
Abstract
In order to perform experiments in the chamber, characterization of physical properties is essential for the evaluation and interpretation of experiments. In this chapter, recommendations are given how to measure physical parameters such as temperature and pressure. For photochemistry experiments, knowledge of the radiation either provided by the sun or lamps is key to calculate photolysis frequencies. Standard protocols are described how to validate the calculation of the radiation inside the chamber using actinometry experiments. In addition, the characterization of loss processes for gas-phase species as well as for aerosol is discussed. Reference experiments can be used to test the state of the chamber. Different types of reference experiments focusing on gas-phase photo-oxidation experiments are recommended and described in detail in this chapter.
You have full access to this open access chapter, Download chapter PDF
Similar content being viewed by others


2.1 Measurements of Temperature, Pressure, and Humidity
Temperature, pressure, and humidity are basic parameters required for the interpretation of almost any experiment carried out in an atmospheric simulation chamber. This is obvious, for example, in cloud studies where small changes in temperature and corresponding relative humidity can lead to cloud activation of aerosol particles, but also for chemical reaction kinetics for which reaction rates can have strong pressure and temperature dependencies. Therefore, we briefly summarize some recommendations on how to measure these parameters in atmospheric simulation chambers. The quality and traceability of such parameters are becoming increasingly important not only allowing for better comparability of experimental results, but especially if data will be used in atmospheric measurement networks like ACTRIS where all data require traceable quality standards. Recently, the European metrological institutions have addressed the issue of traceability. A consortium of national laboratory developed metrological methods for improving atmospheric measurements of pressure, temperature, humidity and airspeed has been carried out in the EURAMET project METEOMET. These methods include corresponding laboratory methods and traceability chains, which are also useful for simulation chambers and are summarized in the METEOMET project report (METEOMET 2020). Measurement procedures, standard operating procedures, good laboratory practices or definitions of traceability chains have been defined by the World Meteorological Organisation (WMO 2018) and the National Institute of Standards (NIST 2019).
Measuring temperatures can be achieved by placing thermocouples (e.g., type J or K), resistant sensors (e.g., PT100 with four wire technique), ultrasonic anemometers or fibre optic sensors (e.g., if electric fields could interfere) at representative locations inside atmospheric simulation chambers. For the selection of the appropriate sensor, the measurement range, temperature, precision, accuracy and time resolution of the sensors required for the different purposes need to be considered. In cases where the simulation chamber is exposed to intense light radiation, the sensor needs to be protected e.g., by a shading cover. If the sensors are exposed to condensable compounds, latent heat release should be considered, especially for fast sensors with low heat capacities. Problems associated with condensation of water can be reduced by coating the sensor with inert e.g., polyfluorinated greases. Furthermore, potential impacts of sensor aging should be avoided by periodic (e.g., annual) calibration. This can be achieved for example in a temperature-controlled liquid bath, in which measurements are compared to those by certified reference sensors which are traceable to national standards. An example overview of potential temperature sensors is given e.g., by Lake Shore Incorporated. Calibrations should include the complete sensor chain including the same wiring as during chamber operation. An example of a temperature measurement inside the AIDA simulation chamber of KIT is shown in Fig. 2.1.

Figure by Harald Saathoff ©, KIT
Gas temperatures measured inside the AIDA aerosol and cloud chamber of KIT using chains of thermocouples (Ni–CrNi). Sensors are placed along the horizontal (left) and vertical (right) axis of the cylindrically shaped chamber. The initial temperature distribution is disturbed by switching on a LED light source on top of the chamber.
The type of sensor (thermocouple Ni–Cr–Ni) used in the AIDA chamber has a high precision and accuracy at a time resolution of seconds can be achieved. In addition to the overall temperature increase due to the illumination, several temperature sensors show impact of direct radiative heating by an average 0.05 K.
The slightly higher temperatures measured by the two sensors (No. 10 and 11) that are placed at the horizontal (left) and a vertical (right) positions of the cylindrically shaped chamber indicate that warm air is trapped at the top of the vessel.
Measuring absolute or differential pressure for the atmospheric pressure range can be done with various types of sensors, which will not be reviewed here. An overview of potential pressure sensors is given for example by Avnet Inc. Some of the most robust and stable sensors are based on measuring changing capacitance (e.g., MKS Baratron). This type of sensor is insensitive to the specific gas mixture, can be temperature-stabilized for high precision measurements, and add typically just a heated stainless steel surface to the simulation chamber. The nature of the chamber environment means that the pressure should be uniform throughout its volume and therefore multiple pressure measurements are not required for most applications.
Measuring absolute or relative humidity in an atmospheric simulation chamber may require different approaches. If temperature and water concentrations are measured, the relative humidity can be calculated using the water vapour pressure over liquid water or ice. For this calculation, the corresponding vapour pressure formulations by Murphy and Koop (2005) are recommended. For temperatures below, 200 K the results by Nachbar et al. (2018a, b) should be used.
For atmospheric measurements often thin-film capacitive humidity sensors are applied. In chambers that work with atmospheric concentrations of reactive species such as the SAPHIR chamber of Forschungszentrum Jülich they offer precise and accurate measurements of relative humidity. However, this type of sensor is not recommended, if high concentrations of oxidizing reactants can get in contact with the sensors as chemical reactions may destroy the thin-film polymer sensors. In this case, metal oxide sensors can be used but they can also suffer from interaction with reactive or condensable compounds.
Absolute water mixing ratios can be measured by dew point mirror sensors which offer an inert e.g., rhodium or gold surface to the chamber contents. However, a successful dew point measurement requires that the major condensing species in the chamber is water and that the mirror is not contaminated e.g., with hygroscopic coatings or particles. Another advantage of the dew point mirror sensors is that they do not need a calibration as long as their temperature measurement is accurate. Among the various dew point mirror instruments several offer traceability to national standards e.g., via transfer standards at the manufacturer (e.g., MBW Calibration Ltd.).
