
Abstract
Cancer is one of the deadliest diseases in the world, causing over half a million deaths a year in the USA alone. Despite recent advances made in the field of cancer biology and the therapies that have been developed [1, 2], it is clear that more advances are necessary for us to classify cancer as curable. The logical question that arises is simple: Why, despite all the technologies and medical innovations of our time, has a complete cure eluded us? This chapter sheds light on one of cancer’s most impactful attributes: its heterogeneity and, more specifically, the intratumoral heterogeneity of cancer metabolism. Simply put, what makes cancer one of the deadliest diseases is its ability to change and adapt. Cancer cells’ rapid evolution, coupled with their irrepressible ability to divide, gives most of them the advantage over our immune systems. In this chapter, we delve into the complexities of this adaptability and the vital role that metabolism plays in the rise and progression of this heterogeneity.
You have full access to this open access chapter, Download chapter PDF
Similar content being viewed by others


Keywords
FormalPara Key Points-
Heterogeneity is a hallmark of cancer.
-
Clonal evolution theory and cancer stem cell theory explain tumor subpopulation growth.
-
Intratumoral metabolic heterogeneity follows intratumoral genetic alterations.
-
Epigenetics alterations lead to intratumoral metabolic heterogeneity.
-
Intratumoral metabolic adaptation and heterogeneity are due to the extreme conditions of the tumor microenvironment.
-
Spatial and temporal heterogeneity provides survival advantages to tumors.
-
Metabolic profile-targeted therapeutics can result in successful clinical outcomes.
1 Introduction
Cancer is one of the deadliest diseases in the world, causing over half a million deaths a year in the USA alone. Despite recent advances made in the field of cancer biology and the therapies that have been developed [1, 2], it is clear that more advances are necessary for us to classify cancer as curable. The logical question that arises is simple: Why, despite all the technologies and medical innovations of our time, has a complete cure eluded us? This chapter sheds light on one of cancer’s most impactful attributes: its heterogeneity and, more specifically, the intratumoral heterogeneity of cancer metabolism. Simply put, what makes cancer one of the deadliest diseases is its ability to change and adapt. Cancer cells’ rapid evolution, coupled with their irrepressible ability to divide, gives most of them the advantage over our immune systems. In this chapter, we delve into the complexities of this adaptability and the vital role that metabolism plays in the rise and progression of this heterogeneity.
2 Multiple Theories Explain Cancer’s Heterogeneous Nature
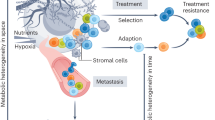
In any observable tumor, there is much more than what meets the eye. In the carcinogenic environment, we can observe a microcosm of the theory of evolution at play. While Darwin’s theory was proposed to explain the evolution of species due to slowly cumulative changes that arise from natural selection, cancer cells, driven by their genetic instability and high reproductive rates, develop and evolve in a fraction of our lifetime, leading to dangerous and unpredictable outcomes. The genetic instability associated with cancerous cells gives rise to a plethora of downstream metabolic phenotypes. These phenotypes offer cancerous cells one of the most valued assets in their battle for survival: their metabolic diversity, which can explain why it is so difficult to find effective therapies for most cancers.
The explanation of intratumoral heterogeneity using the theory of evolution provides a solid basis for understanding why and how tumors possess this medley of metabolic phenotypes. Tumors have different genetic and metabolic phenotypes due to different environmental pressures such as vascularization, oxygen supply, and other factors such as drug treatments. While certain subpopulations with defined metabolic phenotypes may be sensitive to a suitable metabolic inhibitor, other subclones with different metabolic phenotypes may well be resistant to that drug. This explains why patients may become unresponsive to second-round treatment after an initial successful first round in which most of the tumor was targeted and eliminated by the treatment, but small subpopulations were not [3]. These selective pressures promote the survival and propagation of genetically and even epigenetically diverse subclones that lead to the downstream array of distinct metabolic phenotypes in each subclone (Fig. 1). It is important to mention another heterogeneity-emerging theory, namely the cancer stem cell (CSC) theory, which challenges the previously mentioned clonal evolution theory. The clonal evolution theory claims that genetically and metabolically distinct subpopulations arise from a previously larger population of cancer cells due to the expansion of the population, genetic diversification, and selection of certain subclones over others. On the other hand, the CSC theory states that a significant source of heterogeneity in cancer cells is due to CSCs, which are undifferentiated and have high rates of division. These cancer stem cells possess largely variable metabolic phenotypes through their differentiation into different types of cells [4]. They are also capable of differentiating into metabolically and functionally diverse subclones within a single tumor. Moreover, they are usually resistant to many therapeutic methods due to their undifferentiated state. This fact is supported by findings suggesting that more differentiated cancer stem cells tend to lead to better prognoses due to their decreased tumorigenic potential [5]. In fact, the mechanism behind many therapies for cancer patients induces differentiation of CSCs. The origin of these CSCs ranges from tumor cells that acquired stem cell properties to differentiated stem cells that simply accumulated mutations that turn them into CSCs [6].

