
Abstract
To explore ultra-fast dynamics in quantum systems one needs detection schemes which allow time measurements in the attosecond regime. During the recent decades, the pump & probe two-pulse laser technique has provided milestone results on ultra-fast dynamics with femto- and attosecond time resolution. Today this technique is applied in many laboratories around the globe, since complete pump & probe systems are commercially available. It is, however, less known or even forgotten that ultra-fast dynamics has been investigated several decades earlier even with zeptosecond resolution in ion-atom collision processes. A few of such historic experiments, are presented here, where the particle motion (due to its very fast velocity) was used as chronometer to determine ultra-short time delays in quantum reaction processes. Finally, an outlook is given when in near future relativistic heavy ion beams are available which allow a novel kind of “pump & probe” experiments on molecular systems with a few zeptosecond resolution. However, such experiments are only feasible if the complete many-particle fragmentation process can be imaged with high momentum resolution by state-of-the-art multi-particle coincidence technique.
You have full access to this open access chapter, Download chapter PDF
Similar content being viewed by others

1 Introduction
To explore the nature of atomic matter scientists have developed during the last century sophisticated approaches to reveal the microscopic structure of matter and also the dynamics between atoms or even inside atoms and molecules. The resolving power for static structural features of molecular systems, e.g. measured by Cryo-electron microscopy [1] or X-ray spectroscopy [2], is presently in a range of a few 10−10 m, which is about a few times the diameter of a single atom. In these measurement approaches the momenta of electrons or photons scattered on a molecular object are detected. The measured momentum distributions, are converted by Fourier transformation into coordinate space, yielding a spatial image of the molecular structure.
To explore the dynamics of a reaction between quantum objects or to reveal the electron dynamics inside a quantum object the experimenter in general interacts with a fast projectile (photon, electron, ion etc.) in a first step (the excitation or ionization step) with the quantum object and observes after very short time delays (typically attoseconds) electron and ionic fragment emission. Thus, the experimenter obtains information on the dynamically changed final states or even intermediate states of the object.
In order to reveal the “entangled” electron dynamics inside the same molecule, it is typically not sufficient to perform single parameter measurements on the same molecular object at two shortly successive instants in time. To reveal entangled dynamics, the simultaneous detection of the momenta of all fragments emitted from the same single molecule is required. Thus, a multi-fragment coincidence measurement imaging the complete momentum space with high resolution is necessary. Such high-resolution multi-coincidence detection systems are available since about two decades: The COLTRIMS-reaction microscope C-REMI possesses all necessary properties to perform such high-resolution multi-coincidence investigations [3].
What are the time delays Δt of interest? The duration of a chemical reaction is typically in the order of a picosecond, and, accordingly, a nucleus can be considered as locally frozen during a time interval of a femtosecond [4]. Therefore, to explore such nuclear processes a time resolution of about one femtosecond (10−15 s) is required. This is the standard time range where modern femtosecond Laser pump-and probe-schemes can visualize chemical reactions, geometrical changes and their dynamics [5]. Intra-atomic or intra-molecular processes do proceed faster. The duration of a charge transfer process in ion-atom or ion/molecule collisions depends on the projectile velocity and on the quasi-molecular promotion path-way [6]. It can be as short as an attosecond (10−18 s). Hole migration in photon-excited molecules can proceed also in the attosecond range, as well as intra-atomic or intra-molecular Coulombic vacancy or energy transfer. Interatomic (or intermolecular) electronic decay processes occur in a wide range of durations from few femtoseconds to several picoseconds [7]. Energy transfer processes as in photosynthesis of chlorophyll, for example, proceed on the upper femtosecond level.
In fast ion-atom collisions intra-atomic and intra-molecular dynamics can take place even on the lower zeptosecond level (10−19 s), which is about 4 orders of magnitude shorter than the typical femtosecond Laser pulse can resolve [8,9,10,11,12]. In such a short time interval light travels only a distance of 0.3 Å. To visualize these ultra-fast dynamics, one needs detection methods which visualize its time dependence, e.g. in interference structures like in quantum-beats. As will be shown below when dynamical processes proceed via two different pathways they accumulate different phases yielding characteristic interference structures. From these structures phase differences can be determined and, as outlined before, by knowing the velocity of the fast ion, time delays even in the zeptosecond regime can be deduced [9].
It may be a more theoretical and philosophical issue whether ultra-short time scales below one attosecond may be of any relevance in atomic physics. But these time scales are doubtlessly of high interest in quantum physics, in general. For example, fundamental questions arise, as whether the so-called “collapse” of a wave function is a local process and starts in one location inside a molecule and proceeds then with speed of light through the whole molecular system. In this case the “collapse” would last about 300 zeptoseconds to stretch across a simple molecule. Or is the collapse a non-local process instantaneously present everywhere across the molecule? Measurements with 10 zeptosecond time resolution would allow to explore such a fundamental question e.g. in a triatomic molecule with a non-linear geometry (see Sect. 3.3).
2 Ultra-fast Chronometer Mechanisms Using Fast Moving Particles as Clock
Burgdörfer et al. [8] have recently presented a review on the historic development and the present status of attosecond physics performed in the field of ion-atom collisions and short-pulse Laser physics. By discussing the theoretical aspects, they have shown the similarity of ion- and Laser-induced processes. Since the chronometer scheme of the multi-photon pump & probe technique is discussed widely in [5, 8, 12], this paper will concentrate on ionization processes induced by ion impact, where the motion of particles provides the ultra-fast chronometers.
The method of “pumping” a quantum object to an excited or ionized state and “probing” this excited state, i.e. by observing the delayed fragment or photon emission, is in general the principle of any measurement in reactions between quantum particles*. So-called “Pump & Probe” measurements are today commonly identified with Two-pulse Laser Pump & Probe methods where the very short time delay Δt between the two Laser pulses can be well adjusted by two different geometrical path ways yielding a time resolution in the femto- or even attosecond range. In this Laser Pump & Probe approach the probing is processed via a delayed second photon pulse where the delay time can be chosen by the experimenter.
In ion-atom collisions the experimenter can never prepare two projectiles ions such that they interact with an atom or molecule at the same impact parameter with a well-controlled time delay of attosecond precision to undergo like in Laser physics a Pump & Probe process. The “pump & probe” process in ion-atom collisions must be induced by the same ion at two different locations in a molecule. Since the relative locations of atoms in a molecule are known with a precision of about 0.1 a.u., the delay-time between the ion reaction at two different locations can then be varied by changing the ion velocity—and typically achievable ion velocities correspond to 10 zeptosecond-pump-probe-resolution.
2.1 Historic Life-Time Measurements with Nano- and Picosecond Precision
Measurements with time-delay determination have been performed already 100 years ago, e.g. by Stern and Volmer [13], when they measured the mean decay time of photon-excited I2 molecules (Fig. 1). Since many of the articles in these proceedings of the Otto-Stern conference are related to Otto Stern’s scientific work we will shortly discuss here Stern’s and Volmer’s pioneering work of measuring life times of excited molecules, too. Stern and Volmer used the thermal motion of vapor molecules as chronometer. The excited molecules expanded from a tiny interaction spot of a few micrometer diameter, where they have been excited, according to their thermal motion (i.e. the motion of the molecules created streaking). Stern and Volmer observed the excitation and decay positions of the molecules using a light microscope. From the outreaching tails of the excitation spot Stern and Volmer derived the mean life time for the decay process of the excited states with a time resolution of about 2 ns.

