
Abstract
Background
Xp11.2 translocation renal cell carcinoma (RCC) is a rare variety of a kidney neoplasm. We report a case of bilateral Xp11.2 translocation RCC occurring metachronously and discuss this very rare entity with reference to the literature.
Case presentation
The patient was a 56-year-old woman who presented with a right renal tumor. The patient had undergone left radical nephrectomy 7 years previously, which resulted in a histopathological diagnosis of clear cell RCC. Open right partial nephrectomy was performed under the presumptive diagnosis of recurrence of clear cell RCC. The present right renal tumor was pathologically diagnosed Xp11.2 translocation RCC. More than 70% of the tumor cells in the present right tumor were strongly positive for transcription factor E3 (TFE3) expression by immunohistochemical analysis with an anti-TFE3 antibody. A break-apart of the TFE3 genes in the bilateral tumors was identified by fluorescence in situ hybridization analysis. Real time-polymerase chain reaction analysis for the alveolar soft part sarcoma locus-TFE3 fusion gene was performed, which gave a positive result in the bilateral tumors. Pathological comparison of each of the tumors might lead to a final diagnosis of Xp11.2 translocation RCC occurring metachronously.
Conclusions
We present the bilateral Xp11.2 translocation RCC. A combination of immunohistochemical, cytogenetic and molecular biological approaches allowed the final diagnosis of such a rare RCC.
Similar content being viewed by others


Background
Xp11.2 translocation renal cell carcinoma (RCC) is a rare variety of kidney neoplasm that represents approximately 1% of RCC [1]. It is a clinically identified malignant neoplasm of kidney with an advanced stage and a poorer prognosis than conventional clear cell RCC [2]. Xp11.2 translocation RCC results from gene fusions between the transcription factor E3 (TFE3) gene located on chromosome Xp11.2 and various fusion partners. These chimeric gene fusions result in overexpression of fusion proteins that contain the C-terminal portion of TFE3. The TFE3 fusion partner genes have been recently well characterized. A common fusion partner gene is alveolar soft part sarcoma critical region 1 (ASPSCR1), der(17)t(X;17)(p11.2;q25). This unbalanced translocation results in fusion of the TFE3 gene, a member of the basic-helix-loop-helix family of transcription factors, on Xp11.2, to a novel gene named alveolar soft part sarcoma locus (ASPL) on 17q25 [3]. Other common fusion genes are papillary renal cell carcinoma-TFE3 (PRCC-TFE3), t(X;1)(p11.2;q21.2) and PTB-associated splicing factor-TFE3 (PSF-TFE3), t(X;1)(p11.2;p34) [4, 5]. Less commonly observed gene fusions are NonO-TFE3 inv.(X)(p11.2;q12) and clathrin heavy chain-TFE3 (CLTC-TFE3), (X;17)(p11.2;q23) [6, 7].
In this report, we present an extremely rare case of bilateral Xp11.2 translocation RCC occurring metachronously, and discuss the uncommon features of this case as determined by histopathological, cytogenetic and molecular approaches.
Case presentation
A 56-year-old woman was introduced to Kochi Medical School from a private hospital for right renal tumor detected by abdominal computed tomography (CT). She had been undergone radical nephrectomy for left renal cell carcinoma (RCC) 7 years before. An abdominal CT of the present tumor revealed a right renal tumor, 5.3 cm in diameter, showing poorly-defined margins, irregular contrast and no findings of metastases (Fig. 1a, b). An abdominal CT that was performed 7 years ago revealed a left renal tumor, 7.0 cm in diameter, showing well-defined margins, irregular contrast and no findings of metastases, diagnosed clinical stage T1b N0 M0 left RCC (Fig. 1c, d). She did not have any other medical history or family history.