If the sensor cannot be placed inside the chamber and a sampling tube must be used. The sampling tube may require heating to avoid water condensation e.g., if the temperature between the chamber and the instrument is varying. For measuring low water concentrations stainless steel tubing can be used but Teflon tubing should be avoided as it shows memory effects.
If the humidity inside a simulation chamber with condensed water (e.g., cloud droplets or water containing aerosol particles) is to be measured, spectroscopic methods such as FTIR or tuneable diode laser spectroscopy (TDLS) can be useful tools to obtain the condensed water (liquid and ice) and water vapour content. However, for each of these methods the background water concentration e.g., in the spectrometer or transfer optics needs to be treated carefully. An overview of several atmospheric hygrometers, their performance and potential connection to a simulation chamber, is given by Fahey et al. (2014). Tuneable diode laser spectroscopy offers fast and direct humidity measurement with good accuracy, if optical paths of sufficient lengths are available and even allows determination of water isotopes. Another very sensitive absorption method is the cavity ring-down spectroscopy (CRDS). Commercial instruments detecting water vapour in the infrared by CRDS are for example available from Picarro Inc. These instruments do not require calibration. Interferences can occur, if water vapor absorption lines overlap with absorption lines of trace gases that are present in a specific experiment.
It is obvious that the accuracy or precision needed for a certain variable depends on the application. If, for example, the relative humidity is required with an accuracy of 2% at 293 K the temperature needs to be measured with an accuracy of 0.16 K and the water vapour pressure has to be measured with an accuracy of 1%.
2.2 Determination of the Mixing Time and Dilution Rates
Considering the rate of Brownian diffusion of gases, mixing is often wrongly considered as a non-critical characteristic of chamber installation. On the contrary, because of the size of simulation chambers, reaching sufficient chemical homogeneity of the reactive mixture often takes a long time with respect to the rates of many chemical reactions occurring in the atmosphere.
The mixing time of air is a key parameter of a simulation chamber installation that will strongly impact the data analysis, because it is not reasonable to interpret data at a time resolution shorter than the mixing time. For gas phase chemistry, as most of chemical kinetic rate constants are directly proportional to the reactant concentrations, inhomogeneous concentrations can lead to false experimental data and/or strongly complicate the evaluation of experiments (Ibrahim et al. 1987). For particle phase studies, as condensation of semi-volatiles is highly non-linear with concentration, insufficient mixing can lead to an incorrect estimation of secondary organic aerosol yields caused by local supersaturation (Schütze and Stratmann 2008).
Typical mixing times in atmospheric simulation chambers fall in the range of minutes, for example 1 min in the CESAM chamber with 4.2 m3 (Wang et al. 2011) and 2 min in the SAPHIR chamber with 270 m3 (Rohrer et al. 2005). Mixing is often achieved by fans made of inert material operated inside the chamber.
Schütze and Stratmann (2008) analysed the effect of operating one or two fans on the homogeneity of particle concentrations in a cylindrically shaped chamber (12.4 m3 volume) using computational fluid dynamics. They found that inhomogeneities can also be induced by fans in the area, where the air is accelerated. Therefore, it is crucial to carefully choose locations of sampling points for instruments to not be affected by local inhomogeneities.
Apart from the impact on the bulk simulated atmosphere homogeneity, the mixing of air in a simulation chamber also impacts the exchange of energy (Voigtländer et al. 2012) and interactions of matter with the walls. Strong mixing not only increases the level of turbulence in the chamber potentially leading to non-linear effects (Ibrahim et al. 1987), but can also increase the wall loss rate of semi-volatile compounds by increasing the rate of collisions with the chamber wall. Furthermore, in Teflon film chambers, turbulent mixing may lead to movements of the chamber film that can favour the build-up of electrostatic charges and thereby increase the probability that particles are lost on the Teflon film (Wang et al. 2018a, b). Therefore, there is an optimum compromise between homogeneity and wall loss with respect to mixing that is specific for the shape and volume of each chamber.
To determine the mixing time in a simulation chamber, a non-reactive gaseous species that can be measured with a high time resolution can be injected at a single point in the chamber. The species needs to be detected at several positions in the chamber. It is not recommended to use spatially integrated measurements such as in situ spectrometric techniques as they often tend to underestimate the mixing time by spatially averaging the concentration. Moving the sampling point in repeated experiments, simultaneous detection at several points in one experiment and varying the point of injection further increases the precision, with which the mixing time can be determined. As an example, Fig. 2.2 shows the time series of CO concentration detected by an infrared gas filter correlation CO monitor at various sampling points after a point injection in the CESAM chamber (Wang et al. 2011).

(Reused with permission from Wang et al. (2011) Open access under a CC BY 3.0 license, https://creativecommons.org/licenses/by/3.0/)
Time series of CO concentrations sampling at one location after injection of CO at various injection points in the CESAM chamber. Lines are the results of modelling the mixing in the chamber.
