
Abstract
Bacterial population genomics is providing detailed insights into Mycobacterium ulcerans evolution, showing how and when this pathogen emerged and spread across the world and in particular within Africa and Australia, the regions most affected by Buruli ulcer (BU). This chapter summarizes our current understanding of the emergence of M. ulcerans from a Mycobacterium marinum ancestor, and describes how at least two evolutionary bottlenecks, that included acquiring the specific capability to make mycolactones, led to a specific lineage of M. ulcerans causing the majority of human disease. At local scales, detailed longitudinal, comparative genomic studies of M. ulcerans isolates recovered from BU endemic regions have revealed patterns of pathogen spread that are providing useful new understandings around BU transmission.
You have full access to this open access chapter, Download chapter PDF
Similar content being viewed by others


1 First Insights from the Complete Genome of Mycobacterium ulcerans
The first genome sequence of a M. ulcerans isolate was published in 2007 [1]. This finished and completely annotated genome represented an African clinical isolate (strain name Agy99), which was obtained in 1999 from a BU patient living in the Amansie West District of Ghana. An unexpected feature of the genome was the presence of a circular 174 kpb megaplasmid (named pMUM001) [1, 2]. The plasmid harboured three unusually large genes encoding the polyketide synthases (PKS) required for the biosynthesis of the major virulence factor, mycolactone [2, 3]. The 5.6 Mbp circular chromosome of the Agy99 genome also held some surprises, with the architecture of a bacterium undergoing reductive evolution. There was an abundance of pseudogenes (15% of the predicted ancestral protein-coding genes had been inactivated by accumulated mutations), evidence of large chromosome deletions and rearrangements, and extensive proliferation of two insertion sequences (IS2404 and IS2606) [1]. Collectively, these features pointed to a bacterial population that had ‘recently’ passed through an evolutionary bottleneck and was adapting to a changed environment. Scrutiny of the types of genes lost by mutation or DNA deletion in the Agy99 genome, suggested a mycobacterium adapting to an environment where the proteins and pathways required to both survive in diverse aquatic environments and persist intracellularly are no longer required. This assessment also showed that M. ulcerans has lost many of the proteins and cell-wall associated molecules known to be potent antigens in other notable mycobacterial pathogens such as Mycobacterium tuberculosis, Mycobacterium kansasii and Mycobacterium marinum. These observations, combined with the presence of pMUM001 and the specific ability of M. ulcerans to make the immunosuppressive small molecule mycolactone, suggested a mycobacterium adapting to a protected niche environment where extracellular persistence and an immune evasion phenotype provide a survival advantage [4, 5].
2 An Aquatic Origin and Two Bottlenecks for a Recently Emerged, and Globally Distributed Pathogen
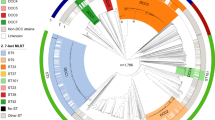
The initial M. ulcerans genome assessment was based on a comparison with the complete genome sequence of a single strain of the fish and human pathogen, M. marinum. From the mid-2000s onwards, scientists began reporting the presence of mycolactone-producing mycobacteria (MPMs) infecting fish, frogs and other ectotherms worldwide [6,7,8,9,10,11]. Subsequent comparative genetic and then comparative genomic studies of these mycobacteria with strains of M. marinum and M. ulcerans confirmed that all MPMs likely emerged during a single evolutionary event when a population of M. marinum-like bacteria acquired a pMUM plasmid and the specific ability to make mycolactones and then spread worldwide (Fig. 1) [12, 13]. The key genetic signatures of extensive pseudogene accumulation, expansion of IS2404 and pMUM plasmid acquisition were present in the ancestor of all MPMs before they radiated around the world, represented by three main lineages, called lineages 1–3 (Fig. 1) [12]. The MPMs have been given a variety of species names including M. pseudoshottsii, M. liflandii, M. shinshuense and (confusingly) M. marinum. Based on their extensive shared genomic features and recent common heritage, it has been proposed that all MPMs should be considered under the single species banner of M. ulcerans [14]. The collective term M. ulcerans–M. marinum complex (abbreviated as MuMC) has also been proposed for all M. marinum and M. ulcerans as they share >97% nucleotide identity across a substantial 4.3 Mbp conserved core genome [12].