Inside the glass tube I2 molecules were evaporated from solid Iodine. A very narrow collimated photon beam (1 µm diameter) excited the molecules (small quadratic box). The fluorescence light emission was observed in a greater halo region due to the thermal motion of the excited molecules. This halo distribution was measured using a lens system. From the halo distribution and the thermal properties, the mean decay lifetime was determined [13]
In the past, numerous methods have been developed and applied to measure decay times and explore dynamics in atomic and molecular systems. For historic reasons we describe here also the so-called “beam-foil” techniques [14]. A fast ion beam (kinetic energy typically 0.1 to 10 MeV/u) penetrates a very thin foil. Inside the foil ions get excited and decay downstream the moving beam. The emitted fluorescence photons are detected with a position-sensitive photon detector. A Soller-slit system allows only photons emitted transversely to reach the detector.
Thus, the exponential decay distribution as function of distance from the foil (i.e. decay time) is measured. From the exponential slope one can calculate the delay time with picosecond precision (“beam-foil” techniques see Fig. 2) [14]. This is a kind of streaking technique where from the observable positions (foil and decay) and from the ion velocity the decay time is deduced by macroscopic methods.

Beam foil spectroscopy with fast ions [14]
If inside the foil two nearby ionic levels can be excited simultaneously, the time-resolved fluorescence light emitted from these coherently excited levels can show a characteristic quantum beat structure.
2.2 Quantum Beat Structures as Ultra-fast Chronometers
Ultra-short time interval measurements can be performed also with fast moving particles. As “clock” the fast motion of ions is used whose trajectory can be considered as classical. Measuring transition probabilities resulting from two spatially localized interaction areas the two transitions amplitudes interfere yielding a characteristic interference pattern. Since the delay-time can be calculated from the classical motion of the ion and from the locations where transitions occur, the phases can be determined from the measured interference pattern and thus information on the dynamics of the reaction process can be derived.
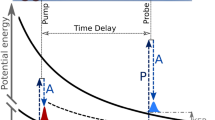
This superposition scheme of two wave amplitudes for moving atoms emitted at time t0 and t1 (see Fig. 3) was already applied in Ramsey’s “Separated oscillating field method”, which is the basis of the atomic clock [15]. In Ramsey’s pioneering experiment a moving object (in Fig. 3 indicated as molecular beam) passes through two cavities and the moving object can be excited either at t0 or t1. Since the experimenter does not know in which cavity the excitation took place both excitation amplitudes at t0 and t1 add coherently. From the interference structure measured in the excitation probability (see right side of Fig. 3) the phase difference ΔΦ of both amplitudes can be determined. From ΔΦ and the known time delay T the transition energy can be deduced with high precision.