Numerical modelling can further help understanding the mixing in the chamber. Wang et al. (2011) divided the volume of the CESAM chamber into 4137 cubic cells each of which has a volume of 1 L and set up a multi-box-model. Time series of trace concentrations were modelled for each box using a kinetic solver (FacsimileTM software package—Curtis 1979) with a non-zero initial concentration in the box in which the injection was located. Assuming isotropic mixing, the exchange rate was adjusted to match the measured concentration time series (Fig. 2.2). For experiments, in which the fan was operated at full speed, these calculations gave a first-order exchange rate of (3 ± 0.5) s−1, which corresponds to the average speed of the gases of 0.3 m s−1. This set-up of model can also be used for the analysis of experiments with complex chemistry as chemical reactions can be added. However, this type of model is not suitable to describe microphysics of the chamber atmosphere. Another approach to gain knowledge of the fluid dynamics in the chamber, is to perform computational fluid dynamics (CFD) calculations. Schütze and Stratmann (2008) used such simulations (FLUENT model, ANSYS Inc., Canonsburg, PA, USA) for a chamber with a volume 12.3 m3 that was divided into 7714 cells. By combining the simulation with the Fine Particle Model (FPM, Particle Dynamics GmbH, Leipzig, Germany, Wilck et al. 2002), they performed simulations of the growth of ammonium-sulphate particles in humid air at room temperature for experiments, in which clouds were generated by gas expansion. Similar calculations with the FLUENT model were performed by Voigtländer et al. (2012) for the CLOUD chamber at CERN that has a volume of 26.1 m3. The authors found that two fans and sufficiently high fan speeds were necessary for a homogeneous mixing of particles and gaseous species.
Trace gases and particles in experiments in atmospheric simulation chamber that have a fixed volume or are kept at a constant pressure are typically diluted over the course of an experiment due to the need to replenish the air that is lost by the consumption of sampling instruments are leakages. Exceptions are chambers where the volume can reduce over the course of an experiment (Carter et al. 2005).
The rate of dilution typically scales with the volume of the chamber. The large EUPHORE (volume 200 m3) and SAPHIR (volume 270 m3) outdoor chambers consist of Teflon film that is kept slightly over-pressurized compared to ambient pressure. The replenishment flow to maintain the pressure leads to a dilution of trace gas and particle concentrations at a low percentage range per hour (Becker 1996; Karl et al. 2004). In the smaller steel CLOUD chamber (volume 26.1 m3) the dilution is typically higher with 6–10% per hour (Hoyle et al. 2016).
Precise and accurate knowledge of the dilution rate is key in the data analysis and modelling of experiments. For this purpose, two strategies can be employed that can be simultaneously applied. In many chambers the flow rates of the replenishment flow are monitored by a mass flow controller from which the dilution rate can that directly calculated, if the volume of the chamber is known (Hoyle et al. 2016; Karl et al. 2004; Wang et al. 2011). An alternative approach is to monitor the concentration of a chemically inert gas that is injected at the start of the experiment. The dilution rate can be calculated from the continuous measurement of its concentration as it decreases over the course of the experiment solely due to dilution. For a chamber equipped with an FTIR spectrometer, SF6 is often used because of its strong infrared absorption lines which gives a clear spectral fingerprint. SF6 can also be monitored with gas chromatography equipped with electron capture detector (GC-ECD) (Fry et al. 2011). For chambers equipped with a Proton-Transfer-Reaction-Mass-Spectrometry (PTR-MS) instrument, hexafluorobenzene (HFB) is a suitable dilution tracer (Hunter et al. 2014). Small alkanes such as ethane or cyclohexane are less inert but measurable with gas chromatography with a flame ionization detector (GC-FID) and have been also used in some studies (Metcalf et al. 2013). However, care has to be taken that the chemistry of these tracers does not disturb the experiment. CO2 which can be precisely measured by cavity ring-down spectroscopy (CRDS) has been used in experiments in the SAPHIR chamber. This is only applicable, if the replenishment flow is free of CO2 and chemical production in the experiment is negligible. This is typically the case for experiments in the SAPHIR chamber, because air is produced from liquid nitrogen of oxygen and trace gas concentrations are within the range of ambient concentrations.

Quantum yield (triangle) and absorption spectrum of NO2 (line); spectral resolution 0.2–0.4 nm (Data from Burrows et al. 1998)
2.3 Determination of Photolysis Frequencies
Photolysis frequencies are important parameters for a quantitative understanding and modelling of photochemical processes in the atmosphere as well as in simulation chambers. For atmospheric measurements of photolysis frequencies, a range of suitable radiometric instruments have been developed and deployed (e.g., Hofzumahaus 2006; Hofzumahaus et al. 2002; Shetter and Müller 1999; Shetter et al. 2003). The most versatile method is spectroradiometry which can monitor spectral actinic flux densities with high time resolution as well as high spectral resolution in the relevant solar spectral range. Thoroughly calibrated instruments provide accurate photolysis frequencies for any photolysis process if the relevant molecular parameters of the molecule X—absorption cross sections \(\sigma\) and quantum yields \(\phi\)—are known.
Recommendations of these parameters can be found in the literature (Fig. 2.3) (Atkinson et al. 2004; Burkholder et al. 2020; Keller-Rudek et al. 2013).
Considering possible radiation inhomogeneity, spectroradiometric measurements are often not able to provide a satisfying absolute light intensity estimation for the whole chamber, especially in indoor chambers. Chemical actinometry is therefore often used to determine the mean light intensity. Chemical actinometry is an independent method to determine photolysis frequencies by monitoring the change of the chemical composition induced by radiation. For atmospheric measurements of photolysis frequencies, chemical actinometry has rarely been applied because the experimental setup is comparatively extensive and process specific. Moreover, chemical actinometry has mostly been confined to the determination of photolysis frequencies j(NO2) and j(O1D). Nevertheless, chemical actinometry plays an important role in the validation of radiometric techniques (e.g., Hofzumahaus et al. 2004; Kraus et al. 2000; Shetter et al. 2003) and it is an integrated measure of the UV light intensity in simulation chambers (Bohn et al. 2005).