A recently emerged and globally distributed pathogen
This first major evolutionary bottleneck that saw the emergence of M. ulcerans has been followed by at least one further population constriction that gave rise to lineage 3. This lineage, also designated the ‘classical’ lineage in the literature [15], represents the M. ulcerans isolates causing Buruli ulcer in Australia, Papua New Guinea (PNG), and Africa, and is thus the lineage that accounts for most of the human cases of Buruli ulcer. This lineage is characterized by additional reductive evolution and expansion of IS 2606 to high copy numbers [12]. Interestingly, the ‘success’ of this lineage as the major human pathogen among all the MPMs is not due to the acquisition of additional genes. Lineage 3 contains no additional DNA sequences compared to isolates in lineages 1 and 2, thus more subtle genetic changes might underlie lineage 3 dominance. One potential genomic region for such changes is the mycolactone PKS gene locus on the pMUM plasmid. The different lineages of M. ulcerans produce different mycolactone sidechain structural variants with varying biological potencies (discussed in more detail in chapter “Mycolactone: More than Just a Cytotoxin” of this book). The genetic basis for some of these variants has been determined. Changes in acyltransferase domain substrate specificity within a particular PKS extension module or loss of an extension module within mlsB, the gene required for synthesis of the mycolactone sidechain, lead to biosynthesis of different mycolactones [16,17,18].
Given the ancestor of all M. ulcerans was an aquatic mycobacterium (M. marinum) and many of the MPMs are recovered from fish, frogs, and turtles, it seems likely that the origin of all MPM (including those lineages associated with Buruli ulcer) had an aquatic animal association. These ideas and inferences should be used to frame thinking around potential reservoirs of M. ulcerans in BU endemic areas. Of note too, the phylogeny of M. ulcerans from Australia and Papua New Guinea is ancestral to that of M. ulcerans from African countries, indicating that the spread of M. ulcerans throughout Australasia likely predates the spread of M. ulcerans across Africa [33].
3 New Understandings from Genomics on the Spread of M. ulcerans Across Africa
African countries carry the highest burden of BU, but until recently, knowledge on the spread of M. ulcerans across the continent has been sparse. Early studies of M. ulcerans populations using traditional genotyping techniques readily identified a highly clonal population structure, showing that M. ulcerans isolates had very conserved genomes that associated strongly with their geographic origin [19,20,21,22,23,24]. However, these techniques sampled only a small proportion of the mycobacterial genome, and thus lacked sufficient discriminatory power to crack the clonal population structure of this species at even the scale of a country, let alone at the scale of a BU endemic village. The advent of low cost, high throughput DNA sequencing has given researchers access to all the potential genetic variation arising within a M. ulcerans population and several studies have capitalised on this advance. Most recently, the genomes of an extraordinary collection of 165 M. ulcerans clinical isolates spanning 48 years and representing 11 endemic countries across Africa were sequenced and compared. Assessment of these genomes has produced the first detailed understanding of the introduction and continental spread of this pathogen. Key findings included the establishment of a molecular clock signal in the sequence data, suggesting that M. ulcerans is accumulating mutations at a rate of 0.33 SNPs per chromosome per year (approx. 6 × 10−8 substitutions/site/year). This rate of genetic change is comparable to M. tuberculosis, but 10–100 times slower than that of other pathogens such as Staphylococcus aureus [25]. Combining these temporal data with the high-resolution phylogeny inferred from genome comparisons and the geographic origins of the 165 isolates has permitted a reconstruction of the spread of M. ulcerans across Africa. The data suggest that there have been two distinct introductions of the bacteria to the African continent; the first occurring around 68 BC, with a likely origin in the area around current day Cameroon and Gabon, and then spreading outwards from these regions [26] (Fig. 2). This has been followed by a second, far more recent introduction during the 1800s. For both the early and recent sub-lineage (called Mu_A1 and Mu_A2 respectively; both representing sub-lineages of lineage 3), there were geographically localized but substantial clonal expansions of M. ulcerans populations in four particular hydrological basins (Congo, Kouffo, Oueme, and Nyong). These expansions occurred contemporaneously from the late 1800s onwards and in waves that mimicked interference in those specific regions by colonial powers during the ‘scramble for Africa’ [26].

