
Abstract
The stressosome is a pseudo-icosahedral megadalton bacterial stress-sensing protein complex consisting of several copies of two STAS-domain proteins, RsbR and RsbS, and the kinase RsbT. Upon perception of environmental stress multiple copies of RsbT are released from the surface of the stressosome. Free RsbT activates downstream proteins to elicit a global cellular response, such as the activation of the general stress response in Gram-positive bacteria. The molecular events triggering RsbT release from the stressosome surface remain poorly understood. Here we present the map of Listeria innocua RsbR1/RsbS complex at resolutions of 3.45 Å for the STAS domain core in icosahedral symmetry and of 3.87 Å for the STAS domain and N-terminal sensors in D2 symmetry, respectively. The structure reveals a conformational change in the STAS domain linked to phosphorylation in RsbR. Docking studies indicate that allosteric RsbT binding to the conformationally flexible N-terminal sensor domain of RsbR affects the affinity of RsbS towards RsbT. Our results bring to focus the molecular events within the stressosome complex and further our understanding of this ubiquitous signaling hub.
Similar content being viewed by others

Introduction
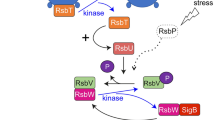
The stressosome is a megadalton signaling complex first described in the Gram-positive model organism Bacillus subtilis, for which a wealth of mutagenesis and functional data are available1,2,3. It consists of multiple copies of a small single STAS (sulfate transporter anti-sigma factor antagonist) domain protein, RsbS, and another STAS-domain protein comprising an additional globular N-terminal sensory domain, RsbR4. Bacillus species encode several RsbR paralogs, differing in their sensory domains, which are also incorporated into the stressosome5,6,7,8,9. However, the nature of the signaling molecule sensed by the RsbR paralogues, with the exception of the blue light responsive YtvA protein, and the nature of RsbR activation remains an open question8,10. RsbT, the third protein of the stressosome complex, was suggested to bind to RsbS in the stressosome core in an inactive state11. Stress signal perception results in the RsbT-dependent phosphorylation of conserved serine and threonine residues in the STAS domains of RsbS and RsbR paralogs, respectively, and finally the detachment of RsbT12,13,14. Free RsbT initiates a signaling cascade resulting in the up-regulation of stress gene transcription; which, in Bacillus and close relatives, is achieved by the activation of the alternative sigma factor, σB, involving the proteins RsbU, RsbV, and RsbW14,15,16,17,18 (Supplementary Fig. 1). The activation state of the stressosome is reset by the removal of phosphates from RsbS and RsbR by the phosphatase RsbX, allowing for a new round of phosphorylation12. Transcription of RsbX is positively controlled by σB and this provides a feedback loop to reset the stressosome to an inactive state19. However, precise knowledge on the feedback mechanism to attenuate RsbT release during continuous stress exposure is still scarce.
Three published single particle cryo-electron microscopy (cryo-EM) reconstructions reveal that the Bacillus and Listeria type stressosomes are assembled in a pseudo-icosahedral scaffold, formed by interaction between the RsbR STAS domain and RsbS11,20,21. The STAS domains of RsbS and RsbR dimers constitute the stressosome core with a proposed 20 RsbR2:10 RsbS2 stoichiometry. This stoichiometry was inferred from the mushroom-like structure of the RsbR sensory domains, termed turrets, which protrude out of the STAS domain core11. From this stoichiometry, D2-symmetry was superimposed to the icosahedral STAS domain for both B. subtilis stressosomes. Interestingly, a recent cryo-EM structure of the L. monocytogenes stressosome complex from the icosahedral core resolved to 3.38 Å and a 4.48 Å map without any symmetry imposed (C1 map) confirms the hetero-assembly of one RsbS dimer with two RsbR dimers along the threefold symmetry axis similar to the B. subtilis stressosome, but differs with respect to the RsbR:RsbS stoichiometry of the pentameric faces of the STAS domain core, resulting in a breakdown of the D2 symmetry21. These differences in stressosome assembly are intriguing, and more structures of stressosome complexes from different organisms are required to understand the role of STAS domain core interactions with RsbT. As STAS domains accommodate the stress-regulated phosphorylation sites, molecular insights into the dynamics of the core are of paramount importance to understand the mechanism of stressosome signal transduction.
Here, we determined a 3.87 Å reconstruction from a D2 symmetrized cryo-EM map of the stressosome of Listeria innocua (LiRsbRS) assembled from co-expressed LiRsbR and LiRsbS proteins. While the LiRsbR complex confirms the 20 LiRsbR2:10 LiRsbS2 stoichiometry found in B. subtilis, it shows pronounced differences in STAS domain secondary structure and assembly compared to the recently published L. monocytogenes structure. Our structure provides insights into the movement of sensor linker helices and STAS domains. We hypothesize a key role for RsbT–RsbT interactions in controlling the allosteric phosphorylation of RsbS and RsbR proteins thereby exploiting the pseudo-icosahedral symmetry of the STAS domain core. Finally, we propose a mechanism of a stress-induced conformational change in the N-terminal domain of LiRsbR, resulting in an altered accessibility of phosphorylation sites.
Results
Architecture of the LiRsbRS complex
Two species of stressosome complex assemblies were eluted at different salt concentrations during ion exchange chromatography: a dimeric and a monomeric form, respectively (Supplementary Fig. 2a). The oligomer complex distribution remained stable at the different salt concentrations during size-exclusion chromatography on a Superose 6 Increase column (Supplementary Fig. 3a), however, complexes self-assembled stronger at higher ion strength judged from negative stain EM (Supplementary Fig. 3b). Only the monomeric complex was subjected to structure determination by single particle cryo-EM after running a size-exclusion chromatography (Supplementary Fig. 2b). Interestingly, the MS analysis on the phosphorylation state of the SEC fraction used for structure determination revealed a phosphorylation of Thr241, while conserved sites Thr175 and Thr209 were not phosphorylated (Supplementary Fig. 2c). Although the stressosome particles were easy to identify in the cryo-EM micrographs, a reconstruction into a 3D volume was challenging due to the pseudo-icosahedral symmetry. 3D classes were selected after classification in RELION not only based on resolution but on the simultaneous appearance of turret densities at a comparably high signal level to the STAS domain core. Classifications that yielded only one or two turrets were rejected independently of the nominal resolution given by the respective programs. Signal subtraction routines for turrets and core were performed for further improvement of the alignment. 3D refinement was performed with the non-uniform refinement method in Cryosparc V222, and yielded a map without imposing any symmetry (C1 map to 4.2 Å, Supplementary movie 1), a map with imposed D2 symmetry (D2 map to 3.87 Å, Fig. 1d–f and Supplementary Fig. 5), and a map with icosahedral symmetry (I map to 3.45 Å, Supplementary Fig. 6). The resolution of the side chains in the C1 map was of sufficient quality to unambiguously identify and model the STAS domains of LiRsbR and LiRsbS (Figs. 1 and 2). The stoichiometry of RsbR:RsbS was identical to the one described for the Bacillus stressosome, 20 LiRsbR2:10 LiRsbS2. In addition, the D2 symmetry axes were visible in the C1 map, which helped improve the turret densities significantly, consistent with the Bacillus stressosome assembly. The 10 LiRsbS dimers form two hook-shaped strips along the core (Fig. 1a, f in yellow and orange color). This assembly leads to pentameric planes with either 1 or 2 RsbS monomers connecting to 4 or 3 RsbR monomers in one of the 12 edges (Fig. 1b, f). A pentameric plane thereby comprises five triangular faces, all of them consisting of one RsbS and two RsbR dimers (Fig. 1c, f). The stressosome icosahedron is comprised of 20 hetero-triangular faces, which most likely is the functional unit for RsbT interaction. This feature is consistently conserved in the recently published cryo-EM C1 map of the stressosome complex from Listeria monocytogenes (EMD-4508; LmRsbRS) determined to a resolution of 4.21 Å (Supplementary Fig. 7). However, in contrast to our LiRsbRS and both BsRsbRS structures, the D2 symmetry is broken in LmRsbRS; as LmRsbS dimers are circularly arranged around two pentagons (Supplementary Fig. 7, yellow and orange RsbS circles). In our unsymmetrized LiRsbRS map, nearly 20 turrets are present simultaneously in a B-factor sharpened map, which highlights the quality of particle alignment. Pronounced differences in the STAS domain dimer interfaces of LiRsbR and LiRsbS can be clearly identified (Fig. 2a, b) and strengthen the assignment of R and S components within the LiRsbRS complex (Fig. 1e, f). For the published LmRsbRS complex, the inconsistent signal intensity of well-resolved STAS domain core vs. weakly resolved LmRsbR turrets (Supplementary Fig. 7) might have hampered the unambiguous assignment of LmRsbR and LmRsbS. Furthermore, the LmRsbRS complex was assembled after purification, which could affect complex assembly, while both Bacillus and LiRsbRS stressosomes were co-expressed and co-purified.