The principle scheme of Ramsey’s “Separated oscillating field” device. A fast-moving object (here indicated as molecular beam) passes two coherent cavities at time t0 and t1 respectively, and the object can be excited at time t0 and t1. From the interference structure measured in the excitation probability (see right side) the phase difference ΔΦ of both amplitudes can be determined [15]
3 Experimental Examples of Quantum-Beat Measurements in Ion-Atom/Molecule Collisions
3.1 Quantum Beats in Quasi-molecular X-Ray Emission
In specially prepared ion-atom collision processes one can use the fast classical motion of an ion as a very fast clock to visualize even electronic dynamics with a time resolution in the lower zeptosecond regime. By measuring the quantum beat structure in the spectra of quasi-molecular X-ray emission Schuch et al. [9] have obtained in fast ion-atom-collisions even a time resolution of nearly 10 zeptoseconds. The X-rays emitted in a reaction visualize the streaking of the quasi-molecular orbitals by the two-center ion-atom nuclear potential, which provides a very fast, with time varying streaking force. The X-ray photon energies encode, thus, the fingerprints of the strength of the streaking force at the moment of emission and yield information on Δt.
The purpose of the experiment by Schuch et al. [9] was to measure the binding energy of the very short living quasi-molecular 1sσ-state which was formed in fast ion-atom collisions for a time duration of only about 1 attosecond. More than 30 years ago several groups (quasi-molecular radiation) [10] and (K-K vacancy sharing) [11] have measured quantum beat structures in ion-atom collisions with oscillations in the atto- and zeptosecond range. From the interference structures of these quantum beats, phase differences were determined yielding energy or time domain information. In Fig. 4 the scheme of such an ion-atom scattering process is presented. An X-ray/scattered-projectile coincidence measurement is required to reveal such quantum-beat structures for a given impact parameter. To probe such a short living quasi-molecular state with the detection techniques of the eighties was extremely difficult since the achieved quasi-molecular X-ray/scattered-projectile coincidence rate was a few true counts per hour. In the laboratory system the projectile ion by-passes an atom on a quasi-straight line (very small deflection angles of about 1°, which are determined by detecting the scattered projectile deflection angle). From the deflection angle the impact parameter b can be deduced. Since the ion velocity is known the internuclear distance R (vector) can be calculated as function of the relative collision time t (t0 = 0 is the time moment at distance of closest approach).

Scheme of an ion-atom scattering process
If R is much smaller than the projectile ion or target atom K-shell radii even the most-inner electronic orbitals steadily approach during this extremely short (sub-attosecond) collision time the united-atom electronic states due to the combined projectile and target nuclear Coulomb potentials. Thus, the combined nuclear potentials “streak” as function of R = R(t) the energy values of the bound quasi-molecular states (see Fig. 5).

Quasi-molecular correlation diagram for Cl16+ on Ar as function of the internuclear distance R. During the collision an electron from the 2pπ orbital can pass over into the 1sσ orbital and an X-ray is emitted. The X-ray transition energy is the energy difference between the 2pπ and the 1sσ orbital [9] at the particular R value
To reveal the streaked quasi-molecular energy values, one has to prepare the projectile in a very special ionic configuration to create observable quantum beat structures. If one bombards a hydrogen-like Cl16+ projectile ion on an Ar atom, thus, a 1sσ vacancy is already present on the incoming part of the collision and identical X-rays can be emitted on the way into the collision (−t values) and on the way out of the collision (+t values). Thus the transition amplitudes on the first half (way into the collision) interfere with those of the second half of the collision. Like in a double slit experiment (see Ramsey’s atomic-clock [15] “separated-oscillating-field method”) one does observe characteristic quantum beat structures in the spectra of quasi-molecular X-rays emitted during the collision (see Fig. 6). Since the quantum beat structures vary with impact parameter the X-rays must be detected in coincidence with scattered projectiles to select one given scattering angle (i.e. a fixed impact parameter-range). The X-ray transition process observed here is the quasi-molecular Kα-transition (electron transition from the 2pπ into the 1sσ quasi-molecular state). The Kα-transition energy in the united-atom limit at very small internuclear distances (ZUA = ZBromium = ZCl + ZAr = 35) is about 12 keV (see Fig. 7). These X-rays are emitted per definition at the time moment t0. X-rays of lower energy are emitted at larger internuclear distances R, i.e. larger -t or +t values (see Fig. 5). Thus, the collision time parameter t = R(t) is zero at Ex of the united atom (Ex = 12 keV) and increases to larger R values, i.e. lower X-ray energies.

Measured X-ray spectra for the 2pπ − 1sσ quasi-molecular transition in Cl16+-Ar collisions [9] at fixed impact parameters. The X-ray energy is directly measured with a Si(Li)-detector and can be transferred into an internuclear distance R via the correlation diagram (Fig. 5). From the internuclear distance R and the ion velocity the time scale can be calibrated

