
Abstract
Background
The morphological identification of different Hippophae species (Shaji) was difficult. This study aims to discriminate between medicinal and non-medicinal Hippophae species by DNA barcodes, the ITS2, psbA-trnH, and a combination of ITS2 and psbA-trnH (ITS2 + psbA-trnH).
Methods
DNA was extracted from the dried fruit samples. Primer pairs ITS2F/3R for ITS2 and psbAF/trnHR for psbA-trnH were used for PCR amplification. The purified PCR products were bidirectionally sequenced. Genetic distances were calculated according to the Kimura 2 parameter model and phylogenetic tree was constructed based on neighbor-joining (NJ) method, barcoding gap was also analyzed to assess identification efficiency.
Results
Amplification and sequencing efficiencies for both ITS2 and psbA-trnH were 100 %. Sequence data revealed that ITS2 + psbA-trnH was the most suitable candidate barcode at the species and subspecies level. The closely related Hippophae species were effectively differentiated in the NJ tree.
Conclusion
The combination of the two loci, ITS2 + psbA-trnH is applicable to the identification of medicinal and non-medicinal Hippophae species.
Similar content being viewed by others

Background
In Hippophae (Fam. Elaeagnaceae) (Shaji), seven species and 11 subspecies have been identified worldwide [1, 2]. In China, there are seven species and seven subspecies of Hippophae, which are mainly distributed from the Hengduan Mountains to the Qinghai-Tibet Plateau [3–6].
Both the fruits and leaves of Hippophae species possess abundant nutritional properties and bioactive compounds [7–9], i.e., high level of vitamin C [10, 11]. Hippophae species have been widely used in food, pharmaceutical, and health care products [12, 13].
Medicinal Hippophae species are used in Chinese medicine (CM) and Tibetan medicine for their antioxidant and anti-tumor activities, to improve lipid metabolism and enhance immunity [14, 15]. The dried fruits are used as remedies for cardiovascular disease; liver, stomach, and spleen disorders; as well as lung and throat phlegm [14–18]. Hippophae species are sometimes misidentified because of the similarities in vegetative morphology [2, 5]. Furthermore, the fruits of different species are labeled with the same name and mainly sold or used in the dried form or as powders. Therefore, different species cannot be identified by only morphological characteristics and accurate identification methods are needed.
With the advantages of high PCR amplification efficiencies, DNA sequencing success rates, and discrimination power, DNA barcoding has become popular with taxonomists and has gained wide acceptance as a standard and effective method in biodiversity research and conservation genetics. It can be applied without the limitation of the samples development stages, parts and gathering time, compared with the conventional identification method [19, 20]. The Consortium for the Barcode of Life (CBOL) Plant Working Group initially recommended the coding plastid regions rbcL and matK as core barcodes for plant species [21]. However, two barcodes are not precise enough because of the low identification rate [22, 23]. The psbA-trnH, ITS, and ITS2 were subsequently suggested [23–25]. Additionally, the amplification efficiency of ITS is lower than that of ITS2, because of the multiple functional copies exist in many taxa [26]. Consequently, more than 6600 plant samples that belong to 4800 species from 753 distinct genera have been barcoded by ITS2, with 92.7 % success at the species level [23, 26–34]. The psbA-trnH intergenic spacer region from plastid DNA has also been recommended as a complementary barcode to ITS2 for a broad series of plant taxa [35].
This study aims to discriminate between medicinal and non-medicinal Hippophae species by DNA barcodes, using the ITS2 and psbA-trnH regions as candidate barcodes.
Methods
Materials
Seventy-five samples (Table 1) representing seven species and seven subspecies were collected from the major distribution areas, including Sichuan, Qianghai, Tibet, Yunnan, Beijing, and Xinjiang (China), between May and November 2013. The native wild samples were identified based on morphological features by Professor Zhang Yi referred to previous Hippophae research [4, 5]. Voucher specimens were deposited in the College of Ethnic Medicine, Chengdu University of Traditional Chinese Medicine. All of the ITS2 and psbA-trnH sequences were submitted to GenBank.
Additional sequences belonging to four subspecies of H. rhamnoides which are only found in Europe were obtained from GenBank. In addition, Elaeagnus angustifolia and E. pungens sequences were downloaded from GenBank for use as outgroups in this study.