a Electronic potential map of the LiRsbRS stressosome with I2 symmetry imposed, with LiRsbS dimers shown in yellow and orange color. The STAS-domain dimers of the LiRsbR are indicated as transparent shapes. A hook-shaped strip of 5 LiRsbS dimers (yellow) is consecutively mirrored along perpendicular D2 axes yielding the second strip of 5 LiRsbS dimers (orange). b, c The STAS-domain dimers represent a leg in the 20 congruent triangular faces. 12 corners connect always five (b) of the triangular faces (c). Protomers from different dimers shown in different colors. d Fitted atomic models of LiRsbR and LiRsbS in the D2 symmetrized 3.87 Å map, with the LiRsbR dimer and LiRsbS dimer shown in blue and yellow color, respectively. The map volume is rendered at two levels, focusing at the STAS-core (black) and showing the sensory domain of LiRsbR (blue, level was set about a factor 5 higher). e In the assembly of the LiRsbRS, the LiRsbS (yellow and orange color) and the LiRsbR subunits (blue color) are each in contact to another. RsbRs in one pentagon are highlighted in similar colors as in (b, c). f A two-dimensional representation of the pentagonal and triangular faces in the STAS icosahedron colored according to (b, c). g, h 3D variability analysis in cryoSPARC v2.923 in C1 created a set of 40 continuous 3D volumes with two distinct conformations of the RsbR sensor domain are visible along the linker helix interface of the RsbR dimer: slightly coiled conformation (g), parallel straight conformation (h), with the former predominantly selected in 3D classification of the D2 map (d).

a LiRsbR and b LiRsbS. The helices of LiRsbR, ɑ0–ɑ4 are shown in different shades of blue and the β-sheets, β0–β4 in shades of green, respectively. In LiRsbS the helices are color-coded in yellow/orange and β-sheets in shades of green. The D2 cryo-EM map is shown as black mesh. The density of ɑ0 was very pronounced and allowed for an unambiguous assignment of the STAS core with the stoichiometry of 20 LiRsbR2: 10 LiRsbS2 described in Fig. 1. c The topology of (a, b) is shown in the sequence alignment of LiRsbR and LiRsbS and Supplementary movie 3. X indicates the phosphorylated threonine residues. The density map and model of the region of ɑ3 containing the conserved threonine adjacent to the phosphorylation site (d) in a phosphorylated state in RsbR (e) in an unphosphorylated state in RsbS.
3D variability analysis in cryoSPARC v2.923 in C1 was used to investigate the conformational heterogeneity in the stressosome cryo-EM data set. From a group of 40 continuous 3D volumes (Supplementary movie 2), two distinct conformations of the RsbR sensor domain are visible (Fig. 1g, h), while the STAS domain adopts one stable conformation within the stressosome core. Conformational changes in the sensor domain occur along the linker helix dimer interface of RsbR. The linker helices switch from a straight orientation to a wedged, slightly intertwined conformation (Fig. 1d). The conformational changes in the linker helices are accompanied by changes in the sensor domain; however, due to the reduced resolution in this domain, it was not possible to model these changes with confidence. However, as the STAS domains show one distinct conformation we conclude that the sensor domain switches conformations stochastically when RsbT is not bound to the stressosome. These conformational changes of the RsbT-unbound N-terminal domains do not seem to affect the conformation of the STAS domain, which suggests an additional element is required for communicating sensor-domain changes to the phosphorylation status of the stressosome STAS domain (Fig. 3a, b).

a The heterotriangle formed from two dimers of RsbR and one dimer of RsbS, with the regulatory phosphorylation sites and their interaction partners indicated by red arrows. b a close-up of the RsbS interaction surface, with Ser56 shown in ɑ2 and the Thr88 in ɑ3. c Relative expression of the SigB-dependent genes lmo2230 (c) and inlA (d) in unstressed L. monocytogenes cells relative to wild type in mutants of rsbR1 and sigB. The mean value is shown by the horizontal line in the boxes, and the height of the box indicates standard deviation, n = 6 for each experiment. e Alignment of STAS domain proteins found in different Gram-positive bacteria, with the conserved T241 residue indicated with a red diamond. The raw data for panels c and d can be found in Supplementary Data 1.
Models of the LiRsbR and LiRsbS STAS domains
Modeling of LiRsbR (Fig. 2a) and LiRsbS (Fig. 2b) confirms the high conservation of the STAS domain fold (Fig. 2c). LiRsbR enters the STAS domain as the linker helix (ɑ0), while a beta-strand (β0) is found at the N-terminal end in LiRsbS. ɑ3 is well-resolved in LiRsbRS (Supplementary Fig. 8d, e), while it is not resolved in LmRsbR. In addition, the models of the STAS domains of LiRsbR and LmRsbR (pdb entry 6QCM) differ by a frame shift of 6 residues (Supplementary Fig. 8a) due to a different positioning of Pro1β56 and Pro159 (Supplementary Fig. 8b, c). The conserved LiRsbS Ser56 and LiRsbR Thr175 and Thr209 are not phosphorylated in our structure. Close to Thr241 in LiRsbR, a spherical density was assigned as phosphoryl group in accordance with the ESI–MS–MS (Fig. 2d; Supplementary Fig. 2c).
Such a density was not observed in LiRsbS at Thr88 (Fig. 2e). Thr241 in LiRsbR and the corresponding Thr88 in LiRsbS are conserved in the STAS domains of RsbR and RsbS of different Listeria species as well as in Bacillus subtilis.