Transition energies between 1sσ and 2pπ quasi-molecular states as function of the internuclear distance R. The dashed-dotted line results from DFS calculations for the 1sσ − 2pπ transition; solid and dashed lines are scaled from H+ + H for the 2pσ − 1sσ and 2pπ − 1sσ transition, respectively [9]. The time scale corresponds to 5 MeV collision energy
From Fig. 7 we derive that each X-ray transition energy corresponds to a well-defined internuclear distance and thus to a well-defined collision moment. Thus, we can visualize the variation of the quasi molecular Coulomb potential with nearly 10 zeptosecond resolution. Furthermore, the observed quantum beat structure yields a phase information with \({\varphi }\left( {\text{t}} \right) =_{{{\text{t}} = 0}} \smallint^{\text{t}} {\text{E}}_{\text{x}} ({\text{R}}({\text{t}}))/\hbar \cdot {\text{dt}}\). The highest X-ray energies correspond to Rmin or t = 0. According to [9] one can now—vice versa—determine the transition energies Ex(R(t)) as function of the collision time, i.e. the internuclear distance R. The final result is shown in Fig. 7, where from all measured spectra (different impact parameters and different collision energies) the quasi-molecular energy values as function of the internuclear distance are displayed.
The analysis of the quantum beats shows that for Ex as function of R an universal curve is obtained independent of the ion-atom collision energy, i.e. independent of the streaking time. For 20 MeV collision energy the R scale (from R = 0 up to 0.1 a.u.) in Fig. 7 corresponds to 500 zeptoseconds, for 5 MeV to 1 attosecond. Such measurements [9,10,11] show that using ion-atom collisions the dynamics of quasi-molecular states could be explored with 10 zeptosecond resolution.
3.2 Young-Type Interference Structures in Slow H2+ +He Collisions
Schmidt et al. have [6] investigated “Young-type interference structures” in 10 keV H2+ + He => H *2 + He+ => H + H + He+ collisions (relative velocity v = 0.45 a.u.). They measured the momentum vectors of all reaction fragments in the final state in coincidence using the C-REMI approach. Thus, the orientation of the H2+ molecule with respect to the projectile momentum vector was determined for each event yielding the He+ scattering distribution in dependence of the angle θ (Fig. 8). In the moving projectile system, the He atom can be scattered by both H2+ projectile nuclei (double slit) (see Fig. 8). The scattered He+ momentum wave function is then the coherent sum of the two amplitudes emerging from the two H nuclei scattering centers. From the resulting interference structures in the He+ scattering distribution the phase shifts between the two amplitudes can be deduced as function of Θ visualizing the tiny time delay between the interaction of the He atom with the first (t1) and second (t2) H atom (see Fig. 8).

Nuclear scattering scheme. In inverse kinematics the He target atom collides with the H2+ molecular ion and transfers one electron to the H2 molecule which breaks up into neutral H atoms. The two H atoms are detected at small scattering angles in forward direction. The He+ ion is detected with a C-REMI under 90°. The He is scattered into the azimuthal angle Φ. The relative orientation of the molecule to the He impact direction is defined by the angle Θ. The time difference t2 − t1 can be determined from the measured angle Θ and the ion velocity
The phase difference between both amplitudes due to the molecular orientation θ is proportional to the measurable delay time (Δt = t2 − t1). In a multi-particle coincidence measurement one can also calculate it directly from the measured angle θ and from the ion velocity v.
Figure 9 clearly shows that the interference pattern varies with θ and even with the KER value (electronic excitation), too. Schmidt et al. presented a model calculation (red dashed line in Fig. 10) for the superposition of the scattering amplitudes at the two H atoms. The differential scattering cross sections can be expressed as dσ/dφ ~ cos2(β/2) where the phase shift ß is ß = π + R · ΔpHe/ħ + ΔE · Δt/ħ. The phase jump π accounts for the inversion of the molecular symmetry in the electronic transition and the second term for the change of the He momentum ΔpHe due to the scattering. which leads to a change of the de Broglie wave length of the scattered He and for the term ΔE · Δt/ħ. It accounts for the correction of the so-called translation factor with Δt = t2 – t1 (see Fig. 8) as time difference of the interaction of the He projectile with the two H atoms

Two-slit interference pattern in the plane perpendicular to the projectile momentum vector for three different θ angles. a Events for molecular orientation angles (with respect to the beam direction) from 80° to 90° and KER* values 1 to 2 eV. This KER corresponds to R values from 2.3 to 2.9 a.u. b Events for molecular orientation angles from 50° to 60° and KER 3 to 4 eV. This KER corresponds to R values from 2.3 to 2.9 a.u. c Events for molecular orientation angles from 80° to 90° and KER 3 to 4 eV. This KER corresponds to R values from 1.7 to 2.0 a.u. [6]