Spread of two distinct M. ulcerans lineage 3 genotypes across Africa
Interestingly, the Mu_A2 genotype was also found in PNG, and phylogenetic inference suggests PNG may have been the origin of the Mu_A2 genotype. How the bacteria were transported across oceans and continents from south east Asia to Africa in the mid 1800s is not clear, although again, several European colonial powers were active in both these regions of the world at that time.
The presence of the Mu_A2 genotype in Africa indicates M. ulcerans can be mobilised and displaced across large distances, but the dominant characteristic of M. ulcerans populations is their strong, geographically restricted genotypes. In Africa, these constrained genotypes align with different hydrogeological basins [26]. This observation is consistent with the well-described epidemiology of BU, where human disease is strongly associated with lentic and lacustrine environments, and also supports the notion of an aquatic reservoir for the bacterium (or at least a reservoir species restrained somewhat by these basins), with the hydrogeography likely providing a major barrier to reservoir (and therefore pathogen) movement [26, 27].
In South Eastern Australia, the native possum is a wildlife reservoir of M. ulcerans and there is a strong association between possums harbouring the bacteria and human Buruli ulcer [28, 29]. Genomics has shown that possum and patient M. ulcerans isolates have identical genotypes [29] indicating that humans and possums are part of the same transmission network.
4 Genomic Approaches to Micro-Molecular Epidemiological Investigations of BU
There were expectations at the dawn of the genomic revolution that comparisons of M. ulcerans genomes collected from BU patients in endemic areas would reveal striking patterns of bacterial spread that in turn would lead to a substantially deeper understanding of how this enigmatic disease is transmitted [15]. Several M. ulcerans population genomic studies have now been conducted in Africa and Australia at the descending scales of country, region and village. Important new insights have been made, whilst also raising new questions about how M. ulcerans is spreading.
Several teams have explored genomic variation of M. ulcerans populations in West Africa and they have all reported and confirmed the strong association between genotype and region as described above. This relationship has a fractal quality, where genotypes continue to associate with region across large spatial scales; from continental, to country, to regional levels. However, somewhat unexpectedly, microepidemiological observations powered by the extreme resolution offered by whole genome sequence comparisons have shown that this association breaks down at very local scales (buffer size <50 km2), where the distribution of M. ulcerans genotypes appears to become random. This finding is illustrated in several independent studies from Ghana and Cameroon that report clonal complexes aligned with specific river systems as predicted from earlier pre-genomic studies, but there is also mixing of genotypes at district and village levels [30,31,32]. Conclusions from these observations include the possibility that (1) the disease is being vectored by a somewhat mobile entity, perhaps an insect or (2) people are moving to different local areas and acquiring the infection. While the data are not conclusive for any specific hypothesis, there are examples of very young infants with no history of any travel developing BU and becoming infected with genotypes present in more distant villages, thus suggesting something(s) is vectoring M. ulcerans to susceptible human hosts [32].
In addition to the genotypic mixing at local scales, there are also several examples now of very distinctly different genotypes co-circulating with a ‘local’ endemic M. ulcerans clone. This was first described in the Asante Akim North district of Ghana, where a genotype reminiscent of strains originating from Nigeria was detected alongside strains representing a local endemic clone [32]. The same phenomenon was recently described in south east Australia, with a distinct M. ulcerans clone from the far east of the country detected in a highly BU endemic area, co-circulating with a local clone [33]. These studies demonstrate the potential for M. ulcerans to be not recent at all mobilized and spread over large distances. The mechanisms of pathogen dispersal remain to be discovered, but perhaps it could be linked to the movement of human or other mammalian reservoirs [30, 31, 33].
M. ulcerans population genomic studies of highly active BU endemic areas in south east Australia have revealed some interesting features of the pathogen and the disease [33]. A study of 178 M. ulcerans genomes, collected over 70-years has provided a compelling reconstruction of the temporal and spatial spread of the pathogen in that region. The disease appears to have emerged in the early 1800s in the east of the country and then spread suddenly westward, moving into areas around the major population centre of Melbourne in the 1980s. Comparison of the temporal phylogeny with epidemiological data indicates that arrival of the bacteria in a specific region predates the appearance of human disease by 7–10 years. Similar to the African genomic studies, these observations and inferences suggest strongly that M. ulcerans is spreading, rather than awakening a reservoir of quiescent bacteria, perhaps following environmental disturbance. This information could help inform control strategies for BU, where environmental surveillance of the pathogen might help predict the occurrence of disease in humans.
