
Abstract
Background
Malaria continues to be a major threat to global public health. Whole genome sequencing (WGS) of the underlying Plasmodium parasites has provided insights into the genomic epidemiology of malaria. Genome sequencing is rapidly gaining traction as a diagnostic and surveillance tool for clinical settings, where the profiling of co-infections, identification of imported malaria parasites, and detection of drug resistance are crucial for infection control and disease elimination. To support this informatically, we have developed the Malaria-Profiler tool, which rapidly (within minutes) predicts Plasmodium species, geographical source, and resistance to antimalarial drugs directly from WGS data.
Results
The online and command line versions of Malaria-Profiler detect ~ 250 markers from genome sequences covering Plasmodium speciation, likely geographical source, and resistance to chloroquine, sulfadoxine-pyrimethamine (SP), and other anti-malarial drugs for P. falciparum, but also providing mutations for orthologous resistance genes in other species. The predictive performance of the mutation library was assessed using 9321 clinical isolates with WGS and geographical data, with most being single-species infections (P. falciparum 7152/7462, P. vivax 1502/1661, P. knowlesi 143/151, P. malariae 18/18, P. ovale ssp. 5/5), but co-infections were identified (456/9321; 4.8%). The accuracy of the predicted geographical profiles was high to both continental (96.1%) and regional levels (94.6%). For P. falciparum, markers were identified for resistance to chloroquine (49.2%; regional range: 24.5% to 100%), sulfadoxine (83.3%; 35.4– 90.5%), pyrimethamine (85.4%; 80.0–100%) and combined SP (77.4%). Markers associated with the partial resistance of artemisinin were found in WGS from isolates sourced from Southeast Asia (30.6%).
Conclusions
Malaria-Profiler is a user-friendly tool that can rapidly and accurately predict the geographical regional source and anti-malarial drug resistance profiles across large numbers of samples with WGS data. The software is flexible with modifiable bioinformatic pipelines. For example, it is possible to select the sequencing platform, display specific variants, and customise the format of outputs. With the increasing application of next-generation sequencing platforms on Plasmodium DNA, Malaria-Profiler has the potential to be integrated into point-of-care and surveillance settings, thereby assisting malaria control. Malaria-Profiler is available online (bioinformatics.lshtm.ac.uk/malaria-profiler) and as standalone software (https://github.com/jodyphelan/malaria-profiler).
Similar content being viewed by others

Background
Malaria is a life-threatening disease caused by Plasmodium parasites that are transmitted to humans by infected female Anopheles mosquitoes [1]. There were 247 million cases of malaria and 619 thousand deaths in 2021 alone, with the vast majority affecting children and pregnant women in Sub-Saharan Africa [1]. There are six parasite species that cause malaria in humans (P. falciparum, P. vivax, P. ovale ssp., P. malariae, P. knowlesi; genome sizes 23–36 Mbp). P. falciparum is the deadliest malaria parasite and the most prevalent on the African continent. P. vivax is the most geographically widespread malaria parasite [2], found in Europe, Asia, South America, and Africa due to its adaptation for temperate climatic conditions. Whilst zoonotic P. knowlesi is found primarily in Southeast Asia due to the presence of the macaque population, which acts as a reservoir for the parasite. P. ovale ssp. and P. malariae malaria cases have been predominantly reported in Africa and can occur with co-infections with P. falciparum, potentially affecting elimination strategies that target prevalent species [3].
Malaria treatment is guided by the knowledge of the infecting Plasmodium species and clinical severity. There are currently fourteen medicines for the treatment of malaria and four for preventative treatment listed by the World Health Organization (WHO) [4]. Global efforts to control and eliminate malaria are hampered by the emergence of P. falciparum parasites resistant to antimalarial drugs. There is a high prevalence of chloroquine and sulfadoxine-pyrimethamine (SP) resistance across continents [5], and partial resistance or slow parasite clearance to artemisinin, used in current treatment combinations, spreading in Southeast Asia [6], with some recent cases appearing in Africa [7]. Similarly, P. vivax isolates resistant to chloroquine have been reported in parts of Asia and South America [2].
Prompt malaria diagnosis either by microscopy or rapid diagnostic tests (RDTs) is recommended by the WHO for all patients with suspected malaria before they are given treatment [4]. Early and accurate diagnosis is essential both for effective management of the disease and for strong malaria surveillance. Measures targeting the treatment of persistent malaria infections, such as P. ovale ssp. and P. vivax with dormant liver stages and P. malariae with possible latent blood infections, will need to comprise all human malaria species. Neither microscopy nor RDTs can detect low-density malaria infections, common in both low and high transmission settings, but nucleic acid amplification tests (NAATs) such as Polymerase chain reaction (PCR), real-time PCR (rt-PCR), loop-mediated isothermal amplification (LAMP), and quantitative nucleic acid sequence-based amplification (QT-NASBA) assays can overcome this limitation. The 18S ribosomal RNA gene has unique sequences that enable the identification of all six malaria species infecting humans and is therefore commonly targeted for amplification. Similarly, the mitochondrial genome (6 kbp) has species-specific markers, and has the added advantage of being present in high copy numbers in Plasmodium cells [8]. A number of studies have revealed P. falciparum genetic markers linked to antimalarial drugs, such as chloroquine, SP and artemisinin [9,10,11], which are being included within NAATS, but the underlying mechanisms for P. vivax chloroquine resistance are unclear [2].