Relative cross sections as function of the angle φ in comparison with model calculations (red dashed lines) and full theory (solid lines) [6]. The left column represents collisions where θ is 85–90°, the middle column where θ is 55–60° and the right column where θ is 45–50°. The rows show data for different transverse momenta and slightly different KER values
In Fig. 10, the measured interference pattern are compared with these model calculations (red dashed lines) and the full theory (black lines) [6]. The data are presented for a given polar scattering angle (i.e. impact parameter) as function of the azimuthal angle Φ. In the left column (a + d + g) of Fig. 10 the data are shown for θ = 90° and Δt = 0. In the middle column (b + c + h) θ = 60° and the right column (c + f + i) data are shown for θ = 45° where the influence of the delay Δt on the scattering as well as electronic excitation becomes strongly visible. The data of Schmidt et al. [6] prove in a convincing manner that the electronic excitation processes as function of the delay Δt does vary.
The full theory (only for the phase variation, not including electronic dynamics for transition probabilities) describes rather well (besides g and h) the measured phase variations. The model calculations (red dashed lines) do not include the nuclear Δt phase effect. They are shifted by about 20° to higher angles and indicate the large effect of Δt on the phases. The good agreement in phase of the full theory proves that the time delay calculated from the geometry and ion velocity are properly taken into account. The time delay Δt (derived from the collision geometry, see Fig. 8) is for θ angles between 45° to 50° Δt = R · sin(45°)/vHe = 2.3 a.u. · sin(45°)/0.45 a.u. = 3.6 a.u. = 87 attosecond. The disagreement in the absolute height shows, that the electron dynamics varies with Δt too. The measured data contain also information on the electon dynamics in such reactions.
The limits of the resolution for Δt determined from the experimentally observed phase shifts can be estimated from the data of Fig. 10 and from the comparison with the theoretical calculations. The resolution in determing phases is about 3°. This corresponds for the 10 keV He on H2 collision system to 10 attosec time resolution in such collisions. If the ion velocity increases the ion-motion based clock would gain resolution. The He on H2 collision system investigated by Schmidt et al. [6] clearly demonstrates that the effect of time delay between both scattering amplitudes is nicely visible in the interference structure. The absolute scattering intensities in the different final excitation states of the two H atoms are, however, only in modest agreement with the data. It is to notice, that only such channels were measured by Schmidt et al. where the final H fragments remained in the ground state. To observe more significant differences in the excitation of both atoms of the dipolar molecule one should investigate molecular species with higher Z values.
Since the overall momentum resolution is so excellent, the different channels of electronic excitation in these scattering processes can be resolved event by event, as well, and one can identify different electronic promotion channels during the collision. From Fig. 11 one can deduce that for each event the different electron promotion channels (different molecular orbitals (see Fig. 12 and 13)) with KER and Q value are fully separated. The different electronic promotion pathways are marked by the letters “a” to “f” and “A” plus “B”. In the three-body (He and two proton nuclei) scattering process the He+ ion is mostly scattered out of plane into the angle Φ.


In the 10 keV H2+ + He => H *2 + He+ => H + H + He+ electron transfer process one He electron is captured to metastable H *2 vibrational states (H(1s) + H(2l)) state with an energy minimum at about R = 2 a.u. [6]. Since the internuclear distance R is a function of time, the H *2 fragmentation process provides a fast clock, too. Thus, one can estimate that the time period for the capture (in the He-H2 system) lasts only about a few hundred attoseconds. In this reaction channel a fraction of these states He+ => H + H + He+ decays after the collision during the ongoing Coulomb explosion (duration about some tenth of femtoseconds) into the H(1s) + H(1s) ground state by emitting photons (green oscillation ħω in Fig. 13) with a variable KER energy (see channel A in Fig. 11). The remaining small amount of energy [relative to the ground state H(1s) + H(1s)] is detectable as final KER value.

In the collision 10 keV H2+ on He, one He electron can also be captured to the H(1s) + H(1s) ground state (red arrows in Fig. 12) (region B in Fig. 11) [6]. Like for region A, this electron transfer occurs during the ongoing Coulomb explosion into the H(1s) + H(1s) ground state. The remaining amount of energy (relative to the ground state H(1s) + H(1s)) converts to KER in the final state.
Similar experiments for molecules with higher Z-values have been performed recently by Iskandar et al. [16]. Iskandar et al. investigated collisions of low energy Ar9+ ions on Ar2 dimer targets and measured all ionic fragments in coincidence. From the measured recoil-ion momenta the dimer orientation with respect to the projectile direction, the nuclear transverse momentum transfer (impact parameter) and the KER values could be measured for each event. They found clear evidence that the capture from the first hit atom in the dimer is favored. Because of the large distance of both atoms in the dimer the highly charged ion interacts preferable only with one atom with a high probability of multiple capture. The subsequent intra-molecular vacancy sharing probability between the two Ar atoms in the dimer is low because of the large distance of both atoms in the dimer. They analyzed their data in the “Over-barrier Model” and found reasonable agreement between theory and experiment describing the capture processes. Since their momentum resolution was not good enough to distinguish quasi-molecular promotion path-ways they were not able to explore the fast electronic dynamics.
3.3 A Proposal: Scheme of an Ion-Atom/Molecule Pump & Probe Technique Approaching 10 Zeptoseconds Time Resolution
As already pointed out above the ion-atom/molecule pump & probe scheme has little in common with the Laser pump & probe scheme, in which two Laser pulses can be created as an interlocked pair. No experimenter can produce an ion beam where always two of these ions move as group with a time delay adjusted to 1 attosecond precision and passing through a molecule on an identical trajectory. Even if one could prepare such an ion pair, these two ions would immediately repel each other by their nuclear Coulomb force. Thus, a pump & probe process with ions can only be performed by a single (solely) moving ion, interacting at two different locations in the same molecule. These locations (e.g. two different atoms in the same molecule) must be detectable with subatomic precision. Thus, the experimenter must be able to measure the position (impact parameters => transferred momenta) of the projectile-molecule reaction and the orientation of the ion trajectory with respect to the structure of the molecule. Measuring all ionic fragment momenta in the final state by a multi-coincidence-approach with high momentum resolution allows for a deduction of both—the orientation and the impact parameters. Thus, the experimenter knows in which time sequence and time delays the ion interacted with the different atoms in the molecule. E.g. using the C-REMI approach these requirements can indeed be satisfied.
Since the relative distances between atoms in a molecule can be calculated with about 0.1 a.u. precision, and as a fast ion (particularly a relativistic moving heavy ion) follows a perfect classical straight-line trajectory, the relative time delays between the impact at the two different atoms in a molecule can be determined with a few zeptoseconds resolution. But how can one utilize this kind of timing to investigate electronic dynamics in ion-molecule collisions?
An ion moving with a relativistic velocity (vion => c speed of light) interacts with atoms or molecules via a very sharp retarded dipole-like electric field where the opening angle scales with 1/γ, where γ = 1/sqrt(1 − (v/c)2). For γ = 20 the opening angle is about 20° yielding a short interaction time in the lower zeptosecond regime. As shown in [17] the virtual photon field of relativistic heavy ions as projectiles is very strong. It interacts simultaneously with nearly all electrons in the molecule resulting in multiple ionization of the ionic fragments. To control the electron dynamics in such a collision, the momenta of all ejected electrons must be measured in coincidence, too. Thus, the scheme of an ion induced pump & probe measurement presented here requires a multi-coincidence detection approach which must have a high detection efficiency and excellent momentum resolution for ions and electrons.
In Fig. 14 that scheme of an ion-molecule pump & probe measurement is shown. The direction of the impacting ion beam is precisely prepared, however, the molecules (occurring in the gas phase) are randomly oriented.