5 Distinguishing Relapse from Reinfection and Familial Studies
An application of genomics has been to try and establish if a patient presenting twice with BU might have suffered a relapse of the previous infection or has been unlucky enough to be reinfected with a different population of bacteria. Distinguishing between these two scenarios is important for informing treatment options. Work in this area is just beginning, but in a study of four patients with recurrent BU episodes three were concluded to have relapsed, with 0–1 SNP differences between first and last obtained isolates from each patient, while the fourth patient was a possible example of re-infection with 20 SNPs different between first and last isolates [34]. This was the first study to deploy genomics in this way for informing BU treatment. The correct interpretation of these comparisons is obviously dependent on the availability of isolates from the patient over time, but equally important is a detailed understanding of the local population structure of M. ulcerans in that region. This understanding is necessary to give the proper context and therefore interpretation to any SNP differences discovered over time within a single patient. Another focused genomics study with similar aims was reported from Australia, where the research team sequenced and compared six M. ulcerans genomes recovered from three familial clusters [35]. The sequence data, combined with epidemiological information argued against person-to-person transmission and suggested that there was a relatively short time window of 1–2 months when family members were exposed to the pathogen [35].
6 Summary and Future Perspectives
M. ulcerans is likely a niche adapted mycobacterium. Its ability to make mycolactone is almost certainly critical to the persistence of the bacterium in that niche. The bacterium’s primary host and reservoir is likely aquatic. This reservoir is probably restrained within lentic systems. BU spreads when the bacterium moves from one region to another. Pathogen spread can occur rapidly. These are some of the useful insights that genomic approaches have yielded. From glimpses into the unusual biology of the pathogen when the genome was first revealed, to the large, population-based studies that reconstruct pathogen spread, genomics has been the key tool to drive these new understandings. Future research should capitalize on the advent of cost-effective long-read sequencing to focus on establishing and comparing complete M. ulcerans genomes. The additional DNA sequence variation that potentially lies within complex regions of the M. ulcerans genome could prove very informative for efforts to track pathogen spread. More effort should be made too, to interrogate and predict metabolic pathways from genome sequence data that can then be exploited to improve efforts to detect and isolate M. ulcerans in pure culture from environmental sources. The pMUM plasmid in particular should be a focus for complete sequence assembly, as assessment of the mycolactone PKS genes will inform on the basis for the biosynthesis of mycolactone structural variants and differential pathogenesis.
References
Stinear TP, Seemann T, Pidot S, Frigui W, Reysset G, Garnier T et al (2007) Reductive evolution and niche adaptation inferred from the genome of Mycobacterium ulcerans, the causative agent of Buruli ulcer. Genome Res 17(2):192–200
Stinear TP, Mve-Obiang A, Small PL, Frigui W, Pryor MJ, Brosch R et al (2004) Giant plasmid-encoded polyketide synthases produce the macrolide toxin of Mycobacterium ulcerans. Proc Natl Acad Sci U S A 101(5):1345–1349
Stinear TP, Hong H, Frigui W, Pryor MJ, Brosch R, Garnier T et al (2005) Common evolutionary origin for the unstable virulence plasmid pMUM found in geographically diverse strains of Mycobacterium ulcerans. J Bacteriol 187(5):1668–1676