Bottleneck effect in tumors. Metabolically different subclones of a tumor, each represented by a different color. Survival and growth rates of subclones depend on the various selective environmental pressures applied such as blood supply shortage, low oxygen levels, and drug treatment
Cancer is further complicated by the fact that the different sources of heterogeneity, namely CSC-derived heterogeneity, evolution-derived heterogeneity, and heterogeneity related to environmental factors, can all coexist at once [7]. This makes it a much more arduous feat to eradicate all subclones within any given tumor which then leads to the following question: Why do cancer cells employ various biological processes, even within a single tumor from a single patient? The ultimate advantage of intratumoral heterogeneity of cancer cell metabolism is that it confers on the cancer cells an ability to survive and proliferate within the tremendously variable, and often harsh, tumor microenvironment. The diversity of the tumor microenvironment—characterized by areas of poor oxygenation, increased acidity, sparse nutrients, or growth factors—is the challenge that cancer cells must overcome in order to achieve the goals of survival and continued rapid cell proliferation.
How do these diverse metabolic phenotypes arise? We know that the different microenvironments in any given tumor provide different selective pressures that lead to the propagation of specific advantageous mutations in each respective cancer subclone. We also know that CSCs can contribute to the heterogeneous aspect of a tumor by providing differentiated subpopulations with varied genetic expressions and metabolic pathways. These changes result in a variety of proteins and, most importantly among them, enzymes necessary to effectively convert locally available nutrients into energy and useful products suited for each microenvironment to obtain what is required for the production of a specific metabolic phenotype for each subclone.
3 Intratumoral Metabolic Heterogeneity Follows Intratumoral Genetic Alterations
The intricate relationship between genetics and metabolism in cancer is arguably the main reason why the diverse metabolic phenotypes within a given tumor can arise. All of the genetic changes, if occurring in different regions of a tumor, can lead to a diverse array of differently regulated metabolic processes in a tumor.
Genetic alterations, which are often the result of a response to the tumor microenvironment, are the means by which cancer cells are able to produce the enzymes necessary to effectively convert locally available nutrients into energy and useful products to achieve their goals. Oxygen and nutrient supply varies across individual tumors. Thus, intratumoral gene expression is diverse, and it is this genetic heterogeneity that allows cancer cells to adapt to the diverse and taxing conditions of the tumor microenvironment. These adaptations in nutrient uptake and biosynthesis, which have been linked to particular genetic mutations, must follow from altered gene expression in cancer cells. As such, the enzymes produced are the proximate causes of the adoption of alternative metabolic pathways, which contribute to the cancer cells’ successful survival and growth.
In light of the evidence of intratumoral genetic heterogeneity, along with the fact that changes in cancer cell metabolism are the consequences of alterations in gene expression, cancer metabolism must be vastly diverse across a single tumor. A recent study noted the coexistence of various genetically different subclones in advanced tumors, challenging the previously held notion that a dominant subclone usually appears in a given tumor [8]. Furthermore, based on the expression of 110 genes, another study showed that different subpopulations in one clear cell carcinoma could be classified as either clear cell A (associated with good prognosis) or B (associated with poor prognosis) [8, 9]. These results emphasize not only how varied gene expression within a single tumor can be, but also the need to accurately use prognostic markers due to the different genotypes within each subclone, as not doing so could potentially lead to erroneous prognoses (Fig. 2).

The intratumoral heterogeneity of cancer metabolism. Clear cell carcinoma tumor (shown in blue) with subclones (shown in orange and gray). The orange subclone consists of clear cell A cells (associated with good prognoses), and the gray subpopulation consists of clear cell B cells (associated with poor prognoses). Single biopsies taken from one population may indicate misleading prognoses
Another study has shown that intratumoral genetic diversity is also widely prevalent within tumor cell populations in breast carcinomas [10, 11]. These tumor cell populations are composed of stem cell-like or more differentiated cell populations expressing different clusters of differentiation and antigens on their surface [10]. These subpopulations were further found to exhibit highly heterogeneous genetic compositions, implying different biological and metabolic functions and, most likely, different responses to the same treatments [10].
4 Epigenetics Alterations Lead to Intratumoral Metabolic Heterogeneity
It is important to note that not all heterogeneity arises from genetic alterations. New studies point to the important role of epigenetics in tumor heterogeneity. Epigenetics studies have recently uncovered increased methylation in promoters of a variety of important genes in tumor progression, such as tumor-suppressor genes [12]. Other findings also showed similar roles of epigenetics in cancer evolution. We can observe an example of the effect of intratumoral epigenetic heterogeneity in a study which revealed that in a given glioblastoma [14] tumor, 40% transcriptional heterogeneity was observed in a gene encoding a DNA repair enzyme: O6-methylguanine DNA methyltransferase (MGMT) [13]. Furthermore, 14% of the heterogeneity was attributed to the methylation levels of the promoter of that gene, whose methylation status has been used for clinical purposes as a marker that correlates with therapeutic response [13]. However, these variations in expression across a single tumor pose a threat to the effectiveness of this enzyme as a clinical marker. In addition to the genetic heterogeneity observed, researchers and clinicians need to keep in mind the variability displayed on an epigenetic level across a single tumor. Therefore, it is fair to keep in mind the potential effect epigenetics could have on metabolism.