Ion-molecule pump & probe scheme
As an example a triatomic molecule is considered here, where the different atoms A, B, and C are bound in a non-linear formation. The distances between the atoms in the molecule are typically in the order of 2 to 3 a.u. In the example shown here, an ion travelling with the speed of light, interacts first with atom A (pump process) and induces the “collapse” of the molecular ground state wave function. After a time delay of about Δt = Δs/c ≈ 3 a.u./137 a.u. ≈ 0.022 a.u. ≈ 480 zeptoseconds the same ion approaches atom C. One interesting question is, when does atom C “know” that atom A suffered the collapse of its wave function? If the collapse is instantly present everywhere in the molecule then atom C is very likely in an excited state, when the ion interacts after about 460 zeptosecond with the molecule at position C. If, however, the collapse emerges from atom A with the speed of light, then the transport of collapse information via atom B to atom C will arrive at best at the same time, but most likely a little later than the relativistic projectile ion. Thus, the observed final state of fragment C may depend on the collapse expansion time.
To explore the nature of the collapse expansion one has to measure the momenta of all ionic fragments and all emitted electrons in the final state in coincidence for all orientations of the molecule with respect to the projectile ion flight direction. A C-REMI provides for each fragment detection a nearly 4π solid angle with about 50% detection efficiency for each fragment. Thus, the total multi-coincidence detection efficiency is rather high (for three ionic fragments and 3 electrons is the total coincidence efficiency still about 2%). However, this multi-particle detection efficiency can be increased dramatically, by optimizing the single particle detection efficiency: enhancing the transmission and open area of the micro channel plates up to 90% [18]. The overall final state of the reaction process may strongly depend in which sequence the ion interacted with the different atoms in the molecule. The overall final state may differ e.g. by mirroring the projectile velocity vector.
A first, “simple” experiment is proposed here, where the final ionic states A* and C* are compared, when the projectile is impinging from “left” or from “right”. Reducing the ion velocity far below the speed of light, the time delay range can be extended. This kind of measurement could already now provide new inside into the range of zeptosecond electronic dynamics. The method proposed here to use ions moving with the speed of light as ultra-fast clocks allows the investigation of fundamental features of “Locality” or “Non-Locality” in quantum systems, i.e., whether the information exchange occurs in such systems instantaneous or only by speed of light.
Theorists may be convinced that the questions raised here are already answered. But nevertheless one should experimentally verify any fundamental theoretical prediction. When Otto Stern decided in 1920 to perform the “Stern-Gerlach-Experiment” [19] and later in 1933 to measure the magnetic moment of the proton [20] theorists tried to convince him, such difficult experiments should not be done since theory had already answered these questions. Nevertheless, he performed his milestone measurements and could disprove theory.
4 Conclusion
This paper does show that since more than 100 years the motion of atoms, molecules or ions was successfully used to measure dynamical features (like lifetimes, phase shifts etc.) in quantum systems. Already more than 30 years ago quantum beat structures with 10 zeptoseconds resolution could be measured about factor 100 to 1000 shorter than present pump & probe two-pulse laser techniques can achieve. The future heavy ion facility FAIR [21] will provide relativistic heavy ion beams with which the here proposed ion-atom/molecule pump & probe technique can be performed to explore dynamics in the zeptosecond regime.
References
W. Kühlbrandt, The resolution revolution. Science, 343, 1443 (2014) and other references therein