Pre-operative diagnostic imaging of the present and the previous tumor. Abdominal CT images of the present right renal tumor (a, b) and the previous left renal tumor (c, d). The present right renal tumor was 5.3 cm in diameter and showed poorly-defined margins and an irregular contrast. The previous left renal tumor was 7.0 cm in diameter, and showed well-defined margins and an irregular contrast
Open right partial nephrectomy was performed under a presumed diagnosis of clinical stage T1b N0 M0 right RCC, recurrent or due to metastasis from the previous left tumor. The tumor was a macroscopically well-circumscribed solid mass. The cross-sectional surface was lobulated and heterogenously yellow to brown with bleeding and necrosis (Fig. 2). Microscopically, the tumor showed an alveolar growth pattern admixed with eosinophilic and clear cytoplasm. Papillary architecture was also focally seen. In some areas, eosinophilic coarse granules were identified in the tumor cytoplasm. Pathological stage was pT1b pN0 with negative surgical margin. Nuclear Grade corresponded to largely Fuhrman Grade 3 and partly Grade 4. Hyaline nodules and psammoma bodies were observed in the stroma. Immunohistochemically, the tumor cells showed diffuse positivity for renal cell carcinoma-maker (RCCMa, PN-15, 1: 100, Cell Marque, CA, USA) and cluster differentiation (CD)10 (56C16, prediluted, Novocastra Laboratories Ltd., Newcastle, UK) and negativity for Cathepsin K (3F9, Abcam, Tokyo, JP), Melanosome (Human melanoma black; HMB45, prediluted, DAKO, Glostrup, Denmark), Melan A (A103, 1: 100, Novocastra Laboratories Ltd., Newcastle, UK), and alpha smooth muscle actin (data not shown). Seventy percent of neoplastic cell nuclei stained positive for TFE3 (MRQ-37, prediluted, Ventana Medical Systems, Inc., Tucson, AZ), with a staining intensity of (moderate) 2+ to (strong) 3+ (Fig. 3). Staining for transcription factor EB (TFEB, polyclonal, V-17, 1: 400, Santa Cruz, Biotechnology, Inc., Dallas, TX) was generally negative (data not shown).

Macroscopic findings of the present right tumor. The present right tumor resected by partial nephrectomy was macroscopically a well-marginated solid mass. The cross-sectional surface was lobulated and heterogenously yellow to brown with bleeding and necrosis

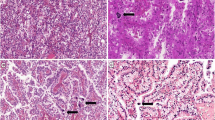
Microscopic findings of the present right tumor and previous left tumor. HE staining of the present right tumor mostly showed an alveolar growth pattern (× 100; a) with cells composed clear cytoplasm (× 100; b). Very large tumor cells (× 100; c) and a papillary growth pattern (× 100; d) were focally observed. Moderate to strong immunostaining of TFE3 in the nuclei of tumor cells was seen (× 200; e). HE staining of the previous left tumor showed an alveolar growth pattern (× 100; f), pale eosinophilic cytoplasm (× 100; g) and very large tumor cells (× 100; h). Dedifferentiated sarcomatoid features were partially observed (× 100; i). Moderate to strong immunostaining of TFE3 in the nuclei of tumor cells was seen (× 200; j)
Hematoxylin and eosin, and immunohistochemical stains from the previous tumor were retrospectively reviewed. In H and E staining, tubular, papillary, and alveolar growth patterns were noted admixed with eosinophilic and clear cytoplasm. Additionally, very large tumor cells were seen and dedifferentiation with a discohesive area and rhabdoid features was also noted. Necrosis and hemorrhage were present. Pathological stage was pT1b pN0. Nuclear Grade corresponded to Fuhrman Grade 4. Small venous invasion by carcinoma cells was seen. Neoplastic cells showed diffuse immunohistochemical expression of RCCMa, CD10, Alpha-Methylacyl-CoA Race (AMACR; P504S, 13H4, 1: 100, DAKO, Glostrup, Denmark) and negative results for cytokeratin 7, Carbonic Anhydrase IX (CA9, D47G3, Cell Signaling, MA, USA), HMB45, Melan A and Cathepsin K (data not shown). TFE3 was positively stained in the nuclei of 5% of neoplastic cells with a staining intensity of 2+ to 3+ (Fig. 3).