Stinear TP, Seemann T, Harrison PF, Jenkin GA, Davies JK, Johnson PD et al (2008) Insights from the complete genome sequence of Mycobacterium marinum on the evolution of Mycobacterium tuberculosis. Genome Res 18(5):729–741
Demangel C, Stinear TP, Cole ST (2009) Buruli ulcer: reductive evolution enhances pathogenicity of Mycobacterium ulcerans. Nat Rev Microbiol 7(1):50–60
Mve-Obiang A, Lee RE, Umstot ES, Trott KA, Grammer TC, Parker JM et al (2005) A newly discovered mycobacterial pathogen isolated from laboratory colonies of Xenopus species with lethal infections produces a novel form of mycolactone, the Mycobacterium ulcerans macrolide toxin. Infect Immun 73(6):3307–3312
Ranger BS, Mahrous EA, Mosi L, Adusumilli S, Lee RE, Colorni A et al (2006) Globally distributed mycobacterial fish pathogens produce a novel plasmid-encoded toxic macrolide, mycolactone F. Infect Immun 74(11):6037–6045
Rhodes MW, Kator H, McNabb A, Deshayes C, Reyrat JM, Brown-Elliott BA et al (2005) Mycobacterium pseudoshottsii sp. nov., a slowly growing chromogenic species isolated from Chesapeake Bay striped bass (Morone saxatilis). Int J Syst Evol Microbiol 55(Pt 3):1139–1147
Suykerbuyk P, Vleminckx K, Pasmans F, Stragier P, Ablordey A, Tran HT et al (2007) Mycobacterium liflandii infection in European colony of Silurana tropicalis. Emerg Infect Dis 13(5):743–746
Tobias NJ, Doig KD, Medema MH, Chen H, Haring V, Moore R et al (2013) Complete genome sequence of the frog pathogen Mycobacterium ulcerans ecovar Liflandii. J Bacteriol 195(3):556–564
Sakaguchi K, Iima H, Hirayama K, Okamoto M, Matsuda K, Miyasho T et al (2011) Mycobacterium ulcerans infection in an Indian flap-shelled turtle (Lissemys punctata punctata). J Vet Med Sci 73(9):1217–1220
Doig KD, Holt KE, Fyfe JA, Lavender CJ, Eddyani M, Portaels F et al (2012) On the origin of Mycobacterium ulcerans, the causative agent of Buruli ulcer. BMC Genomics 13:258
Yip MJ, Porter JL, Fyfe JA, Lavender CJ, Portaels F, Rhodes M et al (2007) Evolution of Mycobacterium ulcerans and other mycolactone-producing mycobacteria from a common Mycobacterium marinum progenitor. J Bacteriol 189(5):2021–2029
Pidot SJ, Asiedu K, Kaser M, Fyfe JA, Stinear TP (2010) Mycobacterium ulcerans and other mycolactone-producing mycobacteria should be considered a single species. PLoS Negl Trop Dis 4(7):e663
Roltgen K, Stinear TP, Pluschke G (2012) The genome, evolution and diversity of Mycobacterium ulcerans. Infection Genet Evol 12(3):522–529
Hong H, Spencer JB, Porter JL, Leadlay PF, Stinear T (2005) A novel mycolactone from a clinical isolate of Mycobacterium ulcerans provides evidence for additional toxin heterogeneity as a result of specific changes in the modular polyketide synthase. Chembiochem 6(4):643–648
Hong H, Stinear T, Skelton P, Spencer JB, Leadlay PF (2005) Structure elucidation of a novel family of mycolactone toxins from the frog pathogen Mycobacterium sp. MU128FXT by mass spectrometry. Chem Commun 34:4306–4308
Pidot SJ, Hong H, Seemann T, Porter JL, Yip MJ, Men A et al (2008) Deciphering the genetic basis for polyketide variation among mycobacteria producing mycolactones. BMC Genomics 9:462
Portaels F, Fonteyene PA, de Beenhouwer H, de Rijk P, Guedenon A, Hayman J et al (1996) Variability in 3’ end of 16S rRNA sequence of Mycobacterium ulcerans is related to geographic origin of isolates. J Clin Microbiol 34(4):962–965
Stinear TP, Jenkin GA, Johnson PD, Davies JK (2000) Comparative genetic analysis of Mycobacterium ulcerans and Mycobacterium marinum reveals evidence of recent divergence. J Bacteriol 182(22):6322–6330
Hilty M, Yeboah-Manu D, Boakye D, Mensah-Quainoo E, Rondini S, Schelling E et al (2006) Genetic diversity in Mycobacterium ulcerans isolates from Ghana revealed by a newly identified locus containing a variable number of tandem repeats. J Bacteriol 188(4):1462–1465