The phosphoryl-group brings Thr241 in interaction range to Thr209 (Fig. 3a, b). At this point we can only speculate that TPO241–Thr209 is mimicking a potential interaction between both residues, which would have taken place with the vice versa phosphorylated Thr209 (TPO209–Thr241). A similar bridged interaction, Thr209–PO3–Thr241, would be conserved by Thr205–PO3–Thr237 in B. subtilis. Substitution of either T209 or T241 with alanine in L. monocytogenes resulted in the same phenotype, with increased transcription of two σB regulated genes, lmo2230 and inlA24,25, during exponential growth in a complex broth medium (Fig. 3c, d). This result suggested that loss of this T209–PO–T241 interaction resulted in an inability to suppress σB activity in the absence of environmental stress. In addition, exploring the sequences of STAS domain proteins of related bacterial species indicates that T241 is a conserved residue, at least in Gram-positive bacteria (Fig. 3e).
Interdimeric interactions within the STAS core
We observe different phosphorylation states of Thr241 depending on their interaction with their STAS domain neighbors within the pseudo-icosahedral assembly (Fig. 4 and Supplementary Fig. 10). Phosphorylated Thr241 was only observed when RsbR was interacting with RsbS (Fig. 4c, d) not in an RsbR–RsbR interaction (Fig. 4g, h), which was instrumental in resolving the phosphoryl density during processing. The interaction between the two LiRsbR dimers (Fig. 4g, h) is comparable, but not similar to the interaction of LiRsbS with LiRsbR at the R–S and S–R interface, respectively (Fig. 4c–f). While the interactions between R–R and R–S comprise similar structural elements, the interactions at the S–R interface are reduced just to a hydrophobic coordination of Leu224 by Leu100 and Ile90 in LiRsbS (Fig. 4h). Therefore, the R–S and S–R interface differ with respect to the coordination and flexibility of ɑ3 and loop ɑ3–β4. Direct consequence of Thr209–TPO241 is a conformational change in ɑ3 and subsequently in symmetry break in the hetero-triangular face (Fig. 4).

The three unique interaction surfaces can be seen from the bottom in (a) and from the side in (c), (e) and (g). A surface rendering of RsbS and the sidechain in the hydrophobic pocket can be seen in (b). Leu71 from the ɑ2 helix of RsbS can insert into the hydrophobic pocket formed by ɑ3 and β4 of RsbR (d), the interaction further strengthened by hydrogen bonds formed between Glu91 of RsbS and Gln217 on RsbR. A very similar interaction occurs between ɑ2 of RsbR and the adjacent RsbS (f) or RsbR (h) protomer, where Leu224 inserts into the hydrophobic pocket former by ɑ3 and β4 of the adjacent STAS domain protein.
RsbT binding and release in LiRsbRS
We have purified LiRsbT after expression in E. coli and performed an in vitro phosphorylation of LiRsbRS complex (Supplementary Fig. 11a, b). We observe a bi-phasic phosphorylation time-course. LiRsbS phosphorylation increases linearly in the first time points during 1 min, while phosphorylation of LiRsbR starts only after this linear phase. Interestingly, when LiRsbR increases LiRsbS phosphorylation jumps into saturation suggesting that RsbT binding to LiRsbR and LiRsbS is not independent to each other. We performed an additional size-exclusion chromatography run with the LiRsbRS complex incubated with RsbT (Supplementary Fig. 11c). Although we obtained a ternary complex the amount of RsbT was varying between different experiments reflecting a highly dynamic interaction in vitro like what was observed for LmRsbRST binding21. A negative stain analysis of the co-eluted ternary complex suggested additional density for RsbT (Supplementary Fig. 11d), however, we were not able to obtain a stable LiRsbRST complex suitable for high-resolution structure determination.
To investigate the interaction of RsbT with the RsbRS complex we performed a docking study using two structurally well described STAS domain complexes, the crystal structures of SpoIIAB–SpoIIAA (pdb entry code 1TIL and 1TH8) and of RsbV–RsbW (pdb entry code 6M37)26, as blueprint. A homology model of ATP and ADP bound LiRsbT was generated and docked to LiRsbR (Fig. 5a, b) and LiRsbS (Fig. 5c) using the spatial orientation of SpoIIAA to SpoIIAB and RsbV and RsbW, respectively, as a template: the STAS domain of LiRsbR/S aligned with SpoIIAA/RsbV and ATP/ADP-bound LiRsbT aligned with SpoIIAB/RsbW. The LiRsbT models were docked to LiRsbR (Fig. 5a, b) and LiRsbS (Fig. 5c, d) using the spatial orientation of SpoIIAA to SpoIIAB as a template: the STAS domain of LiRsbR/S aligned with SpoIIAA and ATP/ADP-bound LiRsbT aligned with SpoIIAB. The superposition of SpoIIAA and LiRsbR (Fig. 5b) revealed that due to the interaction of T209–TPO241, ɑ3 has moved towards ɑ2 in an ‘up-conformation‘ compared to the ɑ3-‘down-conformation‘ in a non-phosphorylated RsbS (Fig. 5c, d). The ‘up-conformation‘ will result in a steric clash of ADP-bound LiRsbT and ɑ3 (Fig. 5b; clash indicated by red star). By contrast, docking of ATP-bound LiRsbT to non-phosphorylated LiRsbS (Fig. 5c) shows that Ser56 is accessible for phosphorylation by LiRsbT, as no interaction between Ser56 and Thr88, the structural equivalent of Thr241 in LiRsbR (Fig. 5d), takes place and ɑ3 thus remains in a ‘down-conformation’. In summary, ɑ3 and loop ɑ3–β4 must move during RsbT release. We suggest that phosphorylation of RsbR affects the conformational flexibility of ɑ3 via Thr241.

a Superposition of LiRsbR model (in blue color) with a crystal structure of SpoIIAA (gray) in complex with the ADP-bound SpoIIAB (pdb entry 1TH8 gray color). A homology model of ADP-bound LiRsbT (in green color) is aligned matching the orientation in the SpoIIAB complex, while SpoIIAA is aligned to the STAS domain of LiRsbR. b Close-up view of the STAS domain superposition in (a) rotated by 90° (color coding as in a). Phosphorylation sites and the respective interacting sidechains are shown as stick models (carbon atoms in blue or yellow, respectively, oxygen atoms in red, and nitrogen atoms in dark blue). The relative conformational difference between the not phosphorylated SpoIIAA from B. subtilis and the phosphorylated LiRsbR is indicated by the dashed lines, and the model clash indicated by a red star. c Superposition of LiRsbS model (in yellow color) with the SpoIIAA in complex with ATP-bound SpoIIAB (in gray color). A homology model of ATP-bound LiRsbT (in green color) is aligned to LiRsbS matching the orientation of SpoIIAB in the SpoIIAA:SpoIIAB in complex. A 90˚ rotated view can be seen in (d).