We performed a dual-color, break-apart fluorescence in situ hybridization (FISH) assay to identify the chromosomal break point of TFE3 in paraffin-embedded tissue [8]. Briefly, the break-apart FISH assay with probes upstream and downstream to TFE3 showed red and green signals. A fused or closely approximated green-red signal pattern was interpreted as a normal result, whereas a TFE3 fusion resulted in a split-signal pattern. Signals were considered to be split when the green and red signals were separated by a distance of more than 2 signal diameters. For each tumor, a minimum of 100 tumor cell nuclei were examined under fluorescence microscopy at × 1000 magnification. Only nonoverlapping tumor nuclei were evaluated. Positive findings were defined as more than 10% of the tumor nuclei showing the split-signal pattern [9]. The TFE3 gene showed gene splitting in 71.55% of 130 neoplastic cells and in 76.82% of 233 neoplastic cells in the present and the previous tumor, respectively. Typical TFE3 break-apart signals of the present and previous tumors are presented in Fig. 4.

FISH analysis of TFE3 gene splitting of the present (a and b) and previous (c and d) tumor cells. A pair of split signals of TFE3 genes are shown as red and blue fusion fluorescence at high magnification (white arrow head). A green signal shows fused normal fluorescence of red and blue (white arrow)
Total RNA was extracted from formalin fixed paraffin embedded tissue of the previous tumor and from frozen tissue of the present tumor using a standard organic extraction method (MACHEREY-NAGEL, Germany and QIAGEN, Germany, respectively). ASPL-TFE3 fusion transcripts were detected using an ASPL forward primer: 5’-AAAGAAGTCCAAGTCGGGCCA-3′ and a TFE3 exon 4 reverse primer: 5’-CGTTTGATGTTGGGCAGCTCA-3′. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) transcripts were detected using the forward: 5’-CGGATTTGGTCGTATTGG-3′ and reverse: 5’-TCCTGGAAGATGGTGATG-3’ GAPDH primers [2]. The ASPL-TFE3 fusion gene was detected in the tissue from the present and the previous tumor but was not detected in the normal tissue. GAPDH that was used as a loading control was detected in each reaction (Fig. 5).

RT-PCR of ASPL-TFE3 fusion genes of the previous and present tumor tissue. Previous and present tumor expressed ASPL-TFE3 fusion gene, but not normal kidney tissue of the present. GAPDH expression of each tissue was confirmed as housekeeping gene
There is a no evidence of recurrence at 8 months postoperatively.
Discussion and conclusions
Xp.11.2 translocation RCC is a rare variety of RCC that was first described in 1995 by Dijkhuizen et al [10]. It is categorized as a separate entity in the 2004 World Health Organization classification of tumors of the urinary system [11]. This type of RCC frequently affects children and adolescents. Our patient was diagnosed as Xp11.2 translocation RCC at the ages of 49 and 56 years-of age. Patients of middle age and over with Xp11.2 translocation RCC have rarely been reported [12]. There is variation in the histological features of Xp11.2 translocation RCC such as clear cell, papillary, alveolar, and nested. Seventy five percent of adult Xp11.2 translocation RCC is predominately the clear cell histological type, whereas most pediatric cases consist of papillary histological features [13]. In our present left tumor, clear cell features were the predominant type, followed by alveolar and papillary. Also, characteristic findings such as eosinophilic, voluminous and clear cytoplasm led to the diagnosis of adult Xp11.2 translocation RCC with ASPL-TFE3 fusion.
Positive immunostaining of TFE3 and negative staining of TFEB excluded 6p21 translocation RCC. The results of positive immunostaining of RCCMa and CD10, and negative staining of Cathepsin K, HMB45 or Melan A also led to a diagnosis of ASPL-TFE3 fusion. Most previous cases of Xp11.2 translocation RCC showed positive staining of RCCMa and CD10. Negative staining of Cathepsin K supported ASPL-TFE3 fusion, while tumors with PRCC-TFE3 fusion mostly display positive staining of Cathepsin K [14]. Melanin may be upregulated in Xp11.2 translocation RCC with PSF-TFE3 and CLTC-TFE3 [7, 15]. Melanosome and Melanin A staining have not been reported in Xp11.2 translocation RCC with ASPL-TFE3 and PRCC-TFE3 fusion.