DNA extraction, PCR amplification, and sequencing
Total genomic DNA was extracted from 50 mg of fruit dried in silica gel. DNA extractions were performed by a Plant Genomic DNA Kit (Tiangen Biotech Co., Beijing, China). Plant material was ground for 2 min at 50 Hz by a DNA Extraction Grinder (Xinzhi Biotech Co., Ningbo, China) as previously described [36]. Primer pairs ITS2F (5′-ATGCGATACTTGGTGTGAAT-3′)/ITS3R (5′-GACGCTTCTCCAGACTACAAT-3′) for ITS2 and psbAF (5′-GTTATGCATGAACGTAATGCTC-3′)/trnHR (5′-CGCGCATGGTGGATTCACAATCC-3′) for psbA-trnH were used for PCR amplification. PCRs were performed in a 25-μL volume, containing 2–3 μL of genomic DNA, 12.5 μL of 2 × EasyTaq PCR MasterMix (Aidlab Biotechnologies Co., Ltd., Beijing, China), 1.0 μL of each primer, and the total volume was adjusted to 25 µL with sterile deionized water. The reaction conditions used were the same as described previously [21, 37]. The PCR products were visualized on agarose gels (the electrophoresis was run in 1 × TBE for 20 min at a constant voltage 120 V). After electrophoresis, purified PCR products were bidirectionally sequenced by the same primers that were used for PCR in a 3730XL sequencer (Applied Biosystems, Foster, CA, USA).
Data analysis
Proofreading and contig assembly of sequencing peak diagrams were performed by CodonCode Aligner 3.7.1 (CodonCode Co., Centreville, MA, USA). The ITS2 region was obtained by the HMMer annotation method based on the Hidden Markov model to remove the 5.8S and 28S sections at both ends of the sequences [38–40]. The psbA-trnH intergenic spacer boundary was determined according to the annotation of similar sequences in GenBank. All sequences were aligned (MUSCLE option) by MEGA 6.0 (Center for Evolutionary Medicine and Informatics, Tempe, AZ, USA) [41], and the genetic distances were calculated according to the Kimura 2 parameter (K2P) model. The distribution of intra- vs. inter-specific variability was assessed by DNA barcoding gaps. A neighbor-joining (NJ) tree was constructed and bootstrap resampling (1000 replicates) was conducted to assess the confidence in phylogenetic analysis by MEGA 6.0. The combination of ITS2 and psbA-trnH (ITS2 + psbA-trnH) was also evaluated by these methods.
Results
Efficiency of DNA extraction and PCR amplification
DNA was successfully extracted from all 75 samples. The PCR amplification success rates for both ITS2 and psbA-trnH were 100 %. All PCR products in correspondence to the ITS2 and psbA-trnH regions were successfully sequenced, and high-quality bidirectional sequences were obtained (Table 2).
Sequence and inter-/intra-specific variation analysis
The sequence characteristics are summarized in Tables 2 and 3. The average G-C contents of the ITS2 and psbA-trnH regions were 52.72 and 25.62 %, respectively. ITS2 sequences ranged from 221 to 223 bp with 43 variable sites; 23 haplotypes were identified, and four indels that were 1–2 bp in length within the aligned 227 bp. The psbA-trnH intergenic spacer region ranged from 300 to 313 bp and showed less variation, with only 19/320 variable sites among 23 haplotypes.
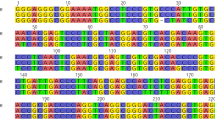
With these ITS2 sequences, both variable sites and deletions provided insight into the identification of H. salifocilia, H. tibetana, and three H. rhamnoides subspecies (Fig. 1). By comparing the sequences, all species except H. salifocilia have deletions from the sites 201–202; in H. tibetana, there were 15 variable sites from site 2 to site 223 which could be used for identification and discrimination from other species. Other important variable sites also provided useful information for species identification and discrimination, such as H. rhamnoides subsp. yunnanensis at site 80, H. rhamnoides subsp. turkestanica at site 153 and site 155, and H. rhamnoides subsp. wolongensis at site 34, site 207, and site 219. With psbA-trnH sequences, the variable sites and insertions enable the identification and differentiation of H. goniocarpa, H. gyantsensis, H. salicifolia, H. tibetana, and two H. rhamnoides subspecies (Fig. 2). When the sequences were compared, most species had no insertions except H. goniocarpa, which had insertions between site 90 and site 91, and H. gyantsensis, which had insertions at site 37 and from site 221 to site 229. Stable sequence variations, which provided useful information for species identification, were found in three species and two subspecies: H. salicifolia at site 38, site 94, and site 211; H. gyantsensis at site 7; H. tibetana at site 65, site 77, and site 302; H. rhamnoides subsp. mongolica at site 64; and H. rhamnoides subsp. turkestanica at site 24.