In a study by Okegawa et al., the characterization of kidney tumors revealed distinct metabolic profiles in different regions of the same tumor [15]. The study identified two distinct tumor metabolic clusters, MC1 and MC2, where MC2 displayed upregulated pyruvate metabolism, which was confirmed using isotope tracing in tumor slices. This suggests that pyruvate metabolism may be a potential therapeutic target due to some clones’ reliance on it. However, genetic differences between subpopulations did not match the metabolic profiles of such subclones, suggesting that factors other than genetics, such as epigenetics, may play a role in developing distinct metabolic phenotypes.
5 Intratumoral Metabolic Adaptation and Heterogeneity Are Due to the Intemperate Conditions of the Tumor Microenvironment
Now we will take a closer look at how a tumor can metabolically adapt to its ever-changing environment. These adaptations also reflect an evolutionary advantage in cancer cells and give rise to the heterogeneity found in cancer. As a tumor grows in size, it develops hypoxic regions that are beyond the diffusion limits of oxygen in existing vasculature. Tumor hypoxia, in addition to its role in the mutation of oncogenes and tumor-suppressor genes, plays a major role in the overexpression of hypoxia-inducible factor-1α (HIF-1α) in cancer. HIF-1α is part of a heterodimeric protein that acts as a transcriptional regulator for many genes involved in angiogenesis, erythropoietin production, and cell survival. While HIF-1α usually degrades quickly under normal conditions, degradation is suppressed in hypoxic environments. Therefore, increased HIF-1α level upregulates the expression of genes that code for adaptive metabolic changes, switching cancer cell metabolism from oxidative phosphorylation (OXPHOS) to glycolysis, increasing the conversion of glucose to glycogen as a glucose reservoir, and using glutamine as the major substrate for fatty acid synthesis [16, 17]. Furthermore, HIF-1 directly transactivates lactate dehydrogenase A (LDHA) expression under hypoxia [17], which explains how hypoxia further promotes glycolysis [18,19,20].
In order for the tumor to metastasize and grow beyond a few millimeters, angiogenesis is necessary [21]. HIF-1α also upregulates the expression of genes that code for angiogenesis. One of the most notable of such genes is the gene encoding proangiogenic vascular endothelial growth factor (VEGF), which induces the proliferation of endothelial cells (ECs), a key process in angiogenesis [22,23,24]. Surprisingly, several studies found that ECs mainly rely on glycolysis rather than oxidative phosphorylation (OXPHOS) despite their ideal location that promotes their function as endothelium and in maintaining vascular barrier homeostasis and bioenergetics [25,26,27,28,29,30]. Similar to cancer cells, ECs choose aerobic glycolysis over OXPHOS due to their rapid growth, which is necessary to fulfill the demands of forming new blood vessels [25]. Reducing glycolysis by silencing its stimulator phosphofructokinase-2/fructose-2,6-bisphosphatase 3 (PFKFB3) decreased angiogenesis [25]. Moreover, PFKFB3-deficient ECs display poor vessel growth in several in vivo models of angiogenesis. VEGF, in turn, also promotes glycolysis through the upregulation of glucose transporter type 1 (GLUT-1), which facilitates glucose uptake [31].
The tumor microenvironment is tremendously dynamic and diverse regarding nutrient and oxygen supply, both spatially and temporally within a single tumor. Temporal variations of the partial pressure of oxygen within a specified region of the tumor referred to as intermittent or cyclic hypoxia occur in different regions throughout the tumor [32]. The occurrence of cyclic hypoxia is attributed to variations in red blood cell flux, which is thought to be a result of changes in blood flow resistance that arise from angiogenesis and other structural changes to the vasculature [32, 33]. Regions of the tumor with adequate vasculature are much more resistant to intermittent hypoxia than regions with insufficient vasculature [34]. Although reduced oxygenation to select either regions or the entire tumor can induce hypoxia, an increase of equal magnitude in oxygen consumption is disproportionately more effective in inducing hypoxia [35,36,37,38,39]. These variations in oxygen and nutrient delivery, as well as in oxygen consumption within a tumor, are fundamental to the pervasive metabolic heterogeneity exhibited by different types of cancers, patients with the same cancer type, and, most notably, a single tumor from any given patient.
6 Metabolic Heterogeneity Leads to Unpredictable Outcomes
Now that we have a basic understanding of how the various metabolic phenotypes in a given tumor arise and the different processes driving it, we can take a look at some specific examples and cases of intratumoral metabolic heterogeneity.