Docking of multiple LiRsbT to the LiRsbRS complex
Multiple copies of LiRsbT were docked to the entire stressosome complex (Fig. 6a) to investigate steric clashes. In the functional hetero-triangular face, one LiRsbT protein can bind to every STAS domain monomer without steric clashes (Fig. 6b). LiRsbR would have sufficient space to capture LiRsbT. LiRsbR shows a mainly negatively charged surface, while one side of LiRsbT is mainly positively charged (Fig. 6d; coulombic surface Supplementary Fig. 9a). The observed conformational change of the linker helices observed by the 3D variability study (Fig. 1g) would most likely affect the binding pose and binding affinity of RsbT and RsbR. A comparison of the electrostatic surfaces obtained from homology models of the other three RsbR paralogs in L. innocua, which are found outside the σB-operon, reveals a significant variation (Supplementary Fig. 12a–d) despite their high degree of conservation in the STAS domains (Supplementary Fig. 12e). The sensory turrets and STAS domain in LiRsbR form a clamp-like binding pocket in which LiRsbT would fit in a fixed orientation, but with the ATP-binding site not accessible to Thr209. Having LiRsbT docked at this position to LiRsbR would coordinate LiRsbT binding to LiRsbS (Fig. 6d). For the proposed LiRsbST complex, the target Ser56 and the potential catalytic base Glu45 align for phosphorylation (Fig. 6e). In contrast to SpoIIAB, a positively charged residue (SpoIIAB-Arg105) for electrostatic stabilization of the transition state is not conserved in LiRsbT. In the proposed LiRsbRS–RsbT assembly the ATP-lid is stabilized by the interaction with a neighboring LiRsbT (bound to LiRsbS). The order of binding to LiRsbR might be restricted such that LiRsbT can only bind to LiRsbR if LiRsbT is either not yet bound to LiRsbS or has been released. The observed conformational changes in the sensor domain and linker helices might help re-orient LiRsbT on LiRsbS for productive phosphorylation.

a View on the triangular faces of LiRsbRS (RsbR in blue and RsbS in yellow colors), and with LiRsbT homology models (shown in dark green color docked to LiRsbR and in light green color docked to LiRsbS). b Two-dimensional representation of the pentagonal and triangular faces in the STAS icosahedron colored according to (a). The dashed box indicates roughly the composition shown in (a). c Sideview (as indicated by the gray, dashed line in a) on the LiRsbRS–RsbT assembly. In this assembly, only the LiRsbT bound to an LiRsbS can dissociate from the complex; LiRsbTs bound to LiRsbRs are sterically confined by the linker helices. d Vicinity of the LiRsbTS (bound to LiRsbS, light green) binding pose shown as surface colored according to the electrostatic potential. The LiRsbT-binding site is created by equal contributions of one LiRsbS and two LiRsbTs, some contact to the sensory domain of LiRsbR is also feasible. e Vicinity of ATP in the proposed LiRsbS–LiRsbT complex. The target Ser56 and the potential catalytic base Glu45 are indicated. In the proposed LiRsbRS–RsbT assembly the ATP-lid is stabilized by the interaction with a neighboring LiRsbTR (bound to LiRsbR).
Discussion
The stressosome transduces environmental stress signals to one single cellular output, which is the phosphorylation-dependent release of the kinase RsbT to start the downstream sigmaB-signaling cascade14,17. This represents a straightforward ‘actio-reactio’ principle: increased amount of stress results in a higher number of released RsbT proteins. For B. subtilis, it was suggested that RsbS preferably sequesters RsbT in the BsRsbRS complex23. In the BsRsbRST structure, 20 BsRsbT molecules were thus positioned to the 20 BsRsbS molecules11. However, environmental stresses lead to an increase in RsbT kinase activity against RsbR, too13, which is not accounted for in an only RsbS–RsbT interaction. The LiRsbRS complex now presented in this manuscript provides a unique opportunity to observe conformational changes in response to differences in phosphorylation especially in RsbR. Different to the recently published structures of the Listeria monocytogenes complex21, we were able to resolve helix 3 and its significant conformational changes. In contrast to the recent B. subtilis structure20, we present a more dynamic picture to the STAS–STAS domain interface and its role in signal propagation in the complex. The high quality of the D2 map with a completely resolved STAS domain and linker helix allow for insights on how the stress signal is translated into a conformational change upon activation. We suggest that RsbS still acts as a primary binding site, lacking the steric hindrance of the RsbR turrets, which is supported by the in vitro phosphorylation of the LiRsbRS complex (Supplementary Fig. 11). It requires a certain threshold of phosphorylated LiRsbS before phosphorylation of LiRsbR is detected. We assume that at least one RsbT is bound to the RsbR protomer in direct contact with RsbS. Without stress RsbT is not able to phosphorylate RsbR. The electrostatic surface potential representation of LiRsbR suggests an ionic interaction of the negatively charged LiRsbR dimer with the positively charged LiRsbT, coordinated by the RsbR STAS domain of one protomer and the turret of the other protomer within the RsbR dimer in a clamp-like fashion. The candidate LiRsbR paralogs Lin0204, Lin1683, and Lin1956, display less pronounced negative charge patches in the same region (Supplementary Fig. 12). Differences in electrostatic attraction between RsbR and RsbT and variation of the affinity and/or number of RsbT molecules might cause the modulation of the stress downstream signal when the stressosome is assembled from different RsbR paralogs.
RsbT might also have an impact on stabilization and signal transduction within the whole stressosome complex. The high resolution of the Listeria innocua stressosome STAS domain shows that in the up conformations of ɑ3 (in RsbR) RsbT cannot be docked in way that its ATP-binding site would be accessible to the phosphorylation sites. Vice versa, in the down conformations of ɑ3 in the de-phosphorylated state (in RsbS) docking is possible. The ɑ3–β4 delineated pocket is the essential interface for interaction between the STAS domains of adjacent dimers, and the nature of the interactions between the domains is very similar to that found in B. subtilis, i.e. the major interaction is between hydrophobic residues, with additional, less conserved residues mediating electrostatic interactions between the protomers between the ɑ3–β4 of one domain and α2 of the other20.The two distinct conformations of the sensor domain and linker helices (straight and intertwined in Fig. 1g) could be attributed to different phosphorylation states of RsbR. However, the STAS domains do not change conformation in the 3D variability study (Supplementary movie 2), which would suggest a rather rigid stressosome core of L. innocua. It is more likely that without RsbT bound, the N-terminal sensor domains display an intrinsic flexibility.
Thr205 in BsRsbR (corresponding to Thr209 in LiRsbR) is only phosphorylated after severe stress in order to limit the stress transmission process27. Therefore, we assume that RsbT can bind to RsbR in different conformations without having the ATP-site positioned for phosphorylation. Our docking studies on LiRsbS suggest that an activation of LiRsbS Ser56 requires neighbor RsbTs to stabilize ɑ3 and loop ɑ3–β4 in RsbS and the ATP-lid in RsbT. RsbT is missing the positive charges to stabilize the binding pocket for ATP by its own. A computational study on the BsRsbRST complex supports this notion and concluded that a model in which phosphorylation by RsbT is increased by the presence of phosphorylated neighbors provided the best fit to the experimental data28. We propose that a direct interaction of RsbT with RsbR already in the down-regulated state will increase the RsbT kinase activity on RsbS and represents the first step in the allosteric phosphorylation process. As a consequence, neighboring RsbR dimers decorated with RsbT would provide additional coordination for the RsbT–RsbS binding increasing the affinity of RsbS towards RsbT.
Different RsbR paralogs with different RsbT affinities, because of their electrostatic potential, would provide an elegant way to modulate efficiency of the RsbT kinase reaction. Most interesting, recent data on B. subtilis RsbR paralogs revealed a similar spectrum of stress stimuli sensed by the paralogs RsbRA-D, however with varying response profiles29.