Most simulation chambers are commonly equipped with instruments for the detection of nitric oxide (NO), nitrogen dioxide (NO2) and ozone (O3) that are suitable to perform j(NO2) and j(O1D) actinometry experiments. In-situ spectrometric techniques such as Differential Optical Absorption Spectroscopy (DOAS), Tunable Diode Laser Spectroscopy (TDLAS) or Fourier-Transfer Infrared (FTIR) spectroscopy are highly recommended as they provide unambiguous and direct quantification of these species and can give integrated values over a large fraction of the chamber volume. On-line gas analysers for ozone (absorption technique) and NOx (chemiluminescence technique) can also be used with confidence provided that the chamber is well mixed and that care is taken to ensure that sampling is performed at a point that is representative of the whole chamber. For NO2 detection by chemiluminescence a photolytic conversion of NO2 to NO is recommended, because molybdenum-converters can be affected by other species such as HONO, HNO3 and organic nitrates (Dunlea et al. 2007). However, photolytic conversion is potentially affected by a negative interference at high VOC levels due to the efficient NO/NO2-conversion through peroxy radicals formed in the photolysis of photolabile VOCs (Villena et al. 2012). The choice of instrumentation therefore depends on the type of experiment. As an alternative to chemiluminescence instruments cavity-based absorption methods can be used for the direct detection of NO2.
Dedicated experiments under suitable conditions are required to obtain useful results. Moreover, a determination of j(NO2) or j(O1D) by chemical actinometry alone is not sufficient to characterize the photolytic properties of a simulation chamber. Rather a combination of techniques is required: spectroradiometry can provide actinic flux density spectra of the light source (artificial or the sun) which can then be scaled up or down to match the photolysis frequencies determined by chemical actinometry. Therefore, actinometry can be used to track the changing chamber radiometric conditions over time. Here, we focus on j(NO2) actinometry.
For sunlit chambers the radiation field inside can become inhomogeneous by shadows cast by structural elements of the chamber or instrumental set-ups, the influence of chamber walls through reflection, scattering and absorption, as well as by internal reflections. A radiometric point measurement inside the chamber, even with an ideal 4π sr field of view, may therefore not be representative for the entire chamber volume, an effect which is irrelevant for most other atmospheric measurements. Moreover, the chamber effects will depend on atmospheric conditions, most importantly on solar zenith and azimuth angles and the presence or absence of clouds. Therefore, sunlit chambers require both a continuous monitoring by radiometric devices, ideally a spectroradiometer (inside or outside the chamber) and suitable corrections accounting for specific in-situ chamber effects. These potentially time-dependent corrections can be determined relatively easily by chemical actinometry.
For chambers using artificial light sources the situation is simpler at a first glance. Unless lamps are dimmed or switched on and off, the radiation field can be considered independent of time (except for lamp aging effects on longer timescales). On the other hand, spatial inhomogeneity can be more pronounced compared to sunlit chambers dependent on the illumination technique, e.g., the use of single lamps, a collimated beam in tube-shaped chambers or all-around systems of tubular fluorescent lamps. In the best case, it is sufficient to occasionally record lamp spectra at a selected point within the chamber and perform chemical actinometry to derive adequate scaling factors. In contrast to sunlit chambers, spectral irradiance measurements are suitable as well. However, if different lamp types are combined, it is necessary to determine the corresponding photolysis frequencies separately.

Concentrations of NOx, NO2, NO, O3 and the difference NO–O3 during an actinometric experiment in SAPHIR at Forschungszentrum Jülich on a mostly clear-sky day. The chamber roof was opened around 06:30 and closed shortly after 17:30
Gradients in the radiation fields can result in gradients in short-lived species concentrations, which need to be considered for any chamber. Active mixing is a means to reduce such concentration gradients. In the following, we assume that trace gas concentrations are homogeneous, so that the chemical composition probed at any location is representative for the entire chamber. In this case, results represent a chamber-mean of actinic radiation.
2.3.1 Chemical Actinometry in Air
The photolysis of NO2 is mostly UV-A driven and leads to the formation of nitric oxide (NO) and ground state oxygen atoms (O(3P)):
Under tropospheric conditions, the photolysis is followed by a fast and quantitative formation of ozone (O3) in the reaction with an oxygen molecule (O2):
M is a third-body reaction partner. A chemical actinometer for atmospheric measurements of j(NO2) typically consists of a quartz flow-tube where a known concentration of NO2 (mixing ratio in the ppm range) in synthetic air or O2 is exposed to sunlight for a short period of time (ca. 1 s) (Shetter et al. 2003). The NO concentration produced during the exposure time is then a direct measure for j(NO2):
The presence of O2 avoids interferences from the fast reaction O(3P) + NO2 → NO + O2. Moreover, at short exposure times the influence of the comparatively slow NO + O3 back-reaction is negligible:
For simulation chambers this concept of actinometry is not applicable because short exposure times are not feasible. However, under typical simulation chamber conditions, Reactions R2.3.1–R2.3.3 lead rapidly to a photochemical equilibrium or photo-stationary state (PSS). The relaxation time constant of this equilibrium is on the order of minutes depending on the values of j(NO2) and concentrations of trace gases. In the atmosphere, this equilibrium can be strongly affected by the presence of peroxyl radicals (HO2 and RO2), which also convert NO into NO2 without consuming O3. The so-called Leighton ratio φ = j(NO2)[NO2]/(k3[NO][O3]) (Leighton 1961) is a common measure for the deviation from the purely NOx/O3 determined equilibrium owing to peroxyl radical perturbations. On the other hand, in the absence of interfering reactions, the Leighton ratio is unity under steady-state conditions. Accordingly, j(NO2) can be calculated from the equilibrium concentrations of O3, NO2 and NO, and the (temperature dependent) rate constant of the NO + O3 (2.07 × 10–12 exp(−1400/T), IUPAC) Reaction R2.3.3:
Figure 2.4 shows an example of an actinometric j(NO2) experiment in the sunlit simulation chamber SAPHIR at Forschungszentrum Jülich. Around 45 ppbv of NO2 was injected into the dark chamber shortly before 6:00. In the illuminated chamber, a fast decrease of NO2, and a corresponding rapid increase of NO and O3 concentrations were observed. The NOx (=NO + NO2) concentration remained nearly constant during this quick initial adjustment of the photochemical equilibrium, as expected. The chamber roof remained open for approximately 12 h. During this time, the NOx concentration slowly decreased mainly caused by dilution. However, this decrease is slow compared to the relaxation time constant of the photochemical equilibrium. Accordingly, at any time NO2, NO and O3 concentrations are in a steady-state equilibrium and j(NO2) can be calculated according to Eq. (2.3.3), taking into account the measured gas-phase temperature.

