
Abstract
Background
Hereditary spherocytosis (HS) is a type of hemolytic anemia caused by abnormal red cell membrane skeletal proteins with few unique clinical manifestations in the neonate and infant. An ANK1 gene mutation is the most common cause of HS.
Case presentation
The patient was a 11-month-old boy who suffered from anemia and needed a regular transfusion therapy at an interval of 2–3 months. Hematological investigations showed moderate anemia (Hb80 g/L). Red cells displayed microcytosis (MCV76.4 fl, MCH25.6 pg, MCHC335 g/L). The reticulocytes were elevated (4.8%) and the spherocytes were increased (10%). Direct antiglobulin test was negative. Biochemical test indicated a slight elevation of bilirubin, mainly indirect reacting (TBIL32.5 μmol/L, IBIL24 μmol/L). The neonatal HS ratio is 4.38, obviously up the threshold. Meanwhile, a de novo ANK1 mutation (exon 25:c.2693dupC:p.A899Sfs*11) was identified by next-generation sequencing (NGS). Thus, hereditary spherocytosis was finally diagnosed.
Conclusions
Gene detection should be considered in some hemolytic anemia which is difficult to diagnose by routine means. We identified a novel de novo ANK1 heterozygous frameshift mutation in a Yi nationality patient while neither of his parents carried this mutation.
Similar content being viewed by others

Background
Hereditary spherocytosis (HS) results from defects in erythrocyte membrane proteins characterized by hemolysis, anemia, jaundice, gallstones and splenomegaly [1, 2]. The severity depends on rate of hemolysis, degree of compensation of anemia by reticulocytosis. The clinical manifestations vary widely, ranging from nearly asymptomatic to transfusion-dependent or severe life-threatening anemia. In the neonatal period, the major clinical manifestations are jaundice and anemia. Splenomegaly and spherocytes are rarely observed [3]. Therefore, it’s difficult to diagnose in neonates. Even during the first year of life, approximately 34% affected infants are diagnosed [4].
Previous researches have shown that mutations in ANK1 (ankyrin 1), SPTB (spectrin, beta, erythrocytic), SPTA1 (spectrin alpha, erythrocytic 1), SLC4A1 (solute carrier family 4, member 1, or band 3), and EPB42 (erythrocyte membrane protein band 4.2) are associated with HS [5]. The mutations of these genes lead to the normally double-concave disc-shaped red cells become spherical, fragile red cells [6]. ANK1 located on 8p11.21, its mutations include nonsense, splicing or frameshift and affect about half of patients with HS [7].
In most cases, HS is usually diagnosed on the basis of a positive family history, increased osmotic fragility, hyperbilirubinemia, reticulocytosis, splenomegaly and spherocytes on peripheral blood smears [8]. The neonatal HS ratio which is calculated by dividing the mean corpuscular hemoglobin concentration (MCHC) by the mean corpuscular volume (MCV) provides valuable information for the physicians. In the index infant, the ratio was > 0.36, which points towards a diagnosis of HS (97% sensitivity, 99% specificity) [3]. However, mild or atypical cases are difficult to identify because of the limitations of the classical approaches. It has been reported that approximately 10% patients of HS may be misdiagnosed due to the lack of the typical sphere-shaped erythrocytes in the peripheral blood [9].
In this report, the next-generation sequencing (NGS) was used to analyze a Chinese family with an infant with unknown causes of hemolysis, and we identified a de novo ANK1 mutation responsible for HS.
Case presentation
The patient came from a Chinese family in Yunnan province. He showed anemia and jaundice without other pathological symptoms or signs when he was born. Gallstones were identified by B-ultrasound scanning. The results of blood tests before transfusion were shown in the Table 1 which indicated that the child suffered from neonatal moderate hemolytic anemia and hyperbilirubinemia. He had received two blood transfusions in neonates. Autoimmune antibody tests were negative. The neonatal HS ratio is 3.67, only slightly up the threshold.
At the age of 3 months, the patient received transfusion because of anemia (Hb: 72 g/L). Before blood transfusion, the following tests were performed. A glucose-6-phosphate dehydrogenase (G-6-PD) screening test and Coombs’ test were negative. Hemoglobin electrophoresis, α and β globin genetic analysis excluded α and β thalassemia. Bone marrow aspiration smears indicated normoblastic hyperplasia. The erythrocyte osmotic fragility wasn’t increased (hemolysis begins: 4.8 g/L referencing 4.4–4.8 g/L; hemolysis completes: 3.2 g/L referencing 2.8–3.2 g/L). Hepatosplenomegaly and spherocytes in peripheral blood smear weren’t observed. The parents were devoid of anemia, jaundice, splenectomy, or early gallstones.
