Natl Acad. Sci. USA 107, 11295–11300 (2010).
Minamino, T. et al. Interaction of a bacterial flagellar chaperone FlgN with FlhA is required for efficient export of its cognate substrates. Mol. Microbiol. 83, 775–788 (2012).
Kinoshita, M., Hara, N., Imada, K., Namba, K. & Minamino, T. Interactions of bacterial chaperone-substrate complexes with FlhA contribute to co-ordinating assembly of the flagellar filament. Mol. Microbiol. 90, 1249–1261 (2013).
Xing, Q. et al. Structure of chaperone-substrate complexes docked onto the export gate in a type III secretion system. Nat. Commun. 9, 1773 (2018).
Kinoshita, M. et al. Rearrangements of α-helical structures of FlgN chaperone control the binding affinity for its cognate substrates during flagellar type III export. Mol. Microbiol. 101, 656–670 (2016).
Furukawa, Y. et al. Structural stability of flagellin subunit affects the rate of flagellin export in the absence of FliS chaperone. Mol. Microbiol. 102, 405–416 (2016).
Inoue, Y. et al. Structural insight into the substrate specificity switching mechanism of the type III protein export apparatus. Structure 27, 965–976 (2019).
Gillen, K. L. & Hughes, K. T. Molecular characterization of flgM, a gene encoding a negative regulator of flagellin synthesis in Salmonella typhimurium. J. Bacteriol. 173, 6453–6459 (1991).
Minamino, T., Morimoto, Y. V., Hara, N. & Namba, K. An energy transduction mechanism used in bacterial type III protein export. Nat. Commun. 2, 475 (2011).
Erhardt, M., Mertens, M. E., Fabiani, F. D. & Hughes, K. T. ATPase-independent type-III protein secretion in Salmonella enterica. PLoS Genet. 10, e1004800 (2014).
Jensen, J., Yamini, S., Rietsch, A. R. & Spiller, B. W. The structure of the type III secretion system export gate with CdsO, an ATPase lever arm. PLoS Pathog. 16, e1008923 (2020).
Abdiche, Y., Malashock, D., Pinkerton, A. & Pons, J. Determining kinetics and affinities of protein interactions using a parallel real-time label-free biosensor, the Octet. Anal. Biochem. 377, 209–217 (2008).
Inoue, Y., Morimoto, Y. V., Namba, K. & Minamino, T. Novel insights into the mechanism of well-ordered assembly of bacterial flagellar proteins in Salmonella. Sci. Rep. 8, 1787 (2018).
Moore, S. A. & Jia, Y. Structure of the cytoplasmic domain of the flagellar secretion apparatus component FlhA from Helicobacter pylori. J. Biol. Chem. 285, 21060–21069 (2010).
Saijo-Hamano, Y., Minamino, T., Macnab, R. M. & Namba, K. Structural and functional analysis of the C-terminal cytoplasmic domain of FlhA, an integral membrane component of the type III flagellar protein export apparatus in Salmonella. J. Mol. Biol. 343, 457–466 (2004).
Minamino, T., Kinoshita, M. & Namba, K. Fuel of the bacterial flagellar type III protein export apparatus. Methods Mol. Biol. 1593, 3–16 (2017).
Minamino, T., Morimoto, Y. V., Kinoshita, M., Aldridge, P. D. & Namba, K. The bacterial flagellar protein export apparatus processively transports flagellar proteins even with extremely infrequent ATP hydrolysis. Sci. Rep. 4, 7579 (2014).
Morimoto, Y. V., Nakamura, S., Kami-ike, N., Namba, K. & Minamino, T. Charged residues in the cytoplasmic loop of MotA are required for stator assembly into the bacterial flagellar motor. Mol. Microbiol. 78, 1117–1129 (2010).
Shimada, M. et al. Functional defect and restoration of temperature-sensitive mutants of FlhA, a subunit of the flagellar protein export apparatus. J. Mol. Biol. 415, 855–865 (2012).
Battye, T. G., Kontogiannis, L., Johnson, O., Powell, H. R. & Leslie, A. G. iMOSFLM: a new graphical interface for diffraction-image processing with MOSFLM. Acta Crystallogr. D Biol. Crystallogr. 67, 271–281 (2011).
Evans, P. R. & Murshudov, G. N. How good are my data and what is the resoluton? Acta Crystallogr. D Biol. Crystallogr. 69, 1204–1214 (2013).
Adams, P. D. et al. PHENIX: a comprehensive Python-based system formacromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 (2010).
Emsley, P., Lohkamp, B., Scott, W. G. & Cowtan, K. Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 (2010).
Hara, N., Namba, K. & Minamino, T. Genetic characterization of conserved charged residues in the bacterial flagellar type III export protein FlhA. PLoS ONE 6, e22417 (2011).
Ohnishi, K., Fan, F., Schoenhals, G. J., Kihara, M. & Macnab, R. M. The FliO, FliP, FliQ, and FliR proteins of Salmonella typhimurium: putative components for flagellar assembly. J. Bacteriol. 179, 6092–6099 (1997).
Kihara, M., Minamino, T., Yamaguchi, S. & Macnab, R. M. Intergenic suppression between the flagellar MS ring protein FliF of Salmonella and FlhA, a membrane component of its export apparatus. J. Bacteriol. 183, 1655–1662 (2001).
Minamino, T. et al. Role of the C-terminal cytoplasmic domain of FlhA in bacterial flagellar type III protein export. J. Bacteriol. 192, 1929–1936 (2010).
Acknowledgements
We thank beamline staffs at SPring-8 for technical help in use of beamlines BL41XU. This work was supported in part by JSPS KAKENHI Grant Numbers JP18K14638 and JP20K15749 (to M.K.), JP16J01859 (to N.T.), 25000013 (to K.N.), 15H02386 (to K.I.), and JP26293097 and JP19H03182 (to T.M.). This work has also been partially supported by JEOL YOKOGUSHI Research Alliance Laboratories of Osaka University to K.N.
Author information
Authors and Affiliations
Contributions
K.N., K.I., and T.M. conceived and designed research; Y.I., M. Kinoshita, M. Kida, N.T., K.I., and T.M. preformed research; Y.I., M. Kinoshita, M. Kida, N.T., K.I., and T.M. analyzed the data; and K.N., K.I., and T.M. wrote the paper based on discussion with other authors.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Inoue, Y., Kinoshita, M., Kida, M. et al. The FlhA linker mediates flagellar protein export switching during flagellar assembly. Commun Biol 4, 646 (2021). https://doi.org/10.1038/s42003-021-02177-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s42003-021-02177-z
- Springer Nature Limited
This article is cited by
-
FliH and FliI help FlhA bring strict order to flagellar protein export in Salmonella
Communications Biology (2024)