The increasing accessibility of advanced high throughput technologies that are cost-effective and with low sequencing error rates, can inform clinical decision making and tracking of infections. Recently, whole genome sequencing (WGS) has gained traction as a diagnostic tool for infections, with the ability to determine strain types of pathogens, characterise transmission patterns, and identify markers linked to antimicrobial resistance [12]. Portable platforms, such as Oxford Nanopore Technology (ONT), are facilitating the real-time generation of sequencing data in the field and clinic. Such platforms can also be used to sequence large numbers of amplicons (~ 500 bp) that cover candidate genes, across many samples, leading to a high throughput low-cost diagnostic tool that can capture new variants in targeted loci [13, 14]. However, one of the main challenges in performing WGS or amplicon-based sequencing studies for clinical malaria parasites is the difficulty in obtaining sufficient high-quality parasite DNA from infected individuals. This difficulty is due to low parasitaemias in infections and human DNA “contamination”. However, recently a selective whole genome amplification (SWGA) strategy has been used to sequence P. falciparum [15], P. vivax [2, 16], P. knowlesi [17] and P. malariae [18] genomes from non-filtered blood and from dried blood spots of clinical samples, leading to the characterisation of single nucleotide polymorphisms (SNPs) and insertions and deletions (indels) for population genomic analyses. More generally, genomic diversity studies using WGS from P. falciparum, P. knowlesi and P. vivax endemic field isolates have provided significant insights into the structure and ancestry of the geographical-based parasite populations, intra- and inter-population genomic diversity, and led to the development of molecular barcodes to determine the geographical source of infections [8, 17, 19,20,21]. Furthermore, population genetic analyses have identified genomic regions under selective pressure, some in drug resistance-associated genes [2, 15, 22,23,24].
As the generation of WGS and amplicon-based sequencing data for Plasmodium parasites continues to increase at a swift pace, including from the portable ONT platform, there is a need for informatics tools for researchers and applied bioinformaticians to rapidly analyse WGS data. Such tools are needed to obtain profiles of (co-)infections and drug resistance markers, thereby supporting clinical decision-making. Further, by additionally identifying likely geographical origin (e.g. country), it could reveal imported parasites, thereby supporting surveillance decision-making too. By monitoring the changes in informative mutations temporally, it will allow an assessment of transmission patterns and the effectiveness of infection control activities. Here, building on the core library used in a similar software for tuberculosis (“TB-Profiler” [25, 26]), we describe the Malaria-profiler standalone and web-based tool, with accompanying dashboard interfaces, for rapid profiling of Plasmodium parasites species, and characterising genetic variants for follow-up studies.
Implementation
Profiling mutation library
The mutation library consists of ~ 100 mitochondrion markers for speciation of P. falciparum, P. vivax, P. malariae, P. knowlesi and P. ovale ssp. (20 per species; Table 1), which also differentiate human from non-human affecting Plasmodium species. In brief, alignments of 75 mitochondrial genomes (51 human and 24 non-human Plasmodium; Fig. 1a) were used to construct a maximum likelihood phylogenetic tree. By annotating the tree branches with ancestral mutations [26], it was possible to define k-mers (31 bp) using kmc software [27], from which 20 SNPs exclusive to each human species were determined. Using the mitochondrial genome has the advantage of ~ 20 more copies than the nuclear genome in cells [8]. In addition, we included a set of established markers (n = 137) that differentiate geographical regions for P. falciparum (61; Eastern, Western and Horn of Africa, Southeast Asia, South America, Oceania), P. vivax (56; East Africa, South Asia, Southeast Asia, Southern Southeast Asia, South America) and P. knowlesi (20; Non-Borneo (Peninsular); Borneo – Macaca fascularis (Borneo-Mf), Borneo – Macaca nemestrina (Borneo-Mn)) [8, 17, 19, 20] (Table 2). In brief, these barcoding markers have been previously determined using the population differentiation FST statistic, and identifying scores of one, which indicate that the SNP allele is fixed in the region of interest and not present outside that location. Lastly, known drug resistance mutations (n = 37) across P. falciparum candidate genes [15] were also included in the library (Table 3) as well as genetic variants in putative drug-associated loci reported for other malaria species (e.g. orthologues of Pfcrt, Pfdhfr, Pfdhps, Pfkelch13 and Pfmdr1) [2, 18, 21]. The mutation libraries are available and hosted on the GitHub open-source site, with versioning capability (https://github.com/jodyphelan/malaria-db). Future changes in the species, geolocation and drug resistance mutation libraries can be discussed, tracked, and visualised as part of the GitHub hosting. This method of hosting also enables multiple users and developers across the malaria genomics community to contribute to the project.