Our case is the first report of bilateral Xp11.2 translocation RCC. The next step was to consider whether the present tumor was due to metastasis from the previous tumor. Microscopic findings of the previous tumor revealed very large tumor cells, a discohesive area and rhabdoid features meaning more dedifferentiation and aggressiveness compared with the present tumor. These data suggest that these tumors occurred metachronously, and that the present tumor was not due to metastasis of the previous tumor.
We demonstrated the presence of the ASPL-TFE3 fusion gene that is the most common chimeric fusion gene resulting from the chromosome translocation that is characteristic of ASPSCR1. By using RT-PCR we also demonstrated that the tumors were negative for the PRCC-TFE3, PSF-TFE3, CLTC-TFE3 or NonO-TFE3 fusion genes. Analysis of von Hippel Lindau tumor suppressor gene mutation by direct sequencing and multiplex ligation-dependent probe amplification methods also gave a negative result (data not shown) [16]. These data supported the final diagnosis of bilateral Xp11.2 translocation RCC with ASPL-TFE3 fusion.
In conclusion, we present a case that may be diagnosed as bilateral Xp11.2 translocation RCC metachronously occurring. Immunohistochemical, cytogenetic and molecular findings allows the differential diagnosis of kidney neoplasms such as Xp11.2 translocation RCC.
Abbreviations
- AMACAR:
-
Alpha-Methylacyl-CoA Race
- ASPSCR1:
-
Alveolar soft part sarcoma critical region 1
- CA9:
-
Carbonic Anhydrase IX
- CD:
-
Cluster differentiation
- CLTC:
-
Clathrin heavy chain
- CT:
-
Computed tomography
- FISH:
-
Fluorescence in situ hybridization
- GAPDH:
-
Glyceraldehyde-3-phosphate dehydrogenase
- MRI:
-
Magnetic resonance imaging
- PRCC:
-
Papillary renal cell carcinoma
- PSF:
-
PTB-associated splicing factor
- RCC:
-
Renal cell carcinoma
- RCCMa:
-
Renal cell carcinoma maker
- RT-PCR:
-
Reverse transcription-polymerase chain reaction
- TFE3:
-
Transcription factor E3
- TFEB:
-
Transcription factor EB
References
Macher-Goeppinger S, Roth W, Wagener N, Hohenfellner M, Penzel R, Haferkamp A, Schirmacher P, Aulmann S. Molecular heterogeneity of TFE3 activation in renal cell carcinomas. Mod Pathol. 2012;25:308–15.
Zhong M, De Angelo P, Osborne L, Paniz-Mondolfi AE, Geller M, Yang Y, Linehan WM, Merino MJ, Cordon-Cardo C, Cai D. Translocation renal cell carcinomas in adults: a single-institution experience. Am J Surg Pathol. 2012;36:654–62.
Argani P, Antonescu CR, Illei PB, Lui MY, Timmons CF, Newbury R, Reuter VE, Garvin AJ, Perez-Atayde AR, Fletcher JA, Beckwith JB, Bridge JA, Ladanyi M. Primary renal neoplasms with the ASPL-TFE3 gene fusion of alveolar soft part sarcoma: a distinctive tumor entity previously included among renal cell carcinomas of children and adolescents. Am J Pathol. 2001;159:179–92.
Sidhar S. The t(X;1)(p11.2;q21.2) translocation in papillary renal cell carcinoma fuses a novel gene PRCC to the TFE3 transcription factor gene. Hum Mol Genet. 1996;5:1333–8.
Mathur M, Das S, Samuels HH. PSF-TFE3 oncoprotein in papillary renal cell carcinoma inactivates TFE3 and p53 through cytoplasmic sequestration. Oncogene. 2003;22:5031–44.
Clark J, Lu YJ, Sidhar SK, Parker C, Gill S, Smedley D, Hamoudi R, Linehan WM, Shipley J, Cooper CS. Fusion of splicing factor genes PSF and NonO (p54nrb) to the TFE3 gene in papillary renal cell carcinoma. Oncogene. 1997;15:2233–9.