Variable sites and deletions for Hippophae species based on ITS2 sequences. The specific variable sites and deletions are highlighted

Variable sites and insertions for Hippophae species based on psbA-trnH sequences. The specific variable sites and deletions are highlighted
The intra- and inter-specific K2P genetic distances for ITS2, psbA-trnH, and ITS2 + psbA-trnH are listed in Table 2. In general, the mean inter-specific distances were higher than the mean intra-specific distances for the single-locus barcodes as well as the 2-locus barcode by the K2P model. ITS2 showed the highest intra- and inter-specific distances among the two DNA regions and the combination of the two regions, whereas the psbA-trnH exhibited the lowest intra- and inter-specific distances.
Assessment of barcoding gaps
Ideal barcode sequences should have a distinct inter-specific distance and relatively little intra-specific variation, and there need to be distinct differences between the sequences to form a spacer region, known as the “barcoding gap”. Figure 3 shows the minimum inter-specific K2P distances vs. maximum intra-specific distances, and the points that represented species distributed above the 1:1 line indicated that there were barcoding gaps for these species. With psbA-trnH and ITS2 + psbA-trnH, the species located in the area with no barcoding gap was H. rhamnoides. With the ITS2 region, there were two species, H. rhamnoides and H. neurocarpa, that had no barcoding gap. There were four points located on the 1:1 line, indicating that these species also had no barcoding gap. These four points included H. litangensis with ITS2, H. goniocarpa and H. neurocarpa with psbA-trnH, and H. neurocarpa with ITS2 + psbA-trnH.

Barcoding gap between Hippophae species based on intra- and inter-specific distances. Minimum inter-specific K2P distance vs. maximum intra-specific K2P distance for ITS2, psbA-trnH, and ITS2 + psbA-trnH. Each data point represents a species, and each species located above the 1:1 line has a barcoding gap
Neighbor-joining tree analysis
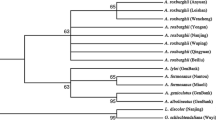
In this study, a phylogenetic tree was constructed by the NJ method, with 1000 bootstrap replicates for ITS2 (Fig. 4), psbA-trnH (Fig. 5), and ITS2 + psbA-trnH (Fig. 6) regions. Using ITS2 + psbA-trnH was the most effective for the species differentiation: all species were clearly identified, including the medicinal and non-medicinal Hippophae species. The ITS2 single-locus barcode was the second-most effective and differentiated five species: H. rhamnoides, H. goniocarpa, H. salicifolia, H. gyantsensis, and H. tibetana. The psbA-trnH region showed relatively poor performance with regard to species identification, as only four species were identified: H. litangensis, H. salicifolia, H. gyantsensis, and H. tibetana.

NJ tree of Hippophae constructed using ITS2. An E. angustifolia sequence downloaded from GenBank was included as an outgroup. The bootstrap scores (1000 replicates) are shown (≥50 %) for each branch. Each color represents one species

NJ tree of Hippophae constructed using psbA-trnH. An E. pungens sequence downloaded from GenBank was included as an outgroup. The bootstrap scores (1000 replicates) are shown (≥50 %) for each branch. Each color represents one species

NJ tree of Hippophae constructed using ITS2 + psbA-trnH. The bootstrap scores (1000 replicates) are shown (≥50 %) for each branch. Each color represents one species
At the subspecies level, four subspecies were identified by psbA-trnH (H. rhamnoides ssp. mongolica, H. rhamnoides ssp. yunnanensis, H. rhamnoides ssp. turkestanica, and H. rhamnoides ssp. wolongensis), three subspecies with ITS2 (H. rhamnoides ssp. yunnanensis, H. rhamnoides ssp. turkestanica, and H. rhamnoides ssp. wolongensis), and four subspecies with ITS2 + psbA-trnH (H. rhamnoides ssp. mongolica, H. rhamnoides ssp. yunnanensis, H. rhamnoides ssp. turkestanica, and H. rhamnoides ssp. wolongensis). Consequently, the 2-locus barcode ITS2 + psbA-trnH showed the highest efficiency for identifying Hippophae at the species and subspecies level. The single-locus barcode psbA-trnH was also suitable for identifying H. rhamnoides subspecies.