6.1 Spatial Heterogeneity Provides a Survival Advantage to Tumors
For a long time, it was believed that cancer cells’ major metabolic footprint was the Warburg effect, which dictates that cancer cells undergo glycolysis to produce lactic acid even in the presence of oxygen, a process termed aerobic glycolysis. Although the Warburg effect is still relevant, it has recently become clear that the metabolic phenotypes of cancer cells are far more varied and intricate. In a recent study published by Le et al. [40], the identification of genetic variability within the same tumor also revealed distinct metabolic profiles of each cell subpopulation within a given tumor. In addition to the hypoxia-inducible factor (HIF) positive and/or cycling cells (Warburg effect-displaying cells), they found that the subpopulation that was HIF negative and non-cycling expressed a distinct set of genes with increased expression of mitochondrial genes as compared to other subpopulations. This subpopulation respires under hypoxia, supported by the fact that it has the highest oxygen consumption rate and mitochondrial capacity. The non-cycling and HIF-negative subclone is able to produce a tumor when purified and injected as a xenograft in vivo. This points to the importance of understanding how cancer metabolism allows for tremendous metabolic variegation.
Hypoxic cells can also coexist with aerobic cells, those that undergo oxidative phosphorylation, in a commensal manner. Hypoxic cells provide lactate that can be converted to pyruvate in the aerobic cells, which use it to run the TCA cycle and undergo oxidative phosphorylation [41]. These aerobic cells are oxygenated due to their proximity to a nearby blood supply. Therefore, they can survive in this manner and are more suited to doing so than hypoxic cells. However, in addition to these two types of cancerous cells, Le et al. recently uncovered the existence of a non-Warburg metabolic phenotype in B lymphoma cells that undergo hypoxic respiration by activating the TCA cycle through glutamine oxidation [19]. Oxidation of glutamine allows it to be used as a source for running the TCA cycle and enables the decrease of reliance on glucose as a primary fuel source for cancer cells [42]. This revelation once again supports the existence of diverse metabolic phenotypes in any given tumor.
The metabolic nature of cancer is muddled. Not only do some cancer subclones form commensal relationships with each other, but cancer cells can also form similar relationships with cancer-associated fibroblasts (CAFs) [43, 44]. CAFs are a subpopulation of cells that reside within the tumor microenvironment and support the proliferation and growth of tumor cells. By providing lactate and ketone bodies, acidic compounds that can form acetyl-CoA in a reversible manner, and by taking up reactive oxygen species that promote glycolytic metabolic pathways, CAFs establish a fundamental relationship with adjacent cancer cells [45]. CAFs are also involved in the maintenance of an acidic extracellular environment, providing suitable conditions for optimal cancer cell growth [45].
Elgogary et al. present another case of spatial metabolic heterogeneity. Pancreatic tumors were targeted by bis-2-(5-phenylacetamido-1,3,4-thiadiazol-2-yl)ethyl sulfide (BPTES), a glutaminase 1 (GLS1) inhibitor, which was encapsulated in a nanoparticle to enhance drug delivery [46]. The drug decreased tumor sizes, but metabolomics analysis [47] revealed that surviving tumors relied on glycolysis and glycogen synthesis instead. Thus, further combination therapy of BPTES and metformin, a drug frequently used to treat diabetes by blocking glucose synthesis, further reduced tumor size. These results support the prevailing consensus that different metabolic phenotypes in any given tumor require specific therapeutic actions based on each subclonal phenotype (Fig. 3).

Depiction of effects of combined therapy based on cancer metabolism. Depicted is an in vivo tumor containing different subpopulations of cancer cells (glycolytic cells presented as blue, glutamine-dependent cells presented as red, and other metabolic pathway-dependent cells presented as green)
As previously noted, hypoxia has been found to play an important role in the development of heterogeneous phenotypes in cancer cells. In a recent study, Fluegen et al. investigated the fate of disseminated tumor cells (DTCs). They revealed that these post-hypoxia DTCs carry an array of upregulated genes, such as dormancy (nuclear receptor subfamily 2 group F member 1 (NR2F1), DEC2, p27) and hypoxia (HIF-1α) genes, in addition to the GLUT-1 gene [48]. This dormant subpopulation, which evades many chemotherapies, as the authors of the paper noted, could explain relapse and the poor survival rates. As a result, heterogeneity in cancer metabolism comes in a variety of forms, and the same factor, in this case, hypoxia, can come into play through different approaches depending on each scenario.
While the different aspects of cancer metabolism may seem to intertwine neatly, the relationship between these parts is far more complex. For instance, while glutamine utilization in the TCA cycle is heavily linked to low oxygen consumption and hypoxia, the latter can sometimes occur independently of the former. Thus, these pathways may overlap when intracellular lactate causes an increase in glutamine uptake and metabolism. However, anaplerosis (pyruvate conversion into oxaloacetate), an alternative use of pyruvate in hypoxic conditions, can sometimes lead to the conversion of glucose to glutamate, taking away glutamine’s role as the glutamate and α-ketoglutarate (α-KG) provider needed to run the TCA cycle [49].
Despite our tendency to separate different metabolic pathways and to assign rigid pathways to cancer metabolism, it must be noted that different pathways often cross-talk and that the correlative nature of many metabolic pathways does not necessarily point to a causative relationship.