F. Calegari, G. Sansone, M. Nisoli, Attosecond Pulses for Atomic and Molecular Physics (2014) Book Lasers in Materials Science, pp. 125–141, Springer International Publishing; A. L. Cavaleri et al., Nature (London) 449, 1029 (2007)
R. Dörner, V. Mergel, O. Jagutzki, L. Spielberger, J. Ullrich, R. Moshammer, H. Schmidt-Böcking, Cold target recoil ion momentum spectroscopy: a ‘momentum microscope’ to view atomic collision dynamics. Phys. Rep. 330, 95–192 (2000); R. Dörner, T. Weber, M. Achler, V. Mergel, L. Spielberger, O. Jagutzki, F. Afaneh, M.H. Prior, C.L. Cocke, H. Schmidt-Böcking, 3-D coincident imaging spectroscopy for ions and electrons, imaging in chemical dynamics, in ACS Symposium Series, vol. 770, ed. by A. Suits, R. E. Continetti, (Oxford Univ. Press, 2001), pp. 339–349; R. Dörner, H. Schmidt-Böcking, V. Mergel, Th. Weber, L. Spielberger, O. Jagutzki, A. Knapp, H.P. Bräuning, From atoms to molecules. Many Part. Quantitative Dynamic Atomic Molecular Fragm., ed. by V.P. Shevelko, J. Ullrich (Springer Verlag, 2002); J. Ullrich, R. Moshammer, A. Dorn, R. Dörner, L.Ph.H. Schmidt, H. Schmidt-Böcking Recoil-ion and electron momentum spectroscopy: reaction-microscopes. Rep. Prog. Phys. 66 1463–1545 (2003)
M. Born, R. Oppenheimer, Zur Quantentheorie der Molekeln. Ann. Phys. 389(20), 457–484 (1927)
A.H. Zewail, Femtochemistry: recent progress in studies of dynamics and control of reactions and their transition states. J. Phys. Chem. (Centennial Issue) 100, 12701 (1996); A.H. Zewail, Femtochemistry: atomic-scale dynamics of the chemical bond†. J. Phys. Chem. A. 104(24), 5660–5694 (2000), adapted from the Nobel Lecture; F. Krausz and M. Ivanov, 2009 Rev. Mod. Phys. 81, 163; M. Drescher, M. Hentschel, R. Kienberger, G. C. Tempea, C. Spielmann, G.A. Reider, P.B. Corkum, F. Krausz, Science 291, 1923 (2001)
L.Ph.H. Schmidt, S. Schössler, F. Afaneh, M. Schöffler, K. Stiebing, H. Schmidt-Böcking, R. Dörner, Young-type interference in collisions between hydrogen molecular ions and helium. Phys. Rev. Lett. 101, 173202 (2008)
T. Jahnke, L. Cederbaum T. Jahnke, A. Czasch, M. S. Schöffler, S. Schössler, A. Knapp, M. Käsz, J. Titze, C. Wimmer, K. Kreidi, R. E. Grisenti, A. Staudte, O. Jagutzki, U. Hergenhahn, H. Schmidt-Böcking, R. Dörner, Experimental observation of interatomic coulombic decay in neon dimers. Phys. Rev. Lett. 93, 163401 (2004); T. Jahnke, A. Czasch, M. Schöffler, S. Schössler, M. Käsz, J. Titze, K. Kreidi, R.E. Grisenti, A. Staudte, O. Jagutzki, L.Ph.H. Schmidt, Th. Weber, H. Schmidt-Böcking, K. Ueda, R. Dörner, Experimental separation of virtual photon exchange and electron transfer in interatomic coulombic decay of neon dimers. Phys. Rev. Lett. 99, 153401 (2007); L.S. Cederbaum, J. Zobeley, and F. Tarantelli; Giant intermolecular decay and fragmentation of clusters. Phys. Rev. Lett. 79, 4778 (1997)
J. Burgdörfer, C. Lemmel, X. Tong, Invited Lecture at ICPEAC 2019, arXiv:2001.02900v1 [quant-ph] 9 Jan 2020
R. Schuch, M. Meron, B.M. Johnson, K.W. Jones, R. Hoffmann, H. Schmidt-Böcking, I. Tserruya, Quasimolecular X-ray spectroscopy for slow Cl16+-Ar collisions. Phys. Rev. A 37, 3313 (1988)
I. Tserruya, R. Schuch, H. Schmidt-Böcking, J. Barrette, Wang Da-Hai, B.M. Johnson, M. Meron, K.W. Jones, Interference effects in the quasimolecular K X-ray production probability for 10 MeV Cl16+- Ar collisions. Phys. Rev. Lett. 50, 30 (1983); R. Schuch, H. Schmidt-Böcking, I. Tserruya, B.M. Johnson, K.W. Jones, M. Meron, X-ray spectroscopy of Cl-Ar molecular orbitals from 1sσ-2pπ transitions. Z. Phys. A 320, 185 (1985)
R. Schuch, H. Ingwersen, E. Justiniano, H. Schmidt-Böcking, M. Schulz, F. Ziegler, Interference effects in K-vacancy transfer of hydrogen like S Ions with Ar. J. Phys. B 17, 2319 (1984); R. Schuch, H. Ingwersen, E. Justiniano, H. Schmidt-Böcking, M. Schulz, F. Ziegler, Experiments with decelerated S15+-beams: interferences in K-shell to K-shell charge transfer, atomic and nuclear heavy ion interactions. Centr. Inst. of Phys. (1986); S. Hagmann, J. Ullrich, S. Kelbch, H. Schmidt-Böcking, C.L. Cocke, P. Richard, R. Schuch, A. Skutlartz, B. Johnson, M. Meron, K. Jones, D. Trautmann, F. Rösel, K-K-charge transfer and electron emission for 0.13 MeV/u F8+ + Ne collisions. Phys. Rev. A 36, 2603 (1987); R. Schuch, M. Schulz, Y.S. Kozhedub, V.M. Shabaev, I.I. Tupitsyn, G. Plunien, P. H. Mokler, H. Schmidt-Böcking, Quantum Interference of K Capture in Energetic Ge31+(1s)-Kr Collisions