Discussion
The morphological similarities of Hippophae species caused a high chance of misidentification and misuse. Raw Hippophae products are often sold in dried and powdered forms, making morphological identification infeasible.
DNA barcoding is an important supplement and validation of conventional morphological identification [23]. In the present study, medicinal and non-medicinal Hippophae species were identified by DNA barcoding after a preliminary morphological identification, and remarkable Hippophae variation at the species level was shown. The genomic DNA could be extracted from dried fruits with both ITS2 and psbA-trnH with 100 % amplification and sequencing efficiencies. Two single-locus barcodes, ITS2 and psbA-trnH, as well as their combination were evaluated and validated. All Hippophae species were successfully identified by DNA barcoding, and four H. rhamnoides subspecies were also differentiated. The information obtained from the variable sequence sites and deletions/insertions facilitated the identification of Hippophae species; H. salicifolia, H. tibetana, and three H. rhamnoides subspecies were identified by ITS2 sequences, whereas H. goniocarpa, H. salicifolia, H. gyantsensis, H. tibetana, and two H. rhamnoides subspecies were identified by psbA-trnH sequences.
A relatively high value was observed for ITS2 + psbA-trnH with regard to the barcoding gap analysis: one species was located under the 1:1 line, and one species was located on the 1:1 line. However, three species had no barcoding gap for each of the single-locus barcodes: H. rhamnoides, H. litangensis, and H. neurocarpa for ITS2 barcode; H. rhamnoides, H. goniocarpa, and H. neurocarpa for psbA-trnH barcode. The identification efficiency of single-locus and combined barcodes by the NJ tree method showed that ITS2 + psbA-trnH was the most suitable barcode, with all seven species as well as four H. rhamnoides subspecies clearly identified. None of the selected barcodes were suitable for H. neurocarpa subspecies identification. Although it was hard to identify all H. rhamnoides and H. neurocarpa subspecies by ITS2, psbA-trnH, and ITS2 + psbA-trnH, the medicinal species were successfully distinguished from non-medicinal Hippophae species. While H. rhamnoides is the original medicinal plant according to Chinese Pharmacopeia, H. neurocarpa, H. gyantsensis, and H. tibetana are used in the Tibetan medicine [14, 15, 17, 18]. Thus, all native Hippophae species were identified by DNA barcode and the accurate and standard sequence information was gained. This information would be applicable to commercial products alignment and authenticate Hippophae species origins in the future.
There have been debates over whether H. litangensis was a subspecies of H. goniocarpa and whether H. rhamnoides subsp. wolongensis was a distinct species [3, 4, 42]. In our study, we considered H. litangensis and H. goniocarpa as two separate species, and the results demonstrated that they could be identified separately at the species level; H. rhamnoides subsp. wolongensis was a subspecies of H. rhamnoides based on the K2P genetic distance, NJ tree, and identification efficiency results.
Conclusion
The combination of the two loci, ITS2 + psbA-trnH is applicable to the identification of medicinal and non-medicinal Hippophae species.
Abbreviations
- ITS2:
-
internal transcribed spacer 2
- K2P:
-
Kimura 2-parameter
- NJ tree:
-
neighbor-joining tree
- CM:
-
Chinese medicine
- CBOL:
-
Consortium for the Barcode of Life
References
Chen XL, Lian YS. The geological distribution patterns and its formative factors on the genus Hippophae L. Xibei Zhiwu Xuebao. 1996;9:15–21.
Chen XL, Ma RJ, Sun K, Lian YS. Germplasm resource and habitat types of seabuckthorn in China. Xibei Zhiwu Xuebao. 2003;23:451–5.
Institute of Botany. Chinese Academy of Sciences: Flora reipublicae popularis sinicae. Beijing: Science Press; 1983.
Institute of Botany. Chinese Academy of Sciences: Flora of China. Beijing: Science Press; 2007.
Lian YS, Chen XL. Taxonomy of the genus Hippophae. Shaji. 1996;9:15–24.
Lian YS, Lu SG, Xue SK, Chen XL. Biology and chemistry of the genus Hippophae. Lanzhou: Gansu Science and Technology Press; 2000.
Pop RM, Weesepoel Y, Socaciu C, Pintea A, Vincken JP, Gruppen H. Carotenoid composition of berries and leaves from six Romanian sea buckthorn (Hippophae rhamnoides L.) varieties. Food Chem. 2014;147:1–9.