By including RsbR–RsbT and RsbT–RsbT interactions we describe the activation mechanism as following: A stress stimulus perceived by RsbR turrets triggers the re-orientation of RsbT necessary to phosphorylate the target Ser/Thr, because conformational changes will be transmitted via the linker helices by a RsbR–RsbT interaction affecting subsequently the RsbT–RsbS interaction. Conformational changes in the RsbR turrets and the linker helices might result directly in an altered RsbR–RsbT interaction, which in turn affect the RsbT–RsbS interaction (T–T crosstalk). Alternatively, conformational changes in the STAS domain of RsbR, can be transduced via the RsbR–RsbS interdimeric interactions in order to render Ser/Thr residues accessible for RsbT to phosphorylate (R–S crosstalk). RsbT will subsequently be released from RsbS by a conformational change in the ɑ3 helix and loop ɑ3–β4, as we have observed in the RsbR dimers exhibiting the Thr209-TPO241 interaction. The flexible nature of LmRsbR ɑ3 is in line with the potential regulatory role of this region21. Once RsbT is released from RsbS, a re-arrangement of RsbT bound to RsbR may occur. If stress continues or increases, RsbT is already positioned to phosphorylate RsbR T209 and will be released. In the allosteric context, release of RsbT from RsbR would lead to a decrease of activation of the RsbT positioned to phosphorylate RsbS and an attenuation of the transmission process. The stepwise re-orientation of RsbT would agree with the observation that binding of RsbT to RsbR increases the phosphorylation probability of RsbS30 but that phosphorylation of RsbR at Thr209 decreases stress signal transmission27. Our model can be extended to the recovery following a stress activation event. We assume that RsbT would dissociate from RsbS first, allowing the access of RsbX to dephosphorylate RsbS. A still bound RsbT to the adjacent RsbR would not hinder such a RsbS–RsbX interaction and dephosphorylation of RsbS may occur before RsbT is released from RsbR. The dephosphorylation of RsbS releases the tension on α2-helix of the neighboring RsbR connected to RsbS (Fig. 4f). Dephosphorylation of S56 in RsbS would support the conformational transition of α3-helix in RsbR from the ‘up’ to the ‘down-state’. In further events, RsbX would dephosphorylate RsbR, resetting the stressosome to process imminent stress.
Different mutations in the RsbR N-terminal domain, e.g., BsRsbR E136K significantly increases the basal level of the \(\sigma\)B response, even without stress10. However, an RsbS mutation with S59A negates these effects suggesting that N-terminal mutations in RsbR lead to increased phosphorylation in RsbS10. According to our docking model, Lys141 of LiRsbR, positioned in the RsbR linker helix, would contact Gln20 in RsbT and, in a putative LiRsbR E140K mutation corresponding to BsRsbR E136K, Lys141 would point in a different direction disrupting the RsbT–RsbR interaction. It can be noted that previously described mutations in the BsRsbR N-terminal domain would be positioned in the RsbR–RsbT contact area4. The postulated T–T cross talk would also shed light onto the puzzling symmetry breaks in RsbR–RsbS assembly. We assume that the key to different stressosome activation profiles is a varying arrangement of different RsbR paralogs within the functional important hetero-triangular faces affecting the RsbT binding/release.
Methods
Cloning and heterologous expression of stressosome proteins
The L. innocua genes CAC96120.1 (LiRsbR) and CAC96121.1 (LiRsbS) were cloned into the pET11a expression vector (Novagen, 69436-3) via NdeI and BamHI restriction sites. A ribosome binding site (construct from GenScript) was inserted between the coding sequences. The construct was transformed into E. coli DH5α for plasmid isolation and maintenance.
For over-expression, the pET11a plasmid was transformed into E. coli BL21 Star (DE3) cells. Expression cultures were grown to a starting OD600 of 0.6–0.8, induced with IPTG (final concentration 1 mM) and harvested by centrifugation after 3 h of expression at 37 °C and 120 rpm. The cells were harvested by centrifugation in a JLA-8.1000 rotor (Beckmann Coulter) at 4000 rpm for 30 min, at 4 °C and stored at −80 °C. For LiRsbT a pGEX6P-2 vector (GE Healthcare) containing a GST-tag fused rsbT gene from L. innocua (CAC96122.1) was transformed into E. coli BL21 (DE3) cells and grown in LB media at 37 °C, 120 rpm until OD600 = 0.6, then induced with 0.1 mM IPTG and allowed to express for 3 h. The cells were resuspended in Elution buffer (50 mM Tris–HCl pH 7.5, 150 mM NaCl) and lysis and clarification carried out as above.
Protein purification
To purify the LiRsbRS complex, cells were resuspended in lysis buffer (50 mM Tris–HCl pH 8.3, 10 mM EDTA), broken in a cell disruptor, and cell debris was removed by ultracentrifugation at 150,000 × g. Intact stressosome complexes were purified by two subsequent ion exchange steps (50 mM step gradient (50–750 mM NaCl) on a DEAE sepharose column (Cytiva Life Sciences), 60 mL linear gradient (0–750 mM NaCl) on a Resource Q column), followed by size-exclusion chromatography on a Superose 6 Increase column in 100 mM NaCl, 50 mM Tris–HCl pH 8.5.
To purify LiRsbT, clarified cell lysate was loaded on a Pierce™ Glutathione Agarose (Thermo Fisher Scientific) gravity column, and eluted with buffer containing 10 mM reduced glutathione. The protein was then pooled and mixed with Prescission Protease, and incubated overnight at 4 °C. Afterwards, the protein was concentrated and purified via SEC chromatography on a Superdex S75 10/300 column.
Phosphorylation state and salt dependence of the LiRsbRS stressosome assembly
The purification protocol consisted of anion exchange chromatography using a DEAE resin (Supplementary Fig. 2a) followed by a polishing anion exchange step with Q-resource resin and a final size-exclusion chromatography (SEC) run (Supplementary Fig. 2b). Nearly two-thirds of the LiRsbRS complex eluted at 50 mM NaCl in the first DEAE run (Supplementary Fig. 2a). The two fractions from the DEAE run differed in the oligomerization behavior. Complex formation of the first fraction of LiRsbRS was confirmed by negative stain EM, revealing the dimerization of two stressosome complexes via two turrets (Supplementary fig. 2a, lower panel). To determine whether ionic strength of the buffers used in anion exchange chromatography affected the stability of the complex, SEC was performed at varying salt concentrations and the individual peak fractions were analyzed by negative stain EM (Supplementary Fig. 3). While the elution volume remained constant regardless of salt concentration, we observed increased interactions of LiRsbRS complexes, resulting in string-like assemblies, in response to increasing ionic strength in negative stain EM (Supplementary Fig. 3b).
For comparison of the stability of LiRsbRS at different salt concentrations, the complexes were expressed and purified as described above. However, after the first ion exchange step, suitable fractions were concentrated using a 100,000 Da MWCO cut-off centrifugal spin filter (Merck Millipore). The same spin filter was used to exchange the sample buffer by repeated concentration and dilution in the appropriate buffers with varying salt concentrations (100 mM NaCl–1000 mM NaCl), and analytical size-exclusion chromatography was performed on a Superose 6 Increase 10/300column in the respective buffers.