6.2 Temporal Heterogeneity Provides Cancer with Short-Term Adaptive Capabilities
As discussed earlier, tumors tend to evolve rapidly and produce dissimilar subclones through their interaction with the microenvironment. There exists a similar sort of evolution in single cancer cells: a form of temporal heterogeneity. Cancer cells are also astoundingly plastic regarding their metabolism. For example, they can switch their mitochondrial energy source between glutamine and glucose through the utilization of different transcriptional factors that encode enzymes required for each respective metabolic pathway. Cancer cells achieve this kind of plasticity through a variety of mechanisms. Posttranslational modifications allow a quick and immediate response to changes in the environment, which could be useful in the sense that blood supply changes can be very rapid. Slower modifications do also exist, such as changes in gene expression and epigenetic modifications.
An example of such posttranslational modifications and an illustration of cancer’s remarkable plasticity are seen once again in hypoxic cancer cells. Hypoxic cancer cells increase the transcription of pyruvate kinase muscle isoform 2 (PKM2), an enzyme responsible for the final nonreversible step in glycolysis, the conversion of phosphoenolpyruvate to pyruvate. This is achieved as the first intron of the PKM2 gene contains a hypoxia response element that is a target for HIF-1α. PKM2 is produced through the alternative splicing of the precursor mRNA PKM and is controlled by c-MYC [50, 51]. As such, high PKM2 levels are correlated with poor survival rates in gastric cancers as this upregulation of PKM2 helps cancer cells dedicate most of their glucose towards lactate production quickly and efficiently under hypoxic conditions [52]. Therefore, this posttranslational modification allows cancer cells to switch their metabolic profiles quickly and efficiently when faced with varying environmental conditions.
7 Tailored Clinical Applications and Therapies Targeting Metabolic Pathways Can Lead to Better Clinical Outcomes
Given the different tumor microenvironments, the diversity of cancer cell metabolism, and their genetic and epigenetic composition, various techniques have been developed to visualize the different tumor microenvironments in a given tumor. These imaging techniques have further propelled us in the search for effective cancer therapies targeting different cancer cell metabolism that can be specialized and tailor-made for every different microenvironment.
The most successful methods currently used to identify different tumor microenvironments include positron-emission tomography (PET) and computed tomography (PET-CT) scans. 18F-Fluorodeoxyglucose-positron-emission tomography (FDG-PET) images of individual cervical tumors have revealed varying levels of glucose consumption across different regions of a single tumor [53]. The variation of glucose consumption within a tumor has been associated with increased expression of glucose transporters GLUT-1, GLUT-3, and hexokinase 2 (HK-2), the first key enzyme of glucose metabolism [54]. PET scans can also be used to identify hypoxic microenvironments through the use of isotopically labeled 18F-fluoromisonidazole (FMISO), which is injected and taken up by cells through passive diffusion. In the absence of oxygen, FMISO accumulates in cells to generate an image of the hypoxic regions within a tumor [55]. PET scans can also be used to measure various tumor microenvironments based on other variables such as the partial pressure of oxygen [56].
Intratumoral metabolic heterogeneity in cancer can also serve as a useful tool for prognosis. In a study done by Mena et al., intratumoral metabolic heterogeneity was measured across 105 patients with oropharyngeal squamous cell carcinoma, along with either standardized uptake values of glucose (SUV) or metabolic tumor volume (MTV). These measurements were shown to have effective capacities as prognostic markers (p = 0.026 and 0.022, respectively), with higher levels indicating poorer prognoses, supporting the notion that the more diverse metabolic phenotypes exist within a tumor, the more arduous a task it becomes to eradicate all different subclones in the tumor [57].
Besides prognostication, increased knowledge of cancer’s heterogeneous metabolic nature, and specifically its intratumoral heterogeneity, can enable specific targeting of subclones in a single tumor and has resulted in a surge in specific tailor-made cancer therapies. One of the earliest hallmarks of cancer is its ability to take up increased amounts of glucose, through the utilization of many GLUTs. Cancer cells are also capable of metabolizing glucose at much quicker rates than normal cells. Consequently, this has resulted in increased research addressing the production of commercial GLUT inhibitors and the transporter isoform specificity of inhibition [58]. Other drugs have been developed to target the hypoxic pathways of cancer cells, such as topotecan, which inhibits hypoxia-inducible factor 1 (HIF-1) transcriptional activity and HIF-1α protein accumulation in hypoxia-treated U251 human glioma cells [59]. This has generated increased interest in mRNA-regulating agents that target HIF-1α. Many drugs have followed with variable success that act by blocking mRNA transcription of the HIF-1α gene. Recent research has also provided many other pathways in cancer metabolism that can be targeted with effective results. For example, lactate dehydrogenase A (LDHA), an enzyme involved in the generation of lactate from glucose in Warburg effect-displaying cells, has also been found to be a suitable target for effective tumor reduction through small-molecule inhibition. Decreased expression of LDHA through small-molecule FX11 inhibition elevated oxidative stress levels and ultimately resulted in cell death and tumor volume reduction [60,61,62]. Other methods targeting HIF-1α and hypoxia have been formulated through the integration of different therapies to each specific tumor microenvironment [63], making complete cancer recession very promising. Again, it is vital to realize the complex nature of cancer metabolism and the need for specific therapies to be directed at the individual metabolic phenotypes in order to see effective responses in patients (Fig. 4).