j(NO2) determined by actinometry (photo-stationary approach PSS, time dependent model TD) and predicted by a combination of outdoor measurements of spectral actinic flux densities and a radiation transfer model (SR) scaled to match the actinometric data. The rectangular box (h) indicates the illumination period of the SAPHIR chamber
In Fig. 2.5 the resulting time-dependent photo-stationary state j(NO2) (PSS) is compared to predictions resulting from a radiometric measurements (SR) that are used to calculate j(NO2) inside the chamber with a radiation transfer model. The model is fed by measurements of spectral actinic flux densities of direct and diffuse radiation outside the SAPHIR chamber (Bohn and Zilken 2005). The SR approach correctly predicts the typical shape of the diurnal variation of j(NO2) inside SAPHIR on this clear-sky day. However, absolute values of j(NO2) predictions need to be significantly scaled down to match the actinometric data. This scaling factor also increases with time. This can be explained by an increasing degree of staining and mechanical degradation of the chamber walls caused by many years of outdoor residence and operation. Therefore, the scaling factor needs to be regularly determined in experiments of this type, in order to correctly scale the SR data during other photochemical experiments. The radiometry/model approach for SAPHIR (Bohn and Zilken 2005) is quite specific and not directly transferable to other sunlit chambers for which a simple spectroradiometer measurement inside or outside the chamber may be sufficient depending on the geometry of the setup.
Experiments such as the one shown in Fig. 2.4 can be employed in any chamber where the mean residence time is sufficiently long compared to the relaxation time constant of the photochemical equilibrium. For chambers with artificial lights the concentrations may also change with time because of dilution but the photolysis frequency should remain constant—an additional test for the validity of the approach. If no time dependence of photolysis frequencies is expected, experiments can be kept much shorter. Even under non-equilibrium conditions during periods where j(NO2) changes more rapidly than in the example above, the actinometric approach works reliably, if concentration changes are analysed numerically. This time-dependent (TD) approach is described in more detail elsewhere (Bohn et al. 2005). Moreover, the dark periods after the experiments can be analysed by testing if the decays of NO and O3 are consistent with the rate constant of Reaction 2.3.3. Analytical solutions for the analysis of the decays are described in Bohn et al. (2005) including conditions for which NO and O3 concentrations are not only determined by the initial NO2 concentration and dilution of trace gases are taken into account.
It should be noted that the experiment shown in Fig. 2.4 starts with a small excess of 2 ppbv O3 from a previous experiment and ends with an about 1 ppbv excess of NO that can be explained by the production of NO from photolysis of nitrous acid (HONO) that is formed inside SAPHIR (Rohrer et al. 2005). In the experiment shown in Fig. 2.4, approximately 3 ppb of HONO was generated and photolyzed over the course of the experiment. OH that is also formed in the photolysis of HONO predominantly reacted with NO2 which merely led to a small increase of the total loss of NOx, but this had no significant influence on the steady-state concentrations of the \({{{\text{NO}}_{\text{x}} } / {{\text{O}}_{3} }}\) equilibrium that is established much faster.
Effects on the steady-state equilibrium concentrations can be minimized by:
-
Reducing the HONO source in the chamber (e.g., low humidity in chambers made of Teflon film), because NO produced from HONO photolysis adds to the total concentration of nitrogen oxides in the chamber. Only, if the NO produced from HONO photolysis is small compared to the initial NO2 concentration, it can be neglected in the calculations (Eq. 2.3.3).
-
The absence of additional OH reactants that could produce HO2 or RO2, which reacts with NO, so that the equilibrium between NO2 and NO is shifted to NO2 and ozone is produced.
-
Using high NO2 concentrations that ensure that any OH is scavenged in the reaction with NO2 to prevent the production of HO2 or RO2.
Optimum experimental conditions need to be carefully chosen for a specific chamber to avoid chemical interferences.
2.3.2 Chemical Actinometry in Nitrogen
When minimizing the radical sources that may affect the steady state equilibrium, the photolysis of NO2 in pure nitrogen as a bath gas is often recommended to avoid the complexity arising from secondary chemistry in the determination of the NO2 photolysis frequency in a simulation chamber. This method has long been known in atmospheric research for the calibration of UV sources and is well described by Holmes et al. (1973) and Tuesday (1961).
A fully oxygen-free atmosphere can be difficult to achieve in chambers, because air may enter the chamber by small leakages or permeation. Leakage of air into the chamber can be minimized by operating the chamber at a pressure slightly above atmospheric pressure or by a second wall around the chamber, so that the gap can be flushed with nitrogen. The latter also minimizes permeation.
Similar to the actinometry experiment in air described in the previous section, NO2 is injected into the chamber, but the chamber is filled with pure nitrogen. In the absence of oxygen, no ozone is produced from the photolysis of NO2, so that only NO and NO2 concentrations need to be precisely measured. A high time resolution of instruments is needed, because the time resolved consumption of NO2 and production of NO is observed. An initial NO2 mixing ratio within the range of 0.1–1 ppmv is recommended, as the photolysis of NO2 can be rather fast. A high initial concentration ensures that the time period that can be used for the evaluation is sufficiently long.