Cryo-electron microscopy
For cryo-electron microscopy, 3.5 µL of purified LiRsbRS complex at a concentration of 0.6 mg/mL (after SEC) were vitrified on a glow-discharged Quantifoil R2/2 holey carbon grid in a Vitrobot plunge freezer after 3 s of blotting. Movies were collected on a Titan Krios G3 300 keV electron microscope equipped with a Falcon III direct electron detector at a ×75,000 magnification. In total, 1803 movies were collected with 25 frames per movie, at an electron dose 2e−/Å2 per frame. For data pre-processing, MotionCor2 and CTFFIND4 were used31,32 Afterwards, 105,020 particles were picked, extracted and classified in RELION 3.033,34. After 6 rounds of 2D classification, a total of 53,487 particles were selected for further processing. An initial model was reconstructed using RELION 3.0, and all the particles were first subjected to global angular search three-dimensional (3D) classification in 60 iterations with four classes and step size of 7.5°, then the particles from the best class was subjected to the 2nd round of 3D classification with four classes and 3.75° local angular search step in 60 iterations. At this stage, 32,031 good particles were combined from the best 2 classes for the further 3D auto refinement. The 3D refinement was done by the non-uniform refinement in Cryosparc V222 using the corresponding map from 3D classification as the reference. Details on the processing workflow are given in Fig. S4 and in Table 1.
Model building
For model building of the LiRsbR and LiRsbS STAS domains, the RsbS anti-sigma-factor antagonist from Moorella thermoacetica (pdb 3ZXN) was used as a template, sharing sequence identities of 25% (LiRsbS) and 22% (LiRsbR), respectively. The D2 map was chosen for model building. The model was then built by manually docking the homology models in Chimera, then adjusted using Coot and refined using Phenix.
Cultivation and construction of genetically modified L. monocytogenes
L. monocytogenes EGD-e (serovar 1/2a) and E. coli DH-5α, plasmids and primers used in this study are listed in Supplementary Tables 1 and 2. Strains were grown in BHI broth or agar (LabM) at 37 °C with constant shaking at 150 rpm min−1. Cells were grown for 16 h and further diluted in fresh BHI to an initial OD600 = 0.05 and allowed to grow until mid-log phase (OD600 = 0.5). The following antibiotics were added to the media to a final concentration when required: erythromycin (Ery) at 5 µg ml−1 for L. monocytogenes strains and ampicillin (Amp) at 100 µg ml−1 for E. coli strains.
The gene rsbR1 were mutated to incorporate Thr-to-Ala in the codons 209 and 241. The codon 209 CAA (Thr) in rsbR1 was changed to GCT (Ala) and codon 241 ACA (Thr) was changed to GCT (Ala). In both cases silent mutations were added the two adjunct codons in order to discern mutant from WT codons by PCR during mutagenesis while taking in consideration the codon frequencies in L. monocytogenes EGD-e strain (rsbR1 Thr209 -TTGATACAATGGTTG- to rsbR1 Thr209Ala -GTAGACGCTATGGTA- and rsbR1 Thr241 -GTTGATACAATGGTT- to rsbR1 Thr241Ala -GTAGACGCTATGGTA-). The mutagenic sequences, each with a total length of 612 bp, which include SalI and BamHI restriction sequences in each edge, were artificially synthetized in the vectors pEX-K168::rsbR1 (T209A) and pEX-A128::rsbR1 (T241A) (Eurofins Genomics). The mutagenic sequences were subsequently cloned into shuttle vector pMAD, originating pMAD::rsbR1 (T209A) and pMAD::rsbR1 (T241A). L. monocytogenes electrocompetent cells were created as previously described35 Briefly, cells were grown at 37 °C to an OD600 of 0.2–0.25 in BHI supplemented sucrose (500 mM). Ampicillin (10 µg ml−1) was added and cultures were further grown for 2 h. Cultures were centrifuged at 5000 × g for 10 min at 4 °C and washed twice with ice-cold sucrose–glycerol washing buffer (SGWB; 10% glycerol, 500 mM sucrose). Lysozyme from chicken egg white (Sigma) was added to a final concentration of 10 µg ml−1 and incubated at 37 °C for 20 min. Cultures were centrifuged at 3000 × g for 10 min and washed in SGWB twice and finally resuspended in SGWB, aliquots were made and stored at −80 °C until used. The electrocompetent L. monocytogenes WT strain was separately transformed with pMAD::rsbR1 (T209A), pMAD::rsbR1 (T241A). Electroporated cells were plated in BHI supplemented with erythromicin. Chromosomal integration and subsequent excision was achieved through a two-step recombination as previously described36—the obtained transformants L. monocytogenes colonies were inoculated in BHI broth supplemented with erythromycin and grown at non-permissive temperatures of 40 °C for 24 h. Cultures were diluted and plated in BHI agar supplemented with erythromycin and grown for 24 h at 40 °C. the obtained integrant colonies were inoculated in BHI and grown at permissive temperatures of 30 °C. Dilutions of 1:100 were made in fresh BHI every 12 h. Simultaneously, serial dilutions were made at each passage and plated in BHI agar. Loss of the mutagenic plasmid was assessed by streaking the same colony in both BHI and BHI supplemented with erythromycin. cPCR with the primers rsbR1_upflank_F paired with either rsbR1 (T209A)_R or rsbR1 (T241A)_R, were used to identify the chromosomal mutation in the respective genes rsbR.
RNA extraction and RT-qPCR
Cultures of L. monocytogenes EGD-e and its isogenic mutants were grown until mid-log phase. Cultures were diluted in RNAlaterTM (Sigma) at a 1:5 ratio to stop the transcription. Total RNA was extracted using an RNeasy minikit (Qiagen) according to the manufacturer’s recommendations. Cells were disrupted by bead beating twice in FastPrep-24 at a speed of 6 m s−1 for 40 s. DNA was removed with Turbo DNA-free (Invitrogen) according to the manufacturer’s recommendations. The RNA integrity was verified by electrophoresis in 0.7% agarose gels. SuperScript™ III First-Strand Synthesis System (Invitrogen) was used to synthetize cDNA according to the manufacturer’s recommendations. cDNA was quantified using NanoDrop 2000c (Thermo Scientific) and diluted to a final concentration of 7 ng ml−1. RT-qPCR was performed using a Quanti-Tect SYBR green PCR kit (Qiagen) and primers for the target genes (Supplementary Table 2). Primer for the target genes 16 S, lmo2230 and inlA were previously tested using cDNA. Samples were analyzed on LightCycler 480 system (Roche) with the following parameters: 95 °C for 15 min; 45 cycles of 15 s at 95 °C, 15 s at 53 °C, and 30 s at 72 °C; a melting curve drawn for 5 s at 95 °C and 1 min at 55 °C, followed by increases of 0.11 °C s−1 until 95 °C was reached; and cooling for 30 s at 40 °C. Cycle quantification values were calculated by using LightCycler 480 software version 1.5.1 (Roche) and the Pfaffl relative expression formula37,38. The 16S rRNA gene expression was used as a reference gene. Results are expressed as Log2 relative expression ratios normalized against the expression of L. monocytogenes WT strain in the absence of stress. Three independent biological replicates were performed.