Argani P, Lui MY, Couturier J, Bouvier R, Fournet J-C, Ladanyi M. A novel CLTC-TFE3 gene fusion in pediatric renal adenocarcinoma with t(X;17)(p11.2;q23). Oncogene. 2003;22:5374–8.
Kim SH, Choi Y, Jeong HY, Lee K, Chae JY, Moon KC. Usefulness of a break-apart FISH assay in the diagnosis of Xp11.2 translocation renal cell carcinoma. Virchows Arch. 2011;459:299–306.
Rao Q, Williamson SR, Zhang S, Eble JN, Grignon DJ, Wang M, Zhou XJ, Huang W, Tan PH, Maclennan GT, Cheng L. TFE3 break-apart FISH has a higher sensitivity for Xp11.2 translocation-associated renal cell carcinoma compared with TFE3 or cathepsin K immunohistochemical staining alone: expanding the morphologic spectrum. Am J Surg Pathol. 2013;37:804–15.
Dijkhuizen T, van den Berg E, Wilbrink M, Weterman M, Geurts van Kessel A, Störkel S, Folkers RP, Braam A, de Jong B. Distinct Xp11.2 breakpoints in two renal cell carcinomas exhibiting X;autosome translocations. Genes Chromosomes Cancer. 1995;14:43–50.
Chan TY. World Health Organization classification of tumours: Pathology and Genetics of Tumours of the Urinary System and Male Genital Organs. Urology. 2005;65:214-5.
Argani P, Olgac S, Tickoo SK, Goldfischer M, Moch H, Chan DY, Eble JN, Bonsib SM, Jimeno M, Lloreta J, Billis A, Hicks J, De Marzo AM, Reuter VE, Ladanyi M. Xp11 translocation renal cell carcinoma in adults: expanded clinical, pathologic, and genetic spectrum. Am J Surg Pathol. 2007;31:1149–60.
Renshaw AA, Granter SR, Fletcher JA, Kozakewich HP, Corless CL, Perez-Atayde AR. Renal cell carcinomas in children and young adults: increased incidence of papillary architecture and unique subtypes. Am J Surg Pathol. 1999;23:795–802.
Martignoni G, Gobbo S, Camparo P, Brunelli M, Munari E, Segala D, Pea M, Bonetti F, Illei PB, Netto GJ, Ladanyi M, Chilosi M, Argani P. Differential expression of cathepsin K in neoplasms harboring TFE3 gene fusions. Mod Pathol. 2011;24:1313–9.
Chang I-W, Huang H-Y, Sung M-T. Melanotic Xp11 translocation renal cancer: a case with PSF-TFE3 gene fusion and up-regulation of melanogenetic transcripts. Am J Surg Pathol. 2009;33:1894–901.
Hes FJ, van der Luijt RB, Janssen ALW, Zewald RA, De Jong GJ, Lenders JW, Links TP, Luyten GPM, Sijmons RH, Eussen HJ, Halley DJJ, Lips CJM, Pearson PL, van den Ouweland AMW, Majoor-Krakauer DF. Frequency of Von Hippel-Lindau germline mutations in classic and non-classic Von Hippel-Lindau disease identified by DNA sequencing, southern blot analysis and multiplex ligation-dependent probe amplification. Clin Genet. 2007;72:122–9.
Acknowledgements
None.
Funding
There are no funding sources for this study.
Availability of data and materials
Requests for the study materials and dataset used to support the conclusions of this article should be directed to the corresponding author.
Author information
Authors and Affiliations
Contributions
TK drafted the report, contributed to the concept, and cared for the patient. TK, HS, SF, MC, and IY cared for the patient. NK and MH generated the histopathological and cytogenetic results. CK generated the molecular results. TS and KI contributed to the concept and design, and approved the final version of the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This study has been approved by the Ethics Committee of Kochi Medical School.
Consent for publication
Written informed consent was obtained from the patient for publication of this case report and any accompanying images. A copy of the written consent is available for review by the Editor of this journal.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Karashima, T., Kuno, T., Kuroda, N. et al. Bilateral Xp11.2 translocation renal cell carcinoma: a case report. BMC Urol 18, 106 (2018). https://doi.org/10.1186/s12894-018-0419-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12894-018-0419-3