Suryakumar G, Gupta A. Medicinal and therapeutic potential of sea buckthorn (Hippophae rhamnoides L.). J Ethnopharmacol. 2011;138:268–78.
Zhang R, Sun RY, Yin W, Ma T, Xu LW. Overview of the comprehensive utilization of Hipopophae rhamnoides resources. Shipin Yanjiu Kaifa. 2012;33:229–32.
Bal LM, Meda V, Naik SN, Satya S. Sea buckthorn berries: a potential source of valuable nutrients for nutraceuticals and cosmoceuticals. Food Res Int. 2011;44:1718–27.
Lu CZ, Shan YK, Yang LH, Xue P, Ren BL. Research and application of seabuckthorn industry. Zhongguo Shiwu Yu Yingyang. 2010;3:26–8.
Purushothaman J, Suryakumar G, Shukla D, Jayamurthy H, Kasiganesan H, Kumar R, Sawhney RC. Modulation of hypoxia-induced pulmonary vascular leakage in rats by seabuckthorn (Hippophae rhamnoides L.). Evid Based Complement Alternat Med. 2011;2011:1–13.
Zeb A. Anticarcinogenic potential of lipids from Hippophae-evidence from the recent literature. Asian Pac J Cancer Prev. 2006;7:32–5.
Yutuoningma Y. Si Bu Yi Dian. Beijing: People’s Medical Publishing House; 1983.
Danzengpengcuo D. Jing Zhu Ben Cao. Shanghai: Science and Technology Press; 1983.
Ma SL, Wang ZH, Mao JZ. Yue Wang Yao Zhen. Shanghai: Shanghai Science and Technology Press; 2012.
Chinese Pharmacopoeia Commission. The pharmacopoeia of the People’s Republic of China. Beijing: China Medical Science Press; 2010.
Qinghai Institute for Drug Control. Tibetan medicine research institute of Qinghai province: Tibetan medicine of China. Shanghai: Shanghai Science and Technology Press; 1996.
Hebert PD, Cywinska A, Ball SL, deWaard JR. Biological identifications through DNA barcodes. Proc Biol Sci. 2003;1512:313–21.
Sucher NJ, Carles MC. Genome-based approaches to the authentication of medicinal plants. Planta Med. 2008;74:603.
CBOL Plant Working Group. A DNA barcode for land plants. Proc Natl Acad Sci U S A. 2009;106:12794–7.
Kress WJ, Wurdack KJ, Zimmer EA, Weigt LA, Janzen DH. Use of DNA barcodes to identify flowering plants. Proc Natl Acad Sci USA. 2005;102:8369–74.
Chen SL, Yao H, Han JP, Liu C, Song JY, Shi LC, Zhu YJ, Ma XY, Gao T, Pang XH, Luo K, Li Y, Li XW, Jia XC, Lin YL, Leon C. Validation of the ITS2 region as a novel DNA barcode for identifying medicinal plant species. PLoS One. 2010;5:e8613.
Wong KL, But PP, Shaw PC. Evaluation of seven DNA barcodes for differentiating closely related medicinal Gentiana species and their adulterants. Chin Med. 2013;8:16.
Ma XC, Xie CX, Guan M, Xu XH, Miki E, Takeda O, Xin TY, Zheng SH, Yao H, Shi LC, Song JY, Chen SL. High levels of genetic diversity within one population of Rheum tanguticum on the Qinghai-Tibet Plateau have implications for germplasm conservation. Pharmaceut Crops. 2014;5:1–8.
Han JP, Zhu YJ, Chen XC, Liao BS, Yao H, Song JY, Chen SL, Meng FY. The short ITS2 sequences seves as an efficient taxonomic sequences tag in comparison with full-length ITS. BioMed Res Int. 2013;2013:741476.
Chen SL, Guo BL, Zhang GJ, Yan ZY, Luo GM, Sun SQ, Wu HZ, Huang LF, Pang XH, Chen JB. Advances of studies on new technology and method for identifying traditional Chinese medicinal materials. Zhongguo Zhong Yao Za Zhi. 2012;37:1043–55.
Li YH, Ruan JL, Chen SL, Song JY, Luo K, Lu D, Yao H. Authentication of Taxillus chinensis using DNA barcoding technique. J Med Plants Res. 2010;4:2706–9.