Whole-genome sequencing and single nucleotide polymorphism (SNP) analysis
The gDNA of all mutants’ strains constructed in this study was extracted using DNeasy Blood and Tissue kit (Qiagen) according to the manufacturer recommendations. The obtained genomic material was analyzed via Illumina sequencing by MicrobesNG (Birmingham, UK). The resulting trimmed reads were analyzed using Breseq software39 to identify additional SNP in the mutant strains chromosome. The nucleotide sequence of L. monocytogenes EGD-e chromosome (NCBI RefSeq accession no. NC_003210.1) was used as reference in this analysis.
Homology modeling of RsbR paralogs in L. innocua
The sequences of the paralogs were detected in the L. innocua Clip11262 genome using the DELTA-BLAST algorithm with RsbR as the query. The homology models of the paralogs were then built using the default modeling script of Modeller (version 9.24)40, using the model of RsbR as template. For each sequence, 20 models were generated and picked based on their Z-DOPE score. The structures were manually inspected for clashes and errors using the Chimera and Coot software packages.
Mass spectrometry
Mass spectrometric analysis of SDS-PAGE separated proteins was carried out as follows: protein bands were washed with 50 mM NH4HCO3, 50 mM NH4HCO3/acetonitrile (3/1), 50 mM NH4HCO3/acetonitrile (1/1) and lyophilized. After a reduction/alkylation treatment and additional washing steps, proteins were in gel digested with trypsin (Trypsin Gold, mass spectrometry grade, Promega) overnight at 37 °C. The resulting peptides were sequentially extracted with 50 mM NH4HCO3 and 50 mM NH4HCO3 in 50% acetonitrile. After lyophilization, peptides were reconstituted in 20 µl 1% TFA and separated by reversed-phase chromatography. An UltiMate 3000 RSLCnano System (Thermo Fisher Scientific, Dreieich) equipped with a C18 Acclaim Pepmap100 preconcentration column (100 µm i.d. ×20 mm, Thermo Fisher Scientific) and an Acclaim Pepmap100 C18 nano column (75 µm i.d. ×250 mm, Thermo Fisher Scientific) was operated at flow rate of 300 nl/min and a 90 min linear gradient of 4–40% acetonitrile in 0.1% formic acid. The LC was online-coupled to a maXis plus UHR-QTOF System (Bruker Daltonics, Bremen) via a CaptiveSpray nanoflow electrospray source. Acquisition of MS/MS spectra after CID fragmentation was performed in data-dependent mode at a resolution of 60,000. The precursor scan rate was 2 Hz processing a mass range between m/z 175 and m/z 2000. A dynamic method with a fixed cycle time of 3 s was applied via the Compass 1.7 acquisition and processing software (Bruker Daltonics). Prior to database searching with Protein Scape 3.1.3 (Bruker Daltonics) connected to Mascot 2.5.1 (Matrix Science), raw data were processed in Data Analysis 4.2 (Bruker Daltonics). Search parameters for searching the UniProt Listeria innocua serovar 6a database were as follows: enzyme specificity trypsin with 2 missed cleavages allowed, precursor tolerance 10 ppm, MS/MS tolerance 0.04 Da, variable modifications: deamidation of asparagine and glutamine, oxidation of methionine, carbamidomethylation or propionamide modification of cysteine, phosphorylation of serine, threonine, and tyrosine.
Phosphopeptide fragment spectra with a Mascot peptide ion-score of at least 20 were evaluated manually.
Phosphorylation of RsbRS by RsbT
Buffers of purified proteins were exchanged to kinase assay buffer (50 mM Tris–HCl pH 7.5, 50 mM KCl, 10 mM MgCl2, 0.1 mM EDTA pH 8.0, 1 mM ATP) using centrifugal filtration units (Amicon) of 100 and 10 kDa MWCO for RsbRS and RsbT, respectively. Purified RsbRS and RsbT were incubated at 37 °C for 5 min and mixed at a ratio of 5 to 1 (w/w), to start the reaction resulting in approximately 20 RsbT molecules per RsbRS complex. Reaction was carried out at 37 °C and quenched at different time intervals by mixing with 6× Laemmli sample buffer and heating to 95 °C for 2 min. The samples were the run on a 17.5% SDS–PAGE gel and stained with the Pro-Q™ Diamond Phosphoprotein Gel Stain (Thermo Fisher Scientific) according to the manufacturer’s instructions. The gels were imaged on a GelDoc Go Gel Imaging System (Bio-Rad) using the settings for Ethidium Bromide gel imaging. Gels were quantified by measuring the intensity of the bands in Fiji41, subtracting the measured background, and adjusting for total measured signal intensity between gels in Microsoft Excel.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
The atomic model of the complex presented in this manuscript has been deposited in the wwPDB with the ID 7B0U. The corresponding EM map can be found in the EMDB under the code EMD-11971. The data for Fig. 3 can be found in the Supplementary Data 1 file supplied with the manuscript. All other data available upon reasonable request.
References
Hecker, M., Pané-Farré, J. & Uwe, V. SigB-dependent general stress response in Bacillus subtilis and related Gram-Positive bacteria. Annu. Rev. Microbiol. 61, 215–236 (2007).
Pané-Farré, J., Quin, M. B., Lewis, R. J. & Marles-Wright, J. Structure and function of the stressosome signalling Hub. Sub-Cell. Biochem. 83, 1–41 (2017).
Pané-Farré, J., Lewis, R. J. & Stülke, J. The RsbRST stress module in bacteria: a signalling system that may interact with different output modules. J. Mol. Microbiol. Biotechnol. 9, 65–76 (2005).
Murray, J. W., Delumeau, O. & Lewis, R. J. Structure of a nonheme globin in environmental stress signaling. Proc. Natl Acad. Sci. USA 102, 17320–17325 (2005).
Akbar, S. et al. New family of regulators in the environmental signaling pathway which activates the general stress transcription factor σB of Bacillus subtilis. J. Bacteriol. 183, 1329–1338 (2001).
Delumeau, O., Chen, C. C., Murray, J. W., Yudkin, M. D. & Lewis, R. J. High-molecular-weight complexes of RsbR and paralogues in the environmental signaling pathway of Bacillus subtilis. J. Bacteriol. 188, 7885–7892 (2006).
Reeves, A., Martinez, L. & Haldenwang, W. Expression of, and in vivo stressosome formation by, single members of the RsbR protein family in Bacillus subtilis. Microbiology 156, 990–998 (2010).
Gaidenko, T. A., Kim, T. J., Weigel, A. L., Brody, M. S. & Price, C. W. The blue-light receptor YtvA acts in the environmental stress signaling pathway of Bacillus subtilis. J. Bacteriol. 188, 6387–6395 (2006).
Kim, T. J., Gaidenko, T. A. & Price, C. W. A multicomponent protein complex mediates environmental stress signaling in Bacillus subtilis. J. Mol. Biol. 341, 135–150 (2004).
Gaidenko, T. A., Bie, X., Baldwin, E. P. & Price, C. W. Substitutions in the presumed sensing domain of the Bacillus subtilis stressosome affect its basal output but not response to environmental signals. J. Bacteriol. 193, 3588–3597 (2011).
Marles-Wright, J. et al. Molecular architecture of the ‘stressosome,’ a signal integration and transduction hub. Science 322, 92–96 (2008).