Stragier P, Ablordey A, Bayonne LM, Lugor YL, Sindani IS, Suykerbuyk P et al (2006) Heterogeneity among Mycobacterium ulcerans isolates from Africa. Emerg Infect Dis 12(5):844–847
Kaser M, Hauser J, Small P, Pluschke G (2009) Large sequence polymorphisms unveil the phylogenetic relationship of environmental and pathogenic mycobacteria related to Mycobacterium ulcerans. Appl Environ Microbiol 75(17):5667–5675
Jackson K, Edwards R, Leslie DE, Hayman J (1995) Molecular method for typing Mycobacterium ulcerans. J Clin Microbiol 33(9):2250–2253
Duchene S, Holt KE, Weill FX, Le Hello S, Hawkey J, Edwards DJ et al (2016) Genome-scale rates of evolutionary change in bacteria. Microb Genom 2(11):e000094
Vandelannoote K, Meehan CJ, Eddyani M, Affolabi D, Phanzu DM, Eyangoh S et al (2017) Multiple introductions and recent spread of the emerging human pathogen Mycobacterium ulcerans across Africa. Genome Biol Evol 9(3):414–426
Vandelannoote K, Jordaens K, Bomans P, Leirs H, Durnez L, Affolabi D et al (2014) Insertion sequence element single nucleotide polymorphism typing provides insights into the population structure and evolution of Mycobacterium ulcerans across Africa. Appl Environ Microbiol 80(3):1197–1209
Carson C, Lavender CJ, Handasyde KA, O’Brien CR, Hewitt N, Johnson PD et al (2014) Potential wildlife sentinels for monitoring the endemic spread of human buruli ulcer in South-East Australia. PLoS Negl Trop Dis 8(1):e2668
Fyfe JA, Lavender CJ, Handasyde KA, Legione AR, O’Brien CR, Stinear TP et al (2010) A major role for mammals in the ecology of Mycobacterium ulcerans. PLoS Negl Trop Dis 4(8):e791
Lamelas A, Ampah KA, Aboagye S, Kerber S, Danso E, Asante-Poku A et al (2016) Spatiotemporal co-existence of two Mycobacterium ulcerans clonal complexes in the Offin River Valley of Ghana. PLoS Negl Trop Dis 10(7):e0004856
Bolz M, Bratschi MW, Kerber S, Minyem JC, Um Boock A, Vogel M et al (2015) Locally confined clonal complexes of Mycobacterium ulcerans in two Buruli ulcer endemic regions of Cameroon. PLoS Negl Trop Dis 9(6):e0003802
Ablordey AS, Vandelannoote K, Frimpong IA, Ahortor EK, Amissah NA, Eddyani M et al (2015) Whole genome comparisons suggest random distribution of Mycobacterium ulcerans genotypes in a Buruli ulcer endemic region of Ghana. PLoS Negl Trop Dis 9(3):e0003681
Buultjens AH, Vandelannoote K, Meehan CJ, Eddyani M, de Jong BC, Fyfe JAM et al (2018) Comparative genomics shows Mycobacterium ulcerans migration and expansion has preceded the rise of Buruli ulcer in south-eastern Australia. Appl Environ Microbiol 84(8):15
Eddyani M, Vandelannoote K, Meehan CJ, Bhuju S, Porter JL, Aguiar J et al (2015) A genomic approach to resolving relapse versus reinfection among four cases of Buruli ulcer. PLoS Negl Trop Dis 9(11):e0004158
O’Brien DP, Wynne JW, Buultjens AH, Michalski WP, Stinear TP, Friedman ND et al (2017) Exposure risk for infection and lack of human-to-human transmission of Mycobacterium ulcerans disease, Australia. Emerg Infect Dis 23(5):837–840
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Open Access This chapter is licensed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license and indicate if changes were made.
The images or other third party material in this chapter are included in the chapter's Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the chapter's Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.
Copyright information
© 2019 The Author(s)
About this chapter

Cite this chapter
Vandelannoote, K., Eddyani, M., Buultjens, A., Stinear, T.P. (2019). Population Genomics and Molecular Epidemiology of Mycobacterium ulcerans. In: Pluschke, G., Röltgen, K. (eds) Buruli Ulcer. Springer, Cham. https://doi.org/10.1007/978-3-030-11114-4_6
Download citation
DOI: https://doi.org/10.1007/978-3-030-11114-4_6
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-11113-7
Online ISBN: 978-3-030-11114-4
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)