Various factors associated with intratumoral metabolic heterogeneity
8 Conclusion
Despite the challenges, there is much hope in the field of cancer therapies. The recently discovered and understood aspects of cancer’s metabolic heterogeneity, including its intricate interactions with CAFs, the exchanges between its distinct subclones, and its impressive plasticity, promise to greatly advance this field. The importance of accounting for intratumoral heterogeneity in any given tumor has never been as widely understood as it is now. The latest findings we have discussed in this chapter give us a more solid understanding of cancer complexities, which we can seek to translate into effective and strategic therapies in the near future.
Abbreviations
- α-KG:
-
α-Ketoglutarate
- BPTES:
-
Bis-2-(5-phenylacetamido-1,3,4-thiadiazol-2-yl)ethyl sulfide
- CAF:
-
Cancer-associated fibroblasts
- CSC:
-
Cancer stem cell
- DTC:
-
Disseminated tumor cells
- EC:
-
Endothelial cells
- FBP1:
-
Fructose-1,6-bisphosphatase 1
- FBP2:
-
Fructose-1,6-bisphosphatase 2
- FDG:
-
[18F]-Fluorodeoxyglucose
- FH:
-
Fumarate hydratase
- FMISO:
-
[18F]-Fluoromisonidazole
- GLUT-1:
-
Glucose transporter type 1
- HIF-1α:
-
Hypoxia-inducible factor-1α
- HK-2:
-
Hexokinase 2
- LDHA:
-
Lactate dehydrogenase A
- MC:
-
Metabolic cluster
- MTV:
-
Metabolic tumor volume
- NR2F1:
-
Nuclear receptor subfamily 2 group F member 1
- OXPHOS:
-
Oxidative phosphorylation
- PET:
-
Positron-emission tomography
- PKM2:
-
Pyruvate kinase muscle isoform 2
- SDH:
-
Succinate dehydrogenase
- SUV:
-
Standardized uptake values of glucose
- TCA:
-
Tricarboxylic acid
- VEGF:
-
Vascular endothelial growth factor
References
Dang, C. V., et al. (2011). Therapeutic targeting of cancer cell metabolism. Journal of Molecular Medicine (Berlin), 89(3), 205–212.
Hirschey, M. D., et al. (2015). Dysregulated metabolism contributes to oncogenesis. Seminars in Cancer Biology, 35(Suppl), S129–S150.
Gonzalez-Angulo, A. M., Morales-Vasquez, F., & Hortobagyi, G. N. (2007). Overview of resistance to systemic therapy in patients with breast cancer. Advances in Experimental Medicine and Biology, 608, 1–22.
Prasetyanti, P. R., & Medema, J. P. (2017). Intra-tumor heterogeneity from a cancer stem cell perspective. Molecular Cancer, 16(1), 41.
Jogi, A., et al. (2012). Cancer cell differentiation heterogeneity and aggressive behavior in solid tumors. Upsala Journal of Medical Sciences, 117(2), 217–224.
Bu, Y., & Cao, D. (2012). The origin of cancer stem cells. Frontiers in Bioscience (Scholar Edition), 4, 819–830.
Yang, T., et al. (2014). Cancer stem cells: Constantly evolving and functionally heterogeneous therapeutic targets. Cancer Research, 74(11), 2922–2927.
Gonzalez-Garcia, I., Sole, R. V., & Costa, J. (2002). Metapopulation dynamics and spatial heterogeneity in cancer. Proceedings of the National Academy of Sciences of the United States of America, 99(20), 13085–13089.
Gerlinger, M., et al. (2012). Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. The New England Journal of Medicine, 366(10), 883–892.
Park, S. Y., et al. (2010). Cellular and genetic diversity in the progression of in situ human breast carcinomas to an invasive phenotype. The Journal of Clinical Investigation, 120(2), 636–644.
Tan, J., & Le, A. (2021). The heterogeneity of breast cancer metabolism. Advances in Experimental Medicine and Biology, 1311, https://doi.org/10.1007/978-3-030-65768-0_6
Greger, V., et al. (1994). Frequency and parental origin of hypermethylated RB1 alleles in retinoblastoma. Human Genetics, 94(5), 491–496.
Parker, N. R., et al. (2016). Intratumoral heterogeneity identified at the epigenetic, genetic and transcriptional level in glioblastoma. Scientific Reports, 6, 22477.
Quinones, A., & Le, A. (2021). The multifaceted glioblastoma: From genomic alterations to metabolic adaptations. Advances in Experimental Medicine and Biology, 1311, https://doi.org/10.1007/978-3-030-65768-0_4
Okegawa, T., et al. (2017). Intratumor heterogeneity in primary kidney cancer revealed by metabolic profiling of multiple spatially separated samples within tumors. eBioMedicine, 19, 31–38.
Masson, N., & Ratcliffe, P. J. (2014). Hypoxia signaling pathways in cancer metabolism: The importance of co-selecting interconnected physiological pathways. Cancer & Metabolism, 2(1), 3.
Semenza, G. L. (2013). HIF-1 mediates metabolic responses to intratumoral hypoxia and oncogenic mutations. The Journal of Clinical Investigation, 123(9), 3664–3671.