The decay of NO2 and production of NO is mostly determined by the NO2 photolysis reaction (Reaction R2.3.1), but oxygen atoms formed from in the photolysis significantly accelerate the loss NO2:
The highest impact on the NO2 loss is due to the formation of NO (Reaction R2.3.4) because the reaction rate constant of Reaction R2.3.4 is 5 times higher than that of Reaction R2.3.5 at room temperature and ambient pressure. Both reactions are only relevant in experiments in nitrogen because molecular oxygen is missing as a reaction partner for the oxygen atom (Reaction R2.3.2). For the same reason, also the reaction of oxygen atoms with NO impacts the temporal behaviour of NO and NO2 in this type of experiment:
This reaction together with the NO2 photolysis reaction are responsible that eventually a photo-stationary state is established between NO and NO2 concentrations. Rather small effects are expected from the formation of oxygen in Reaction R2.3.4, which would allow to form ozone (Reaction R2.3.2), but the reaction rate constant is too small for producing significant ozone concentrations at oxygen concentrations formed in the system.
The production of nitrate radicals in Reaction R2.3.5 further complicates the chemistry, because a small fraction of the nitrogen oxides is converted to NO3 and N2O5 right after the start of the NO2 photolysis, but converted back to NO2 and NO at later times of the experiment with increasing NO concentrations due to the following reactions:
Depending on the radiation in the chamber, NO3 may be additionally photolyzed and NO3 chamber wall loss could be significant.
For these reasons, the photolysis of NO2 in nitrogen is not a first-order loss process. In Holmes et al. (1973) the following equation is derived to describe the decay rate of NO2:
The meaning of this equation is that as first approximation each photolyzed NO2 molecule produces 2 NO molecules due to the direct formation of NO (Reaction R2.3.1) and the subsequent reaction of the oxygen atom with NO2 (Reaction R2.3.4). The other terms are corrections that are needed due to other competing reactions of the oxygen atom (Reaction R2.3.2, R2.3.5 and R2.3.6).
Equation 2.3.4 can be used to calculate j(NO2) from the measured [NO2] decay. Figure 2.6 shows measured [NO], [NO2] and NOx ([NO2] + [NO]) concentrations in an experiment for the determination of j(NO2) in the CESAM atmospheric simulation chamber using 460 ppbv of gaseous NO2 diluted in nitrogen. The total NOx concentration is nearly unchanged during the experiment, because NO2 to NO reactions (Reaction R2.3.1 and R2.3.4) dominate the reaction system. The total NOx concentration decreases approximately by a 5% in this experiment. This loss can be attributed to the dilution of the reaction mixture due to sampling by the monitors. It is recommended to monitor the dilution rate, so that measured concentrations can be corrected for dilution.

(© Jean-François Doussin, personal communication)
NO2, NO and NOx concentrations versus reaction time during NO2 photolysis in nitrogen in the CESAM chamber
Figure 2.5 shows the evaluation of the actinometry experiment describing the logarithm of the measured NO2 concentration by integrating Eq. 2.3.4. The decreasing loss rate is due to the competition of the reaction of NO with the oxygen atom (Reaction R2.3.5), which gains in importance due to the increasing NO concentration while NO2 is being photolyzed. Deviations of the calculations are likely due to neglecting further NO3 chemistry (Reaction R2.3.7 and R2.38.). A first approximation of the photolysis frequencies can be calculated by taking only the first data points after the light is switched on. Assuming that the impact of the reaction of NO with oxygen atoms is negligible, because NO has not yet formed from the photolysis of NO2, NO2 decays as first-order loss process, so that a linear behaviour of the logarithm of the NO2 concentration is expected Fig. 2.6). The photolysis rate can be calculated from the slope using Eq. 2.3.4 with [NO] = [O2] = 0 (Fig. 2.7).

(© Jean-François Doussin, personal communication). In the present example, the initial slope method provides a j(NO2) value of (2.3 ± 0.2) × 10–3 s−1, while the use of the Eq. 2.3.5 yields j(NO2) = (2.8 ± 0.1) × 10–3 s−1
NO2 concentration time series in a typical actinometry experiment in nitrogen in the CESAM chamber. The dashed line is the result of a linear regression using the first four data points after the light had been switched on. The solid line is the result of the integration of Eq. 2.3.5 assuming a negligible initial oxygen concentration
Because Eq. 2.3.4 does not consider the impact of NO3 chemistry and the photo-stationary state between [NO2] and [NO] concentrations that is established at later times of the experiment, it is recommend applying this calculation only for the time right after NO2 photolysis has started. As an alternative, box-model calculations may be applied to determine the photolysis frequency by adjusting its value such that the NO2 decay is best described. However, uncertainties may occur, if the oxygen concentration due to leakages is not known or NO3 wall loss leads to a significant loss of nitrogen oxides during the experiment.
Both data analysis procedures exhibit a significant sensitivity to oxygen concentration which can be difficult to control in a large reactor. They are also very sensitive to the precision of the NOx measurement and to the potential interferences of NOy species. This is why, when routinely applied, it is recommended that data analysis is performed only on the first few data points. This avoids giving undue weighting to data acquired at the end of the experiment when NO2 concentrations are close to the detection limit, NOy species arising from secondary chemistry may have accumulated and when O2 concentration may have increased due to leaks. Results which show a deviation from those expected from the analysis described below may indicate that the O2 concentration is not low enough.
jNO2 can be evaluated by considering short time steps and neglecting oxygen equation in Eq. 2.3.4 which becomes
If only NO2 measurements are available, Eq. 2.3.5 may be transformed under the assumption that NO and NO2 contain almost all the NOx at any time (\(\left[\mathrm{NO}\right]={[\mathrm{NO}]}_{0}+{[{\mathrm{NO}}_{2}]}_{0}- [{\mathrm{NO}}_{2}]\)).