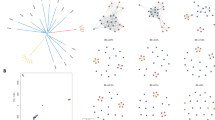
Population structure of Plasmodium. a Circular maximum likelihood tree of 51 human and 24 non-human Plasmodium isolates using mitochondrial sequences shows perfect clustering of species as expected. This indicates the presence of a species-specific sequence which is exploited in the k-mer-based speciation function. Pf P. falciparum, Pv P. vivax, Pk P. knowlesi, Pcyn P. cynomolgi, Pm P. malariae, Poc P. ovale curtesi. b P. falciparum principal component analysis showing clustering by geographic region specifically separation between Southeast Asia and Oceania and Africa. c P. vivax principal component analysis showing clustering by geographic region. d P. knowlesi principal component analysis showing clustering by region (Peninsular (Pen-Pk) vs. Borneo Malaysia), and within Borneo based on host (Macaca fascularis (Mf-Pk) and Macaca nemestrina (Mn-Pk))
In silico profiling
The Malaria-Profiler tool for the in silico analysis of species, geolocation and drug-resistant mutations was developed using the Python language (v3.8) with the pathogen-profiler library [12] and well-established bioinformatic tools such as trimmomatic [28], BWA [29] and SAMtools [30]. The pipeline can be customised (Additional file 1: Fig. S1), but in its default mode, reads are trimmed using trimmomatic (parameters: LEADING:3 TRAILING:3 SLIDINGWINDOW:4:20 MINLEN:36) then mapped to the appropriate Plasmodium reference (e.g. P. falciparum 3D7, P. vivax PvPO1) using bwa (with default parameters). The raw data can be in an Illumina or ONT format (Additional file 1: Fig. S2). With the default settings, variants are called using freebayes [31] (parameters: -F 0.05) and annotated using snpEff [32] (parameters: -noLog -noStats), with the processing parallelised using GNU parallel [33]. Annotated variants are compared to the list of mutations in the Malaria-Profiler libraries. Variants can be filtered using coverage depth, allele frequency and per-strand depth parameters that can be set by the user. Additionally, other variant calling tools can be used instead of freebayes with bcftools [30] and gatk [34] also implemented. A minimum depth of tenfold coverage to call variants is set as the default (consistent with [5, 12]), but this can be changed by the user. Positions below this cut-off will be recorded and presented in the final report. The Malaria-Profiler pipeline calculates the proportion of the reads supporting each allele and reports this information, which can serve as a proxy for multi-infections. The Malaria-Profiler pipeline is available on GitHub (from https://github.com/jodyphelan/malaria-profiler) and can be installed through the bioconda channel [35]. Malaria-Profiler report outputs are written in json, txt and pdf formats, with options to collate data into multi-sample reports in a dashboard (Additional file 1: Fig. S2).
Sequencing data and variants
To test the Malaria-Profiler tool, a dataset of 9321 strains was collated from Illumina WGS raw data in the public domain (see https://www.ebi.ac.uk/ena). This database includes P. falciparum (n = 7462; https://www.malariagen.net/apps/pf6 [36]; PRJEB2136, PRJEB2143, PRJEB4348, PRJEB4410, PRJEB4580, PRJEB4589, PRJEB4611, PRJEB4725, PRJEB5045, PRJNA108699 and PRJNA51255), P. vivax (n = 1661, https://www.malariagen.net/data/open-dataset-plasmodium-vivax-v4.0; PRJEB10888, PRJEB2136, PRJEB2140, PRJEB4409, PRJEB4410, PRJEB44419, PRJEB4580, PRJEB56411, PRJNA175266, PRJNA240366-240531, PRJNA271480, PRJNA284437, PRJNA295233, PRJNA420510, PRJNA432819, PRJNA603279, PRJNA643698, PRJNA65119, PRJNA655141, PRJNA67065, PRJNA67237, and PRJNA67239) [2, 21], P. knowlesi (n = 151; PRJEB10288, PRJEB1405, PRJEB23813, PRJEB28192, PRJEB33025, and PRJNA294104) [17, 20], P. malariae (n = 18; PRJEB33837) [18] and P. ovale (n = 5; PRJEB51041) [18]. Meta data, including the geographical site of sampling, was available from the same sources (e.g. www.malariagen.net/resources/open-data-resources). In addition, the mitochondrion reference genomes were obtained from GenBank for the neglected non-human malaria parasites (n = 24; e.g. P. cynomologi, P. inui, P. reichenowi and P. simiovale) were also included in the analysis. Alignments of mitochondrial genomes to the species library allow for the identification of primary Plasmodium infection and potential co-infections. Species were assigned if half of the 20 specific markers were identified in the data. For intra-species analysis, genome-wide SNPs and indels were called using established bioinformatic pipelines [2, 15, 17, 18]. In brief, the raw Illumina WGS data (fastQ format) were aligned to their respective reference genomes using BWA-mem software (default parameters). SNPs and short indels were called using the SAMtools and GATK software suites (see [19]). For ONT data, a similar pipeline was adopted, except sequence alignment was performed using minimap2 [37] software.