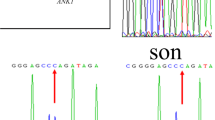
At the age of 6 months, the patient received transfusion again (Hb: 76 g/L). To identify the cause of unexplained hemolysis, we performed genetic analysis by next-generation sequencing according to the methods of He’s [1]. Blood samples were collected before transfusion. Written informed consent for genetic testing was obtained from the parents. DNA was extracted from peripheral blood and 566 genes associated with hematopathy diseases were selected to detect. We detected a mutation in ANK1 (NM_001142446: exon 25:c.2693dupC: p.A899Sfs*11) in the patient that could be implicated in the patient’s phenotype. The variation resulted in an amino acid change and affected protein function. The mutation was a heterozygous mutation. (Fig. 1a). According to the ESP6500 database, the human genome database and the dbSNP database, this mutation hasn’t been reported previously. However, his parents did not carry this mutation (Fig. 1a-b). Therefore, the patient has a de novo mutation in ANK1. In addition, the prevalence of this mutation is extremely low in the population.

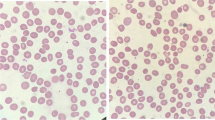
The ANK1 mutation and pedigree. a Sanger sequencing identified an ANK1 c.2693dupC mutation in the patient. An arrow indicates the mutation site. b Family tree and the genotype at the ANK1 c.2693dupC. Squares and circle denote males and female, respectively. Black symbols denote patient with gene mutation. c Peripheral blood smears of the patient. Spherocytes are indicated by arrows
At the age of 11 months, pre-transfusion values of the routine blood examination were shown in Table 2 which indicated that he suffered from moderate hemolytic anemia with hyperbilirubinemia. The spherocytes on peripheral blood smear were 10% (Fig. 1c). The neonatal HS ratio is 4.38, obviously up the threshold. He needed a regular transfusion therapy at an interval of 2–3 months with a hemoglobin level of 70–80 g/L before transfusion. The growth and development of the boy are normal. Partial splenic embolization will be planned (Additional file 1).
Discussion
In this report, we described a Chinese family with a patient affected by HS. A de novo mutation (exon 25: c.2693dupC:p.A899Sfs*11) causing an amino acid change in exon 25 of ANK1 was found through next-generation sequencing followed by Sanger sequencing to verify the relationship between the ANK1 mutation and HS.
HS is an inherited disorder characterized by the presence of spherical-shaped blood cells [10, 11]. Approximately two-thirds of cases are autosomal dominant (AD), and the remaining cases represent autosomal recessive (AR) inheritance or de novo mutations in some sporadic cases [12]. Cases of HS are sporadic in China [1]. However, in some countries and continents, many HS patients have no family history [13].
ANK1 mutations are responsible for the majority of cases of HS. A heterozygous ANK1 IVS3-2A > C mutation that may lead to exon 4 skipping of the ANK1 gene and cause HS was recently identified in a 7-year-old girl [14]. A patient with HS who was diagnosed clinically with only 10% spherical-shaped erythrocytes in the peripheral blood was identified to have a novel de novo ANK1 c.4276C > T (p.R1426*) nonsense mutation, while neither of his parents or his young brother carried this mutation [15]. A 6-year-old girl who was clinically diagnosed with HS carried a de novo nonsense ANK1 mutation (c.796G > T, p.Glu266X), a single-nucleotide change from G to T, which caused a substitution from glutamic acid to a premature stop at codon 266 [16].
ANK1 is an important red cell membrane protein which plays a vital role in the maintenance of erythrocyte membrane integrity [17, 18]. ANK1 consists of three structural domains: a multiple repeats N-terminal domain, a spectrin-binding center region and a regulatory C-terminal domain [7, 19, 20]. Mutations in the spectrin-binding domain and regulatory C-terminal domains result in the most severe anemia compared with those located in the other domains [19, 21].