Shi LC, Zhang J, Han JP, Song JY, Yao H, Zhu YJ, Li JC, Wang ZZ, Xiao W, Lin YL, Xie CX, Qian ZZ, Chen SL. Testing the potential of proposed DNA barcodes for species identification of Zingiberaceae. J Syst Evol. 2011;49:261–6.
Techen N, Parveen I, Pan ZQ, Khan IA. DNA barcoding of medicinal plant material for identification. Curr Opin Biotechnol. 2014;25:103–10.
Li DZ, Liu JQ, Chen ZD, Wang H, Ge XJ, Zhou SL, Gao LM, Fu CX, Chen SL. Plant DNA barcoding in China. J Syst Evol. 2011;49:165–8.
Li XW, Yang Y, Henry RJ, Rossetto M, Wang YT, Chen SL. Plant DNA barcoding: from gene to genome. Biol Rev Camb Philos Soc. 2015;90:157–66.
Song JY, Shi LC, Li DZ, Sun YZ, Niu YY, Chen ZD, Luo HM, Pang XH, Sun ZY, Liu C, Lv AP, Deng YP, Larson-Rabin Z, Wilkinson M, Chen SL. Extensive pyrosequencing reveals frequent intra-genomic variations of internal transcribed spacer regions of nuclear ribosomal DNA. PLoS One. 2012;7:e43971.
Xin TY, Yao H, Gao HH, Zhou XZ, Ma XC, Xu CQ, Chen J, Han JP, Pang XH, Xu R, Song JY, Chen SL. Super food Lycium barbarum (Solanaceae) traceability via an internal transcribed spacer 2 barcode. Food Res Int. 2013;54:1699–704.
Yao H, Song JY, Ma XY, Liu C, Li Y, Xu HX, Han JP, Duan LS, Chen SL. Identification of Dendrobium species by a candidate DNA barcode sequence: the chloroplast psbA-trnH intergenic region. Planta Med. 2009;75:667–9.
Chen SL. Traditional Chinese medicine molecular identification by DNA barcode. Beijing: People’s Medical Publishing House; 2012.
Lahaye R, Van der Bank M, Bogarin D, Warner J, Pupulin F, Gigot G, Maurin O, Duthoit S, Barraclough TG, Savolainen V. DNA barcoding the floras of biodiversity hotspots. Proc Natl Acad Sci U S A. 2008;105:2923–8.
Wolf M, Achtziger M, Schultz J, Dandekar T, Müller T. Homology modeling revealed more than 20,000 rRNA internal transcribed spacer 2 (ITS2) secondary structures. RNA. 2005;11:1616–23.
Keller A, Schleicher T, Schultz J, Müller T, Dandekar T, Wolf M. 5.8S-28S rRNA interaction and HMM-based ITS2 annotation. Gene. 2009;430:50–7.
Koetschan C, Hackl T, Müller T, Wolf M, Förster F, Schultz J. ITS2 database IV: interactive taxon sampling for internal transcribed spacer 2 based phylogenies. Mol Phylogenet Evol. 2012;63:585–8.
Tamura K, Stecher G, Peterson D, Filipski A, Kumar S. MEGA6: molecular evolutionary genetics analysis version 6.0. Mol Biol Evol. 2013;30:2725–9.
Sun K, Chen XL, Ma RJ, Li C, Wang Q, Ge S. Molecular phylogenetics of Hippophae L. (Elaeagnaceae) based on the internal transcribed spacer (ITS) sequences of nrDNA. Plant Syst Evol. 2002;235:121–34.
Authors’ contributions
LX and YZ designed the study. YL, CL, YQZ, YLC and MS performed the experiment. YL analyzed the data and wrote the manuscript. LX, YZ, WS, GF and XL revised the manuscript. All authors read and approved the final manuscript.
Acknowledgements
This study was supported by Science Technology Support Plan Project from Science & Technology Department of Sichuan Province (No. 2013SZ01114)-“Key technology, research and development of distinctive Chinese and Tibetan medicine resources utilization” and The Major Scientific and Technological Special Project for ‘‘Significant New Drugs Creation’’ (No. 2014ZX09304307).
Competing interests
The authors declare that they have no competing interests.
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Liu, Y., Sun, W., Liu, C. et al. Identification of Hippophae species (Shaji) through DNA barcodes. Chin Med 10, 28 (2015). https://doi.org/10.1186/s13020-015-0062-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13020-015-0062-9