J.M. Dahlström, A.I. Huillier, A. Maquet Introduction to attosecond delays in photoionization. J. Phys. B. At. Mol. Opt. Phys. 45, 183001 (2012); J.M. Dahlström and E. Lindroth, Study of attosecond delays using perturbation diagrams and exterior complex scaling, J. Phys. B: Atomic Mol. Opt. Phys. 47, 124012 (2014)
O. Stern, M. Volmer, Über die Abklingungszeit der Fluoreszenz. Physik. Z. 20, 183–188 (1919)
S. Bashkin, Nucl. Instrum. Methods 28: 88 (1964); S. Bashkin, ed., Beam-Foil Spectroscopy (Gordon and Breach, New York 1968); S. Bashkin and I. Martinson, J. Opt. Soc. Am. 61: 1686 (1971); S. Bashkin (ed.), Beam-Foil Spectroscopy Springer, Berlin (1976); H G Berry 1977 Rep. Prog. Phys. 40, 155. View the article online for updates and enhancements. Related content
N.F. Ramsey, A molecular beam resonance method with separated oscillating fields. Phys. Rev. 78, 695 (1950); N. Ramsey, Rev. of Mod. Phys. 62(3) 541–552 (1990)
W. Iskandar, J. Matsumoto, A. Leredde, X. Fléchard, B. Gervais, S. Guillous, Atomic site-sensitive processes in low energy ion-dimer collisions. Phys Rev Lett. 113, 14, 143201
R. Moshammer, J. Ullrich, H. Kollmus, W. Schmitt, M. Unverzagt, H. Schmidt-Böcking, R.E. Olson, The dynamics of target single and double ionization induced by the virtual photon field of fast heavy ions, X-ray and inner-shell processes. AIP Conf. Proc. 389, 153 (1996); R. Moshammer, W. Schmitt, J. Ullrich, H. Kollmus, A. Cassimi, R. Dörner, O. Jagutzki, R. Mann, R.E. Olson, H.T. Prinz, H. Schmidt- Böcking, L. Spielberger, Ionization of helium in the attosecond equivalent light pulse of 1 GeV/nucleon U92+ projectiles. Phys. Rev. Lett. 79, 3621 (1997); R. Moshammer, J. Ullrich, W. Schmitt, H. Kollmus, A. Cassimi, R. Dörner, R. Dreizler, O. Jagutzki, S. Keller, H.J. Lüdde, R. Mann, V. Mergel, R.E. Olson, Th. Prinz, H. Schmidt-Böcking, L. Spielberger, Photodisintegration of atoms in the attosecond equivalent light pulse of highly charged relativistic ions. Phys. Rev. Lett. 79, 3621 (1997); C.F. von Weizsäcker, Z. Phys. 88, 612 (1934); E. J. Williams, Kgl. Danske Videnskab. Selskab Mat.-fys. Medd. 13(4) (1935)
K. Fehre, D. Trojanowskaja, J. Gatzke, M. Kunitski, F. Trinter, S. Zeller, LPhH Schmidt, J. Stohner, R. Berger, A. Czasch, O. Jagutzki, T. Jahnke, R. Dörner, M.S. Schöffler, Absolute ion detection efficiencies of microchannel plates and funnel microchannel plates for multi-coincidence detection. Rev. Sci. Instrum. 89, 045112 (2018)
O. Stern, Ein Weg zur experimentellen Prüfung der Richtungsquantelung im Magnetfeld. Z. Physik 7, 249–253 (1921)
ETH-Bibliothek Zürich, Archive, http://www.sr.ethbib.ethz.ch/, Otto Stern tape-recording Folder» ST-Misc.«, 1961 at E.T.H. Zürich by Res Jost
Acknowledgements
We thank Siegbert Hagmann, Hans Jürgen Lüdde, John Briggs, Mike Prior, and C. L. Cocke for many helpful discussions. Furthermore, we are indebted to the Deutsche Forschungsgemeinschaft and the BMFT for financial support as well as Roentdek company for technical support in performing the experiments.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Open Access This chapter is licensed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license and indicate if changes were made.
The images or other third party material in this chapter are included in the chapter's Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the chapter's Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.
Copyright information
© 2021 The Author(s)
About this chapter

Cite this chapter
Schöffler, M.S. et al. (2021). Ultra-fast Dynamics in Quantum Systems Revealed by Particle Motion as Clock. In: Friedrich, B., Schmidt-Böcking, H. (eds) Molecular Beams in Physics and Chemistry. Springer, Cham. https://doi.org/10.1007/978-3-030-63963-1_17
Download citation
DOI: https://doi.org/10.1007/978-3-030-63963-1_17
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-63962-4
Online ISBN: 978-3-030-63963-1
eBook Packages: Physics and AstronomyPhysics and Astronomy (R0)