In our study, the patient had suffered from unexplained hemolysis and hyperbilirubinemia since the neonatal period. At the age of 3 months, hepatosplenomegaly and spherocytes which is critical to diagnose the HS weren’t observed [22]. Erythrocyte osmotic fragility was negative. It was difficult to diagnose HS which originates from mutations in the genes coding for RBC membrane components. Gene detection is the principle method for cases with no family history of HS, especially in some atypical cases. NGS is able to provide a thorough genetic analysis and identify which candidate gene is responsible for the disease [23,24,25]. Therefore, with this patient we used an NGS panel for the analysis of 566 genes responsible for hematological disorders. The genetic tests showed a de novo ANK1 c.2693dupC (p.A899Sfs*11) frameshift mutation which was not found in the 1000G, ExAC, or HGMD databases. Moreover, this mutation was located in the spectrin-binding domain, which might cause HS. At the age of 11 months, spherocytes on a peripheral blood smear were 10% and the neonatal HS ratio was 4.38. Our report strongly suggests that in infants, it is important for the physicians to monitor the sphere-shaped erythrocytes and the neonatal HS ratio when the patients are at risk for HS. Regrettably, eosin-5′-maleimide binding assay with flow cytometry is the test of choice to diagnose HS but isn’t available in our laboratory.
In conclusion, this report suggests that genetic detecting should be considered for some unexplained hemolytic diseases. Meanwhile, we identified a novel de novo ANK1 c.2693dupC (p.A899Sfs*11) heterozygous frameshift mutation in a Yi nationality patient. However, the pathogenesis of this ANK1 mutation should be explored further to improve the diagnosis and treatment of HS.
Abbreviations
- AD:
-
Autosomal dominant
- AR:
-
Autosomal recessive
- EPB42:
-
Erythrocyte membrane protein band 4.2
- HS:
-
Hereditary spherocytosis
- NGS:
-
Next-generation sequencing
- RBC:
-
Red blood cell
- SLC4A1:
-
Solute carrier family 4, member 1
- SPTA1:
-
Spectrin alpha, erythrocytic 1
- SPTB:
-
Spectrin, beta, erythrocytic
References
He Y, Jia S, Dewan RK, Liao N. Novel mutations in patients with hereditary red blood cell membrane disorders using next-generation sequencing. Gene. 2017;627:556–62.
Perrotta S, Gallagher PG, Mohandas N. Hereditary spherocytosis. Lancet. 2008;372:1411–26.
Christensen RD, Yaish HM, Gallagher PG. A pediatrician's practical guide to diagnosing and treating hereditary spherocytosis in neonates. Pediatrics. 2015;135:1107–14.
Delhommeau F, Cynober T, Schischmanoff PO, Rohrlich P, Delaunay J, Mohandas N, et al. Natural history of hereditary spherocytosis during the first year of life. Blood. 2000;95:393–7.
He BJ, Liao L, Deng ZF, Tao YF, Xu YC, Lin FQ. Molecular genetic mechanisms of hereditary spherocytosis: current perspectives. Acta Haematol. 2018;139:60–6.
Manciu S, Matei E, Trandafir B. Hereditary spherocytosis - diagnosis, surgical treatment and outcomes. A literature review. Chirurgia (Bucur). 2017;112:110–6.
Gallagher PG. Hematologically important mutations: ankyrin variants in hereditary spherocytosis. Blood Cells Mol Dis. 2005;35:345–7.
Bianchi P, Fermo E, Vercellati C, Marcello AP, Porretti L, Cortelezzi A, et al. Diagnostic power of laboratory tests for hereditary spherocytosis: a comparison study in 150 patients grouped according to molecular and clinical characteristics. Haematologica. 2012;97:516–23.
Christensen RD, Henry E. Hereditary spherocytosis in neonates with hyperbilirubinemia. Pediatrics. 2010;125:120–5.
Delaunay J. The molecular basis of hereditary red cell membrane disorders. Blood Rev. 2007;21:1–20.
Llaudet-Planas E, Vives-Corrons JL, Rizzuto V, Gomez-Ramirez P, Sevilla Navarro J, Coll Sibina MT, et al. Osmotic gradient ektacytometry: a valuable screening test for hereditary spherocytosis and other red blood cell membrane disorders. Int J Lab Hematol. 2018;40:94–102.
Iolascon A, Avvisati RA. Genotype/phenotype correlation in hereditary spherocytosis. Haematologica. 2008;93:1283–8.
Da Costa L, Galimand J, Fenneteau O, Mohandas N. Hereditary spherocytosis, elliptocytosis, and other red cell membrane disorders. Blood Rev. 2013;27:167–78.