Using genomic data to inform on Plasmodium parasite speciation and geographical clustering
A maximum likelihood phylogenetic tree for Plasmodium species was constructed using RAxML-NG (v 0.9.0; 1000 bootstraps) software applied to mitochondrial genomes (n = 75; 5592 nucleotides), which were aligned using MUSCLE software [38] and filtered with the Gblocks tool [39] (default settings). The optimal substitution model of nucleotide or amino acid evolution for phylogenetic construction was determined by MEGAX software [40]. Parasite clustering within species (e.g. P. falciparum, P. vivax; total n = 9321), which is typically geographically based, was explored by performing a principal component analysis (PCA) on the isolates using pairwise Manhattan distances based on biallelic SNPs.
Malaria-Profiler performance
To test the performance of the library, the WGS raw data for the 9321 strains were processed through the Malaria-Profiler pipeline to predict species, geolocation, and resistance status (for P. falciparum). The predictions were then compared to primary Plasmodium species and geographical recorded meta information (see www.malariagen.net/resources/open-data-resources), which were assumed to be the gold standard, and thereby allowed the calculation of the predictive accuracy of the Malaria-Profiler library. Phenotypic drug resistance status was not available for most isolates. Samples identified by Malaria-Profiler with potential co-infections were also analysed with Centrifuge software [41] to confirm the main Plasmodium species. When applying Centrifuge, the threshold for potential co-infection was based on the whole genome abundance (minimum 5%) and samples with > 1 Plasmodium species exceeding the threshold were assigned as mixed. To demonstrate the utility of WGS in the clinic, processed DNA (see [42] for protocols) from two isolates (isolate1, isolate2) sourced from two malaria patients at the Radboud University Medical Center were sequenced on the ONT MinION platform (v10) at The Applied Genomics Centre, LSHTM (accession numbers ERR11254081 and ERR11254083).
Results
Species prediction
Using the mitochondrion alignments of 51 human and 24 non-human Plasmodium parasites, a phylogenetic analysis revealed clustering by species (Fig. 1a), as well as the robustness of the species-level barcoding markers used within the Malaria-Profiler library. Across the 9321 isolates with WGS data, the Malaria-Profiler tool predicted the labelled primary species in almost all samples (9300/9321; 99.8%). Mixed co-infections were also detected (456/9321; 4.9%), with P. falciparum (298; 63.4%) and P. vivax (150; 32.9%) being the dominant parasites (Table 1), and most co-infections were supported by a parallel analysis using Centrifuge software (P. falciparum 165/298, 55.4%; P. vivax 116/150, 77.3%). Discrepancies arise due to Centrifuge software excluding genomes with very minor frequencies (< 5%). The 24 non-human related Plasmodium mitochondrion sequences were also processed by the tool, leading to the predicted (and expected) absence of any of the six human-affecting Plasmodium species (Table 1).
Geographical predictions
The geographical-based population structure of P. falciparum, P. vivax, and P. knowlesi was confirmed using a principal component analysis of SNPs which revealed clustering by geographic region (Fig. 1b–d). Using the geographical barcodes on isolates with recorded location (n = 8775), the Malaria-profiler tool predictions were accurate to continental (96.1%) and regional (94.6%) levels (Table 2). The best performance was for P. knowlesi (141/143; 98.6%), known to display high variability between clusters [20]. The accuracy for P. falciparum predictions was high (6834/7130; 95.8%), across all regions (> 91%). The accuracy for P. vivax was lower (1322/1502; 88.0%), especially for the South Asia region (80.5%), due to high similarity between neighbouring countries across regions.
Genotypic drug resistance
Using the known P. falciparum markers for chloroquine, SP and artemisinin, the patterns of predicted genotypic resistance were similar to established patterns. Resistance to pyrimethamine was high across all regions (> 87%), leading to high SP prevalence (> 80%), except in Oceania and South America. Chloroquine resistance was lowest in East and West Africa (< 38%), where the drug was withdrawn as a treatment more than 20 years ago, and there has been some reversion back to wild-type alleles [5, 43]. Mutations linked to (partial) resistance to artemisinin in P. falciparum were found in Southeast Asia (30.6%), in keeping with their known emergence and spread from the Greater Mekong region [6, 11]. For one such P. falciparum isolate with partial resistance to artemisinin, we show the informative nature of the Malaria-Profiler Dashboard output (Fig. 2a). The Thai isolate was sequenced on an Illumina platform (accession no. ERR248945), and Malaria-Profiler predicts that it is from Southeast Asia, and has a complex drug resistance profile involving genotypic resistance to chloroquine, SP, and artemisinin (Fig. 2a).