Bose, S., Zhang, C., & Le, A. (2021). Glucose metabolism in cancer: The Warburg effect and beyond. Advances in Experimental Medicine and Biology, 1311, https://doi.org/10.1007/978-3-030-65768-0_1
Le, A., et al. (2012). Glucose-independent glutamine metabolism via TCA cycling for proliferation and survival in B cells. Cell Metabolism, 15(1), 110–121.
Lee, S. L., & Fanburg, B. L. (1987). Glycolytic activity and enhancement of serotonin uptake by endothelial cells exposed to hypoxia/anoxia. Circulation Research, 60(5), 653–658.
Cox, T. R., & Erler, J. T. (2011). Remodeling and homeostasis of the extracellular matrix: Implications for fibrotic diseases and cancer. Disease Models & Mechanisms, 4(2), 165–178.
Forsythe, J. A., et al. (1996). Activation of vascular endothelial growth factor gene transcription by hypoxia-inducible factor 1. Molecular and Cellular Biology, 16(9), 4604–4613.
Mazure, N. M., et al. (1996). Oncogenic transformation and hypoxia synergistically act to modulate vascular endothelial growth factor expression. Cancer Research, 56(15), 3436–3440.
Brychtova, S., et al. (2008). The role of vascular endothelial growth factors and their receptors in malignant melanomas. Neoplasma, 55(4), 273–279.
De Bock, K., et al. (2013). Role of PFKFB3-driven glycolysis in vessel sprouting. Cell, 154(3), 651–663.
Parra-Bonilla, G., et al. (2010). Critical role for lactate dehydrogenase A in aerobic glycolysis that sustains pulmonary microvascular endothelial cell proliferation. American Journal of Physiology. Lung Cellular and Molecular Physiology, 299(4), L513–L522.
Peters, K., et al. (2009). Changes in human endothelial cell energy metabolic capacities during in vitro cultivation. The role of “aerobic glycolysis” and proliferation. Cellular Physiology and Biochemistry, 24(5-6), 483–492.
Polet, F., & Feron, O. (2013). Endothelial cell metabolism and tumour angiogenesis: Glucose and glutamine as essential fuels and lactate as the driving force. Journal of Internal Medicine, 273(2), 156–165.
Merchan, J. R., et al. (2010). Antiangiogenic activity of 2-deoxy-D-glucose. PLoS One, 5(10), e13699.
Wang. (2011). Q., et al., 2-Deoxy-D-glucose treatment of endothelial cells induces autophagy by reactive oxygen species-mediated activation of the AMP-activated protein kinase. PLoS One, 6(2), e17234.
Yeh, W. L., Lin, C. J., & Fu, W. M. (2008). Enhancement of glucose transporter expression of brain endothelial cells by vascular endothelial growth factor derived from glioma exposed to hypoxia. Molecular Pharmacology, 73(1), 170–177.
Kimura, H., et al. (1996). Fluctuations in red cell flux in tumor microvessels can lead to transient hypoxia and reoxygenation in tumor parenchyma. Cancer Research, 56(23), 5522–5528.
Lanzen, J., et al. (2006). Direct demonstration of instabilities in oxygen concentrations within the extravascular compartment of an experimental tumor. Cancer Research, 66(4), 2219–2223.
Bennewith, K. L., & Durand, R. E. (2004). Quantifying transient hypoxia in human tumor xenografts by flow cytometry. Cancer Research, 64(17), 6183–6189.
Secomb, T. W., et al. (1993). Analysis of oxygen transport to tumor tissue by microvascular networks. International Journal of Radiation Oncology, Biology, Physics, 25(3), 481–489.
Barger, J. F., & Plas, D. R. (2010). Balancing biosynthesis and bioenergetics: Metabolic programs in oncogenesis. Endocrine-Related Cancer, 17(4), R287–R304.
Schafer, Z. T., et al. (2009). Antioxidant and oncogene rescue of metabolic defects caused by loss of matrix attachment. Nature, 461(7260), 109–113.
Samudio, I., et al. (2010). Pharmacologic inhibition of fatty acid oxidation sensitizes human leukemia cells to apoptosis induction. The Journal of Clinical Investigation, 120(1), 142–156.
Buzzai, M., et al. (2005). The glucose dependence of Akt-transformed cells can be reversed by pharmacologic activation of fatty acid beta-oxidation. Oncogene, 24(26), 4165–4173.
Le, A., et al. (2014). Tumorigenicity of hypoxic respiring cancer cells revealed by a hypoxia-cell cycle dual reporter. Proceedings of the National Academy of Sciences of the United States of America, 111(34), 12486–12491.
Zheng, J. (2012). Energy metabolism of cancer: Glycolysis versus oxidative phosphorylation (Review). Oncology Letters, 4(6), 1151–1157.