Chen, C. C., Yudkin, M. D. & Delumeau, O. Phosphorylation and RsbX-dependent dephosphorylation of RsbR in the RsbR–RsbS complex of Bacillus subtilis. J. Bacteriol. 186, 6830–6836 (2004).
Kim, T. J., Gaidenko, T. A. & Price, C. W. In vivo phosphorylation of partner switching regulators correlates with stress transmission in the environmental signaling pathway of Bacillus subtilis. J. Bacteriol. 186, 6124–6132 (2004).
Yang, X., Kang, C. M., Brody, M. S. & Price, C. W. Opposing pairs of serine protein kinases and phosphatases transmit signals of environmental stress to activate a bacterial transcription factor. Genes Dev. 10, 2265–2275 (1996).
Price, C. W. et al. Genome-wide analysis of the general stress response in Bacillus subtilis. Mol. Microbiol. 41, 757–774 (2001).
Petersohn, A. et al. Identification of σ(B)-dependent genes in Bacillus subtilis using a promoter consensus-directed search and oligonucleotide hybridization. J. Bacteriol. 181, 5718–5724 (1999).
Kang, C. M., Vijay, K. & Price, C. W. Serine kinase activity of a Bacillus subtilis switch protein is required to transduce environmental stress signals but not to activate its target PP2C phosphatase. Mol. Microbiol. 30, 189–196 (1998).
Hardwick, S. W. et al. Structural and functional characterization of partner switching regulating the environmental stress response in Bacillus subtilis. J. Biol. Chem. 282, 11562–11572 (2007).
Scott, J. M., Mitchell, T. & Haldenwang, W. G. Stress triggers a process that limits activation of the Bacillus subtilis stress transcription factor σ(B). J. Bacteriol. 182, 1452–1456 (2000).
Kwon, E. et al. Structural insights into stressosome assembly. IUCrJ 6, 938–947 (2019).
Williams, A. H. et al. The cryo-electron microscopy supramolecular structure of the bacterial stressosome unveils its mechanism of activation. Nat. Commun. 10, 3005 (2019).
Punjani, A., Rubinstein, J. L., Fleet, D. J. & Brubaker, M. A. CryoSPARC: Algorithms for rapid unsupervised cryo-EM structure determination. Nat. Methods 14, 290–296 (2017).
Zhang, S., Reeves, A., Woodbury, R. L. & Haldenwang, W. G. Coexpression patterns of σB regulators in Bacillus subtilis Affect σB inducibility. J. Bacteriol. 187, 8520–8525 (2005).
Kim, H., Marquis, H. & Boor, K. J. σ B contributes to Listeria monocytogenes invasion by controlling expression of inlA and inlB. Microbiology 151, 3215–3222 (2005).
Utratna, M., Cosgrave, E., Baustian, C., Ceredig, R. & O’Byrne, C. Development and optimization of an EGFP-based reporter for measuring the general stress response in Listeria monocytogenes. Bioengineered 3, 93–103 (2012).
Pathak, D. et al. Structural insights into the regulation of SigB activity by RsbV and RsbW. IUCrJ 7, 737–747 (2020).
Eymann, C. et al. In vivo phosphorylation patterns of key stressosome proteins define a second feedback loop that limits activation of Bacillus subtilis σB. Mol. Microbiol. 80, 798–810 (2011).
Liebal, U. W., Millat, T., Marles-Wright, J., Lewis, R. J. & Wolkenhauer, O. Simulations of stressosome activation emphasize allosteric interactions between RsbR and RsbT. BMC Syst. Biol. 7, (2013).
Cabeen, M. T., Russell, J. R., Paulsson, J. & Losick, R. Use of a microfluidic platform to uncover basic features of energy and environmental stress responses in individual cells of Bacillus subtilis. PLoS Genet. 13, e1006901 (2017).
Gaidenko, T. A., Yang, X., Lee, Y. M. & Price, C. W. Threonine phosphorylation of modulator protein RsbR governs its ability to regulate a serine kinase in the environmental stress signaling pathway of Bacillus subtilis. J. Mol. Biol. 288, 29–39 (1999).
Zheng, S. Q. et al. MotionCor2: anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nat. Methods 14, 331–332 (2017).
Rohou, A. & Grigorieff, N. CTFFIND4: fast and accurate defocus estimation from electron micrographs. J. Struct. Biol. 192, 216–221 (2015).
Zivanov, J. et al. New tools for automated high-resolution cryo-EM structure determination in RELION-3. eLife 7, 1–22 (2018).
Zhang, K. Gctf: real-time CTF determination and correction. J. Struct. Biol. 193, 1–12 (2016).
Monk, I. R., Gahan, C. G. M. & Hill, C. Tools for functional postgenomic analysis of Listeria monocytogenes. Appl. Environ. Microbiol. 74, 3921 (2008).
O’Donoghue, B. et al. Blue-light inhibition of Listeria monocytogenes growth is mediated by reactive oxygen species and is influenced by σB and the blue-light sensor Lmo0799. Appl. Environ. Microbiol. 82, 4017–4027 (2016).
Pfaffl, M. W. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 29, e45 (2001).
Pfaffl, M. W. et al. Real-time RT-PCR quantification of insulin-like growth factor (IGF)-1, IGF-1 receptor, IGF-2, IGF-2 receptor, insulin receptor, growth hormone receptor, IGF-binding proteins 1, 2 and 3 in the bovine species. Domest. Anim. Endocrinol. 22, 91–102 (2002).
Deatherage, D. E. & Barrick, J. E. Identification of mutations in laboratory-evolved microbes from next-generation sequencing data using breseq. Methods Mol. Biol. 1151, 165–188 (2014).
Eswar, N. et al. Comparative protein structure modeling using Modeller. Curr. Protoc. Bioinform. Chapter 5, Unit-5.6 (2006).
Schindelin, J. et al. Fiji: an open-source platform for biological-image analysis. Nat Methods 9, 676–682 (2012).
Acknowledgements
This project has received funding from the European Union’s Horizon 2020 research and innovation program under the Marie Skłodowska-Curie grant agreement No. 721456. Cryo-EM was carried out in the cryo-EM facility of the Julius-Maximilian University Würzburg. A.M. also thanks Maxi Kunzmann for help with the ProQ diamond gels during her internship.
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Contributions
A.M. purified all protein samples used in the study and performed the biochemical assays, L.F. analyzed the cryo-EM data, M.G.M. built and analyzed the structure models, D.N.G. performed the Listeria in vivo experiments, S.K. cloned the LiRS and LiT proteins, M.C. and S.E. did bioinformatical analysis, and A.B. did the MS analysis. A.M., L.F., C.W., J.M.-W., R.J.L., C.O.B., J.P.-F., and C.Z. interpreted the data and wrote the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Communications Biology thanks the anonymous reviewers for their contribution to the peer review of this work. Primary Handling Editors: Manidipa Banerjee and Anam Akhtar. Peer reviewer reports are available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Miksys, A., Fu, L., Madej, M.G. et al. Molecular insights into intra-complex signal transmission during stressosome activation. Commun Biol 5, 621 (2022). https://doi.org/10.1038/s42003-022-03549-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s42003-022-03549-9
- Springer Nature Limited