2.4 Gas-Phase Wall Losses of Species
Significant wall loss can be observed for gaseous inorganic as well as organic compounds. The wall loss rate can be highly variable and is specific for a compound and the chamber. Therefore, wall loss needs also to be considered in the evaluation of experiments as part of the chamber auxiliary mechanism (Sect. 2.6).
For inorganic compounds, the wall uptake can be observed to be irreversible, reversible and/or even reactive, and is commonly measured for ozone and nitrogen containing compounds specifically for NO and NO2 (Grosjean et al. 1985; Wang et al. 2014). Grosjean et al. (1985) reported little or no significant wall loss for most tested organic compounds. If they observed wall loss the loss appeared to be irreversible. More recent work has shown that this loss can be significant and also reversible for low volatile and/or oxygenated organic compounds (e.g., Loza et al. 2010; Matsunaga and Ziemann 2010). The wall losses of gaseous inorganic and organic compounds can occur on the same timescales as their gas-phase oxidation and gas/particles mass transfer processes and can therefore be competitive (e.g., Grosjean et al. 1985; Krechmer et al. 2016). These wall losses are expected to depend on: (1) the chamber characteristics (e.g., nature of the walls, geometry, age/history, surface to volume ratio, mixing time and procedure), (2) the environmental conditions (e.g., temperature, irradiation, relative humidity) and (3) the physicochemical properties of the compounds themselves.
Atmospheric simulation chambers are extensively used to study the homogeneous and/or multiphasic evolution of gas-phase compounds. Chamber wall losses can thus affect experimental results on (1) the kinetic and mechanistic studies of compounds in the gas phase (e.g., Bertrand et al. 2018; Biermann et al. 1985; Yeh et al. 2014) and (2) the formation and composition of secondary organic aerosols (e.g., Krechmer et al. 2016; Kroll et al. 2007; La et al. 2016; McVay et al. 2014; Pathak et al. 2008; Shiraiwa et al. 2013; Zhan et al. 2014).
In order to determine the wall loss parameters of an individual compound, its concentration in the gas phase is usually measured over time in a clean, dark chamber under constant environmental conditions. The preparation of the clean chamber is done as described in Chap. 3. and the wall loss characterization experiment is similar to the blank experiment described in Sect. 2.6 In the simplest case, the decay can be fitted to a function that describes an irreversible first order loss process, if a decrease of the gaseous concentration is observed after correcting for dilution. A parameterization can also include physical conditions or chemical properties such as equilibrium concentrations or saturation vapour pressure of the specific compound.
Prior to each characterization experiment, the chamber is prepared using usual cleaning, conditioning and filling protocols (Chap. 3). If the chamber allows, experiments should be performed at a fixed temperature and relative humidity but may need to be varied in a series of experiments. The wall losses of gaseous compounds are either studied for compounds that are directly injected into the chamber (e.g., Huang et al. 2018; Loza et al. 2010; Matsunaga and Ziemann 2010; Shiraiwa et al. 2013; Yeh and Ziemann 2014a, b, 2015; Zhang et al. 2014) or produced in the chamber from the oxidation of parent compounds (e.g., Huang et al. 2018; Krechmer et al. 2016; Zhang et al. 2015).
The injection of single compounds or mixtures of different compounds is typically done into the dark chamber using common procedures (Chap. 4). To avoid competition of gas/wall loss with gas/particle partitioning, the injected quantity of the compound should be below its saturation vapour pressure once inside the chamber, thereby preventing particle nucleation occurring. Ideally, a known quantity of the compound is injected into the chamber and is homogeneously mixed instantaneously, so that mixing of the compound and wall loss are separated in time. However, depending on the volatility of the compound, the injection duration can vary from minutes to hours, so that both processes may need to be taken into account for the determination of the wall loss rate.
If the wall loss of oxidation products that are not available as pure compounds need to be characterized, the precursors are first injected into the chamber. Concentrations should again be low enough that aerosol formation does not play a role for the species loss. Depending on the specific chamber, oxidation needs to be initiated for example by injection of ozone or hydroxyl-radical precursors. If photo-oxidation is required to produce oxygenated products, the chamber air needs to be exposed to light (lamps or sunlight). In the ideal case, oxidation is stopped after a few seconds for example by switching off lights, when a sufficiently high concentration of products is formed, and the decay of the target species can be used to determine the wall loss. If oxidation cannot be stopped or if there is a reversible loss of the compound, all processes need to be considered such as gas-phase production and equilibrium between the gas-phase and deposition on the wall.
Depending on the wall loss rate, concentrations need to be monitored over a few hours (2–15 h). It is worth noting that interactions of the compound with the walls can also occur in the inlet line of instruments (Deming et al. 2019; Krechmer et al. 2016; Liu et al. 2019; Pagonis et al. 2017). Delays and underestimations of concentrations can therefore be observed. Therefore, passivation of the inlet lines is recommended.
If the loss of compound A (gas phase concentration [Ag]) is irreversible, the process is described by a first order loss rate constant kgw:
If there is no production or injection of the compound during the time of observation and if wall loss is the only relevant process, the observed decay can be fitted to a single-exponential function that directly gives the first order loss rate constant.