Example of Malaria-Profiler report outputs. a Thai isolate confirmed to be P. falciparum from Southeast Asia, with a complex drug resistance profile (accession no. ERR248945). b A traveller isolate sequenced on Oxford Nanopore Technology and determined to be from East Africa and with chloroquine, Sulfadoxine and Pyrimethamine resistance (accession no. ERR11254081)
Profiling using ONT platform data
Isolates were sourced from two travellers attending the Radboud University Medical Center who tested positive for malaria. Isolate DNA (isolate1: ERR11254081, isolate2: ERR11254083) was sequenced on the ONT platform to establish their likely geographical source and genotypic drug resistance. Isolate1 was sequenced twice with 410,115 and 1,168,719 reads mapped in total, leading to a median coverage of 31- and 94-fold, respectively. Across both sequencing runs, all positions in candidate genes used for profiling were covered by at least 10 reads. The profiles resulting from each sequencing run were identical. Resistance to chloroquine was predicted through mutations in pfmdr1 (Asn86Tyr, Asp1246Tyr) and pfcrt (Lys76Thr, Ala220Ser, Gln271Glu, Arg371Ile). Resistance to SP was predicted through mutations in pfdhfr (Asn51Ile, Cys59Arg, Ser108Asn) and pfdhps (Ala437Gly, Lys540Glu, Ala581Gly). The geographic origin was predicted to be East Africa (Fig. 2b), and consistent with the traveller staying in Uganda. Isolate2 had 1,271,185 mapped reads, leading to 109-fold median coverage and all candidate gene positions covered by at least 10 reads. Resistance to SP was predicted through mutations in pfdhfr (Asn51Ile, Ser108Asn) and pfdhps (Ala437Gly, Lys540Glu). The traveller had been in Rwanda and India, and the predicted geographic origin was Africa (Additional file 1: Fig. S2b), suggesting that the source of infection was the former.
Discussion
Advances in WGS technology have expanded a role for genome analysis in the clinical laboratory and field settings. Determining the profile of Plasmodium species using WGS will guide elimination strategies, including through the monitoring of important mutations temporally and assessing the extent of mixed infections. The sequencing of DNA from malaria infections with low parasite density will be crucial in pre-elimination settings and is possible through low-cost selective whole genome amplification protocols [18]. We have previously shown the robustness of variant calling tools to detect SNPs, small indels and large deletions from WGS data [9, 26, 44]. As WGS is adopted more widely as a diagnostic tool, there is a need for robust and reliable software tools to rapidly process the vast amounts of data generated. Further, the growing application of third and fourth-generation sequencing platforms (e.g. ONT MinION) and linked cost-effective amplicon-based approaches have driven the need to integrate analysis options for these technologies into profiling tools to support their use in a more automated format than currently available.
The Malaria-Profiler framework allows for an adaptive mutation library, where the set of barcoding markers can be extended to cover gaps in our knowledge. As our knowledge of Plasmodium drug resistance mechanisms (e.g. P. vivax chloroquine resistant loci) and geographical-specific markers (e.g. for P. malariae and P. ovale ssp.) grows, prediction software must be flexible and allow for customisation of barcoding databases. The generation of informative genomic data will be facilitated through advances in sequencing platforms, including low-cost applications of amplicon-based assays that target candidate genes. Further, ONT platforms can implement “adaptive” sequencing, where it is possible enrich on-target reads through real-time alignment to specified genomes of interest and eject uninteresting reads, thereby minimising the generation of contaminant sequences in a clinical sample. Whilst human contaminants in blood are typically removed through sample processing protocols (e.g. SWGA, leucocyte depletion), Malaria-Profiler also filters non-Plasmodium sequences using bioinformatic methods. Ultimately, if there is insufficient sequence coverage of Plasmodium parasite DNA, then Malaria-Profiler cannot call variants robustly. A future extension of the software could be to identify potentially informative markers in the human genome [45], such as sickle cell HbS, but this would require extensive evaluation of sequencing protocols and data generated across a range of asymptomatic and clinical blood samples. The increased deployment and availability of such technologies could lead to assessments of Plasmodium genetic diversity in sites with currently limited data and studies. There is a constant need to update, re-evaluate and improve mutation libraries in response to new genomic data and functional evidence, including through the implementation of artificial intelligence approaches [36, 46]. To minimise the risk that mutation libraries become unmaintained and remain static versions of evidence at the time, we have hosted the library on a repository that facilitates user input (https://github.com/jodyphelan/malaria-db). Further improvements can involve the exploration of structural changes, such as copy number variants, as some have been linked with the drug resistance. For example, markers of resistance to mefloquine and piperaquine, include amplifications of Pfmdr1 and PfPlasmepsin2/3, respectively. Similarly, low-density infections, and deletions of the P. falciparum hrp2/3 genes (encoding the HRP2 and HRP3 proteins) [47] present challenges for some rapid diagnostic tests, therefore such deletions could also be included [48]. Whilst analyses of gene coverage are possible through sequencing-based approaches, leading to insights into amplifications and deletions, there may also be SNPs that tag structural variants to facilitate implementation.