Wang X, Mao L, Shen N, Peng J, Zhu Y, Hu Q, et al. An ANK1 IVS3-2A>C mutation causes exon 4 skipping in two patients from a Chinese family with hereditary spherocytosis. Oncotarget. 2017;8:113282–6.
Wang X, Yi B, Mu K, Shen N, Zhu Y, Hu Q, et al. Identification of a novel de novo ANK1 R1426* nonsense mutation in a Chinese family with hereditary spherocytosis by NGS. Oncotarget. 2017;8:96791–7.
Guan H, Liang X, Zhang R, Wang H, Liu W, Zhang R, et al. Identification of a de novo ANK1 mutation in a Chinese family with hereditary spherocytosis. Hematology. 2018;23:357–61.
Han JH, Kim S, Jang H, Kim SW, Lee MG, Koh H, et al. Identification of a novel p.Q1772X ANK1 mutation in a Korean family with hereditary spherocytosis. PLoS One. 2015;10:e0131251.
Eber S, Lux SE. Hereditary spherocytosis--defects in proteins that connect the membrane skeleton to the lipid bilayer. Semin Hematol. 2004;41:118–41.
Park J, Jeong DC, Yoo J, Jang W, Chae H, Kim J, et al. Mutational characteristics of ANK1 and SPTB genes in hereditary spherocytosis. Clin Genet. 2016;90:69–78.
Wang C, Wei Z, Chen K, Ye F, Yu C, Bennett V, et al. Structural basis of diverse membrane target recognitions by ankyrins. Elife. 2014;3:e04353.
Hughes MR, Anderson N, Maltby S, Wong J, Berberovic Z, Birkenmeier CS, et al. A novel ENU-generated truncation mutation lacking the spectrin-binding and C-terminal regulatory domains of Ank1 models severe hemolytic hereditary spherocytosis. Exp Hematol. 2011;39:305–20 20 e1–2.
Arora RD, Dass J, Maydeo S, Arya V, Kotwal J, Bhargava M. Utility of mean sphered cell volume and mean reticulocyte volume for the diagnosis of hereditary spherocytosis. Hematology. 2018;23:413–6.
Del Orbe BR, Arrizabalaga B, De la Hoz AB, Garcia-Orad A, Tejada MI, Garcia-Ruiz JC, et al. Detection of new pathogenic mutations in patients with congenital haemolytic anaemia using next-generation sequencing. Int J Lab Hematol. 2016;38:629–38.
Lim EC, Brett M, Lai AH, Lee SP, Tan ES, Jamuar SS, et al. Next-generation sequencing using a pre-designed gene panel for the molecular diagnosis of congenital disorders in pediatric patients. Hum Genomics. 2015;9:33–41.
Russo R, Andolfo I, Manna F, Gambale A, Marra R, Rosato BE, et al. Multi-gene panel testing improves diagnosis and management of patients with hereditary anemias. Am J Hematol. 2018;93:672–82.
Acknowledgements
We thank the patient and his families for their cooperation.
Funding
This work was supported by the Yunnan Province Education Department Fund Project (grant No: 2014Z053), the Yunnan Health Science and Technology Project (grant No: 2016NS126), and the Kunming Health Science and Technology Personnel Training Project (grant No: sw-78). The funding bodies have the role in the design of the study, collection, analysis, and interpretation of data and in writing the manuscript.
Availability of data and materials
All data used during the current study are included in this published article.
Author information
Authors and Affiliations
Contributions
TLH, XT, and YHY designed the study. BHS, QLL, CYS, YBL and YL treated patients and helped draft the manuscript. CHY and NL participated in the collection of patients’ data. TLH and XWZ analysed the data and wrote the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
This study was approved by the Ethics committee of the Kunming Children’s Hospital. Written informed consent about genetic testing and taking part in this study were obtained from the parents of the patient.
Consent for publication
Written informed consent for publication this case report and accompanying images were obtained from the patients’ parents.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional file
Additional file 1:
Timeline of this case. of a de novo ANK1 mutation associated to hereditary spherocytosis: a case report. (DOCX 16 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Huang, TL., Sang, BH., Lei, QL. et al. A de novo ANK1 mutation associated to hereditary spherocytosis: a case report. BMC Pediatr 19, 62 (2019). https://doi.org/10.1186/s12887-019-1436-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12887-019-1436-4