Li, T., Copeland, C., & Le, A. (2021). Glutamine metabolism in cancer. Advances in Experimental Medicine and Biology, 1311, https://doi.org/10.1007/978-3-030-65768-0_2
Sazeides, C., & Le, A. (2021). Metabolic relationship between cancer-associated fibroblasts and cancer cells. Advances in Experimental Medicine and Biology, 1311, https://doi.org/10.1007/978-3-030-65768-0_14
Jung, J. G., & Le, A. (2021). Targeting metabolic cross talk between cancer cells and cancer-associated fibroblasts. Advances in Experimental Medicine and Biology, 1311, https://doi.org/10.1007/978-3-030-65768-0_15
Martinez-Outschoorn, U. E., Lisanti, M. P., & Sotgia, F. (2014). Catabolic cancer-associated fibroblasts transfer energy and biomass to anabolic cancer cells, fueling tumor growth. Seminars in Cancer Biology, 25, 47–60.
Elgogary, A., et al. (2016). Combination therapy with BPTES nanoparticles and metformin targets the metabolic heterogeneity of pancreatic cancer. Proceedings of the National Academy of Sciences of the United States of America, 113(36), E5328–E5336.
Hoang, G., Udupa, S., & Le, A. (2019). Application of metabolomics technologies toward cancer prognosis and therapy. International Review of Cell and Molecular Biology, 347, 191–223.
Fluegen, G., et al. (2017). Phenotypic heterogeneity of disseminated tumour cells is preset by primary tumour hypoxic microenvironments. Nature Cell Biology, 19(2), 120–132.
Eales, K. L., Hollinshead, K. E., & Tennant, D. A. (2016). Hypoxia and metabolic adaptation of cancer cells. Oncogene, 5, e190.
Dang, C. V., Le, A., & Gao, P. (2009). MYC-induced cancer cell energy metabolism and therapeutic opportunities. Clinical Cancer Research, 15(21), 6479–6483.
Le, A., & Dang, C. V. (2013). Studying Myc’s role in metabolism regulation. Methods in Molecular Biology, 1012, 213–219.
Wong, N., De Melo, J., & Tang, D. (2013). PKM2, a central point of regulation in cancer metabolism. International Journal of Cell Biology, 2013, 242513.
Kidd, E. A., & Grigsby, P. W. (2008). Intratumoral metabolic heterogeneity of cervical cancer. Clinical Cancer Research, 14(16), 5236–5241.
Zhao, S., et al. (2005). Biologic correlates of intratumoral heterogeneity in 18F-FDG distribution with regional expression of glucose transporters and hexokinase-II in experimental tumor. Journal of Nuclear Medicine, 46(4), 675–682.
Farwell, M. D., Pryma, D. A., & Mankoff, D. A. (2014). PET/CT imaging in cancer: Current applications and future directions. Cancer, 120(22), 3433–3445.
Plathow, C., & Weber, W. A. (2008). Tumor cell metabolism imaging. Journal of Nuclear Medicine, 49(Suppl 2), 43S–63S.
Mena, E., et al. (2017). Value of intratumoral metabolic heterogeneity and quantitative 18F-FDG PET/CT parameters to predict prognosis in patients with HPV-positive primary oropharyngeal squamous cell carcinoma. Clinical Nuclear Medicine, 42(5), e227–e234.
Ojelabi, O. A., et al. (2016). WZB117 (2-fluoro-6-(m-hydroxybenzoyloxy) phenyl m-hydroxybenzoate) inhibits GLUT1-mediated sugar transport by binding reversibly at the exofacial sugar binding site. The Journal of Biological Chemistry, 291(52), 26762–26772.
Rapisarda, A., et al. (2004). Topoisomerase I-mediated inhibition of hypoxia-inducible factor 1: Mechanism and therapeutic implications. Cancer Research, 64(4), 1475–1482.
Le, A., et al. (2010). Inhibition of lactate dehydrogenase A induces oxidative stress and inhibits tumor progression. Proceedings of the National Academy of Sciences of the United States of America, 107(5), 2037–2042.
Rajeshkumar, N. V., et al. (2015). Therapeutic targeting of the warburg effect in pancreatic cancer relies on an absence of p53 function. Cancer Research, 75(16), 3355–3364.
Dutta, P., et al. (2013). Evaluation of LDH-A and glutaminase inhibition in vivo by hyperpolarized 13C-pyruvate magnetic resonance spectroscopy of tumors. Cancer Research, 73(14), 4190–4195.
Antonio, M. J., Zhang, C., & Le, A. (2021). Different tumor microenvironments lead to different metabolic phenotypes. Advances in Experimental Medicine and Biology, 1311, https://doi.org/10.1007/978-3-030-65768-0_10.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Open Access This chapter is licensed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license and indicate if changes were made.
The images or other third party material in this chapter are included in the chapter's Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the chapter's Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.
Copyright information
© 2021 The Author(s)
About this chapter

Cite this chapter
Nabi, K., Le, A. (2021). The Intratumoral Heterogeneity of Cancer Metabolism. In: Le, A. (eds) The Heterogeneity of Cancer Metabolism. Advances in Experimental Medicine and Biology, vol 1311. Springer, Cham. https://doi.org/10.1007/978-3-030-65768-0_11
Download citation
DOI: https://doi.org/10.1007/978-3-030-65768-0_11
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-65767-3
Online ISBN: 978-3-030-65768-0
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)