In summary, monitoring genetic markers of resistance can help guide antimalarial therapy and surveillance activities. The introduction of drug resistance markers to new geographical areas may be detected through the WGS of clinical samples and analysis of data using Malaria-Profiler. Routine WGS across time and geographical regions can detect the presence and spread of established or new markers, and inform infection control practice. WGS has the potential to improve the resolution and timeliness of Plasmodium profiling and, in combination with clinical trials and robust experimental work using Plasmodium culture and CRISPR-Cas9 systems [49], can lead to new insights into drug resistance mechanisms. Malaria-Profiler is a flexible software tool that allows users to rapidly obtain useful information from WGS (and amplicon) data generated by Illumina and MinION platforms to predict species, drug resistance and geographical profiles with high accuracy.
Conclusions
We have developed an online software tool and methodology that provides rapid analysis of genome sequence data to describe Plasmodium species and geographical source and predict resistance to antimalarial drugs. The tool utilises a library consisting of ~ 250 mutations that is the most comprehensive and accurate such data source yet reported. The ability to rapidly analyse raw sequence data and extract information of clinical relevance has advantages over current in vitro drug assays, which require parasite culture-based systems [50]. Accelerated access to tailored treatment could improve cure rates and reduce exposure to ineffective drugs, thereby improving the patient experience and facilitating compliance. The analytical methodology described is customisable to allow moderation of the library to encompass novel mutations and incorporate new drugs should the need arise. Overall, we have shown that Malaria-Profiler can be used to reliably predict Plasmodium species, geographical source, and drug resistance from WGS. This pipeline can be applied to data from multiple sequencing platforms and can support informatically the application of WGS as a diagnostic and surveillance tool.
Availability of data and materials
The Malaria-Profiler webtool and source code can be accessed (https://github.com/jodyphelan/Malaria-Profiler). The barcoding mutations for speciation, geography and resistance can be accessed (https://github.com/jodyphelan/malaria-db). The raw sequencing data analysed during the current study are available from the ENA and NCBI databases. The Pf6 database was used for P. falciparum (https://www.malariagen.net/apps/pf6; [51]), and the Pv4 database was used for P. vivax (https://www.malariagen.net/data/open-dataset-plasmodium-vivax-v4.0; [52]). For P. knowlesi, the raw sequence data is available from the ENA (accession numbers: PRJEB10288, PRJEB1405, PRJEB23813, PRJEB28192, PRJEB33025, and PRJNA294104) [17, 20]. Similarly, raw sequence data are available for P. malariae (accession number: PRJEB33837) [19] and P. ovale ssp (accession number: PRJEB51041) [19]. Raw sequence data for the two newly sequenced P. falciparum isolates are available from the ENA (https://www.ebi.ac.uk/ena/browser/view/ERR11254081; www.ebi.ac.uk/ena/browser/view/ERR11254083).
Availability of data and materials
Project name: Malaria-Profiler.
Project home page: https://github.com/jodyphelan/Malaria-Profiler; https://github.com/jodyphelan/malaria-db.
Programming language: Python.
Operating system(s): Platform independent.
Other requirements: None.
License: GNU GPL.
Any restrictions to use by non-academics: None.
Abbreviations
- SP:
-
Sulfadoxine-pyrimethamine
- WGS:
-
Whole genome sequencing
- WHO:
-
World Health Organization
References
World Health Organization. World Malaria Report. 2022. (World Health Organization, 2022).
Benavente ED, et al. Distinctive genetic structure and selection patterns in Plasmodium vivax from South Asia and East Africa. Nat Commun. 2021;12(1):3160.
Fuehrer H-P, Campino S, Sutherland CJ. The primate malaria parasites Plasmodium malariae, Plasmodium brasilianum and Plasmodium ovale spp.: genomic insights into distribution, dispersal and host transitions. Malar J. 2022;21:138.
World Health Organization. Consolidated Guidelines for Malaria. 2023. (World Health Organization, 2023).
Turkiewicz A, et al. Genetic diversity of the Plasmodium falciparum GTP-cyclohydrolase 1, dihydrofolate reductase and dihydropteroate synthetase genes reveals new insights into sulfadoxine-pyrimethamine antimalarial drug resistance. PLoS Genet. 2020;16(12):e1009268.
He Y, et al. Artemisinin resistance-associated markers in Plasmodium falciparum parasites from the China-Myanmar border: Predicted structural stability of K13 propeller variants detected in a low-prevalence area. PLoS One. 2019;14(3):e0213686.
Balikagala B, et al. Evidence of Artemisinin-Resistant Malaria in Africa. N Engl J Med. 2021;385:1163–71.
Preston MD, et al. A barcode of organellar genome polymorphisms identifies the geographic origin of Plasmodium falciparum strains. Nat Commun. 2014;5:4052.
Conrad MD, et al. Evolution of Partial Resistance to Artemisinins in Malaria Parasites in Uganda. N Engl J Med. 2023;389(8):722–32.
Djimdé A, et al. A molecular marker for chloroquine-resistant falciparum malaria. N Engl J Med. 2001;344:299–302.
Ariey F, et al. A molecular marker of artemisinin-resistant Plasmodium falciparum malaria. Nature. 2014;505:50–5.
Phelan JE, et al. Integrating informatics tools and portable sequencing technology for rapid detection of resistance to anti-tuberculous drugs. Genome Med. 2019;11:41.
Acford-Palmer H, et al. Identification of two insecticide resistance markers in Ethiopian Anopheles stephensi mosquitoes using a multiplex amplicon sequencing assay. Sci Rep. 2023;13:5612.
Collins EL, et al. A next generation targeted amplicon sequencing method to screen for insecticide resistance mutations in Aedes aegypti populations reveals a rdl mutation in mosquitoes from Cabo Verde. PLoS Negl Trop Dis. 2022;16:e0010935.
Osborne A, et al. Characterizing the genomic variation and population dynamics of Plasmodium falciparum malaria parasites in and around Lake Victoria, Kenya. Sci Rep. 2021; 11:19809.
Cowell AN, et al. Selective Whole-Genome Amplification Is a Robust Method That Enables Scalable Whole-Genome Sequencing of Plasmodium vivax from Unprocessed Clinical Samples. mBio. 2017; 8:e02257–16.
Benavente ED, et al. Whole genome sequencing of amplified Plasmodium knowlesi DNA from unprocessed blood reveals genetic exchange events between Malaysian Peninsular and Borneo subpopulations. Sci Rep. 2019;9:9873.
Ibrahim A, et al. Selective whole genome amplification of Plasmodium malariae DNA from clinical samples reveals insights into population structure. Sci Rep. 2020;10:1–11.
Diez Benavente E, et al. A molecular barcode to inform the geographical origin and transmission dynamics of Plasmodium vivax malaria. PLoS Genet. 2020;16:e1008576.
Turkiewicz A, et al. Population genetic analysis of Plasmodium knowlesi reveals differential selection and exchange events between Borneo and Peninsular sub-populations. Sci Rep. 2023;13(1):2142.
Ibrahim A, et al. Population-based genomic study of Plasmodium vivax malaria in seven Brazilian states and across South America. Lancet Reg Health Am. 2023;18:100420.
Benavente ED. et al. Genomic variation in Plasmodium vivax malaria reveals regions under selective pressure. PLoS One. 2017;12(5):e0177134.
Ravenhall M, et al. Characterizing the impact of sustained sulfadoxine/pyrimethamine use upon the Plasmodium falciparum population in Malawi. Malar J. 2016;15:575.
Samad H, et al. Imputation-Based Population Genetics Analysis of Plasmodium falciparum Malaria Parasites. PLoS Genet. 2015;11(4):e1005131.
Phelan JE, et al. Integrating informatics tools and portable sequencing technology for rapid detection of resistance to anti-tuberculous drugs. Genome Med. 2019;11:41.
Napier G, et al. Robust barcoding and identification of Mycobacterium tuberculosis lineages for epidemiological and clinical studies. Genome Med. 2020;12:114.
Kokot M, Dlugosz M, Deorowicz S. KMC 3: counting and manipulating k-mer statistics. Bioinformatics. 2017;33:2759–61.
Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:2114–20.
Li H. and Durbin R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics. 2010;26:589–95.
Li H, et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–9.
Garrison E, Marth G. Haplotype-based variant detection from short-read sequencing. arXiv. 2012;1207:3907.
Cingolani P, et al. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly (Austin). 2012;6(2):80–92.
Tange, O. GNU Parallel 2018. (2018) https://doi.org/10.5281/ZENODO.1146014.
Poplin R, et al. Scaling accurate genetic variant discovery to tens of thousands of samples. Pac Symp Biocomput. 2019;24:224–35.
Grüning B, et al. Bioconda: sustainable and comprehensive software distribution for the life sciences. Nat Methods. 2018;15:475–6.
Deelder W, et al. Geographical classification of malaria parasites through applying machine learning to whole genome sequence data. Sci Rep. 2022;12:21150.
Li H. Minimap2: Pairwise alignment for nucleotide sequences. Bioinformatics. 2018;34:3094–100.
Edgar RC. MUSCLE: A multiple sequence alignment method with reduced time and space complexity. BMC Bioinformatics. 2004;5:1–19.
Castresana J. Selection of Conserved Blocks from Multiple Alignments for Their Use in Phylogenetic Analysis. Mol Biol Evol. 2000;17:540–52.
Kumar S, Stecher G, Li M, Knyaz C, Tamura K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol Biol Evol. 2018;35:1547–9.
Kim D, Song L, Breitwieser FP, Salzberg SL. Centrifuge: rapid and sensitive classification of metagenomic sequences. Genome Res. 2016;26:1721–9.
van de Vegte-Bolmer, M, et al. A portfolio of geographically distinct laboratory-adapted Plasmodium falciparum clones with consistent infection rates in Anopheles mosquitoes. Malar J. 2021;20(1):381.
Ocholla H, et al. Whole-genome scans provide evidence of adaptive evolution in Malawian Plasmodium falciparum isolates. J Infect Dis. 2014;210:1991–2000.
Ravenhall, M. et al. An analysis of large structural variation in global Plasmodium falciparum isolates identifies a novel duplication of the chloroquine resistance associated gene. Sci Rep. 2019;9(1):8287.
Osborne A, et al. High throughput human genotyping for variants associated with malarial disease outcomes using custom targeted amplicon sequencing. Sci Rep. 2023;13:12062.
Deelder W, et al. Using deep learning to identify recent positive selection in malaria parasite sequence data. Malar J. 2021;20(1):270.
Grignard L, et al. A novel multiplex qPCR assay for detection of Plasmodium falciparum with histidine-rich protein 2 and 3 (pfhrp2 and pfhrp3) deletions in polyclonal infections. EBioMedicine 2020;55:102757.
Sepúlveda N, et al. Global analysis of Plasmodium falciparum histidine-rich protein-2 (pfhrp2) and pfhrp3 gene deletions using whole-genome sequencing data and meta-analysis. Infect Genet Evol. 2018;62:211–19.
Moon RW, et al. Normocyte-binding protein required for human erythrocyte invasion by the zoonotic malaria parasite Plasmodium knowlesi. Proc Natl Acad Sci U S A. 2016;113:7231–6.
Mohring, F., Hart, M. N., Patel, A., Baker, D. A. & Moon, R. W. CRISPR-Cas9 Genome Editing of Plasmodium knowlesi. Bio Protoc. 2020;10(4):e3522.
MalariaGEN et al. (Pf6). An open dataset of Plasmodium falciparum genome variation in 7,000 worldwide samples. Wellcome Open Res. 2021; 6:42. https://doi.org/10.12688/wellcomeopenres.16168.1
MalariaGEN et al. (Pv4). An open dataset of Plasmodium vivax genome variation in 1,895 worldwide samples. Wellcome Open Res. 2022;7:136. https://doi.org/10.12688/wellcomeopenres.17795.1.
Acknowledgements
We thank members of the LSHTM Malaria Centre for their thoughts on the design of the tool.
Funding
This work was funded by a UKRI MRC Artificial Intelligence for Biomedical and Health Research grant (Ref. MR/X005895/1), a Wellcome iTPA Translational Accelerator Award (214227/Z/18/Z), and a UKRI EPSRC award in Artificial Intelligence innovation to accelerate health research (EP/Y018842/1). SC and TGC are funded by the UKRI MRC (MRC IAA2129, MR/R026297/1, and MR/X005895/1) grants. JG and TGC were funded by a Royal Society FLAIR Collaborative grant (FCG\R1\211029). JGD was supported by fellowships from FAPESP (2019/12068–5 and 2022/02771–3).
Author information
Authors and Affiliations
Contributions
SC and TGC conceived and directed the project. JEP, EM, and JT developed the profiling software and website, and JEP, AT, and EM curated the sequencing data. AT and EM performed bioinformatic and statistical analyses under the supervision of JEP, SC and TGC. JEP, AT, EM, SC and TGC interpreted the results. JEP, AT, SC and TGC wrote the first draft of the manuscript. MvdV-B and TB contributed samples for sequencing, performed by LNV and SC. NTHN, NTHB, NQT, JG, KBB, JGD, SMDS, and CJS tested the software. All authors commented and edited on various versions of the draft manuscript. JEP, AT, EM, SC, and TGC compiled the final manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable for the use of published raw sequence data. Informed consent was provided by the two adult patients treated at the Radboud University Medical Center to sequence the parasite DNA. Permission for the use of LSHTM WGS data has been granted by the UK National Research Ethics Service (Ref: 18/LO/0738) and LSHTM Research Ethics Committee (Ref: 14710). The research conformed to the principles of the Helsinki Declaration. All other data was sourced from published studies, which had obtained ethical approval previously from the appropriate organisations.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Fig. S1.
Schematic highlighting the main steps in the Malaria-Profiler pipeline. Fig. S2. Malaria-Profiler tool.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Phelan, J.E., Turkiewicz, A., Manko, E. et al. Rapid profiling of Plasmodium parasites from genome sequences to assist malaria control. Genome Med 15, 96 (2023). https://doi.org/10.1186/s13073-023-01247-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13073-023-01247-7