In the case of a reversible loss process, the transfers of a gaseous species A to the wall (wall reservoir concentration \(\left[{A}_{w}\right]\)), and back to the gas phase, can be described by the gas phase first order loss rates \({k}_{wg}\) and \({k}_{gw}\), respectively:
In the case that there are no other relevant production or destruction processes concurrently happening, Eq. (2.4.2) can be solved. Boundary conditions are that the sum of concentrations in the gas-phase and the wall reservoir equals the initial concentration \({\left[A\right]}_{0}\) that has been injected or produced by oxidation and \({\left[{A}_{g}\right]}_{eq}\) is the equilibrium gas-phase concentration that is eventually obtained:
The fit of the observed concentration time series results in the effective wall loss rate of \({k}_{Weff}=({k}_{gw}+{k}_{wg})\) and the equilibrium concentration \({\left[{A}_{g}\right]}_{eq}\). The initial concentration may be fixed to measured values or can also be obtained by the fitting procedure.
As an alternative to fitting to an analytic solution, the observed time profile of the measured concentration can be described by optimization of parameters in a numerical box model, which can allow for taking additional loss and production processes into account. The wall loss rate constant can be optimized using standard optimization procedures to minimize the difference between measured and modelled concentration time series.
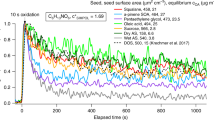
Figure 2.8 shows the wall deposition velocities (i.e., the first order loss rate corrected by the surface (S) to volume (V) ratio of the simulation chamber calculated as vgw = kgw V/S) measured in various EUROCHAMP simulation chambers for several organic compounds as a function of their saturation vapour pressure (Psat). The deposition velocity appears to depend on (i) the chamber characteristics which cover a large diversity of wall materials, surface to volume ratios and mixing times, (ii) the organic species properties, such as the saturation vapour pressure, and (iii) the environmental conditions, such as the relative humidity. For example, the walls of the chambers made of aluminium like the AIDA chamber at Karlsruhe Institute of Technology may induce a constant loss of gas-phase compounds, depending on molecular properties and wall temperature. In chambers made of Teflon, the wall loss rates often correlate with the vapour pressure. However, the effect of wall loss can be minimized, if the chamber has a high volume to surface ratio like the SAPHIR chamber made of Teflon film (270 m3) at Forschungszentrum Jülich.

(Figure from EUROCHAMP-2020 Deliverable 2.7, www.eurochamp.org)
Wall deposition velocities (vgw) of gaseous organic compounds measured in EUROCHAMP chambers as a function of the saturation vapour pressure (Psat). The organic species properties were estimated with the GECKO-A tool (e.g., Valorso et al. 2011) using the Nannoolal method to calculate the saturation vapour pressure at 298 K (Nannoolal et al. 2008). The loss rate also highly depends on the wall material and size of the individual chamber and the type of molecule, so that there is no unique value for the wall deposition velocity for a specific saturation vapour pressure value.
2.5 Particle Wall Losses
Aerosol processes in the atmosphere can have typical timescales ranging from a few seconds up to several days. In order to investigate such processes under reasonably representative atmospheric conditions, simulation chambers used for the investigation of physical, chemical or biological transformations of aerosol particles should enable a sufficiently long particle lifetime. In addition to their interaction with each other and with the gases in suspension, the lifetime of aerosol particle in chambers may be substantially controlled by wall losses resulting from the combination of adsorption, deposition, diffusion and mixing processes, gravitational settling and electrostatic attraction, all depending on particle and wall properties. The physical wall loss of particles in closed vessels such as chambers will vary with the particle size and will depend on (i) the chamber shape, (ii) the mixing regime (especially for small particles), (iii) the density of the considered particles, (iv) the electrostatic state of the wall.
Clearly it is important to understand particle loss rates in any experiments aiming to characterize the dynamic evolution of the distribution of particles, which can range from characterization of formation and transformation processes through to the determination of optical properties and their dependence on particle mixing-state to investigation of gas-aerosol interactions such as in the formation of secondary organic aerosol (SOA). By definition, simulation chambers are volumes enclosed by walls and interactions of particles with their surfaces cannot be assumed negligible. The advantages of knowing the size-dependent wall losses of particles are substantial and various. The losses will determine the reduction in lifetime for contribution of the particles to properties or processes of interest in the experiment. For example, reduction in the particle lifetime will reduce the condensation sink it presents for gas–aerosol interaction (such as SOA formation). Similarly, optical extinction by a diminishing particle population will be commensurately reduced. Consequently, knowledge of the losses will enable more confidence to be ascribed to measured properties and inferred processes. For example, model-measurement comparison of the dynamical evolution of the particle population will enable model processes such as condensational growth, and properties such as vapour pressure, to be constrained so long as particle wall losses (as well as vapour–wall interactions) are known. Conversely, the consequences of neglecting to characterize particle wall losses is a substantially compromised ability to interpret any experiments aiming to capture properties or processes that depend on particle size distributions.
Provided that the aerosol in the chamber is well mixed, in the absence of any other process, the rate coefficient for the wall loss, βi, can be represented as a simple first order loss:
where Ni is the number concentration of particles of size class in the chamber (Crump et al. 1983). The coefficient βi for each size class can be obtained by integrating Eq. (2.4.1) to give (Fig. 2.9):

Size dependent wall losses directly obtained from measurements of the first order decay of polydisperse particles nebulized or mechanically generated and injected into the CESAM chamber (Lamkaddam thesis 2017)
The requirement to quantify and account for the wall losses of particles in simulation chambers used for aerosol experiments is universal, though the extent of the required characterization is, to some extent, application dependent. This has led to a pragmatic variety of approaches to wall loss determination that broadly fit into two classifications:
-
wall loss quantification using deliberate characterization experiments (denoted “seed injection” methodologies)
-
wall loss quantification using chamber experiments (denoted as “In-experiment” methodologies).
Moreover, the methodology is dependent on both the chamber geometry and materials used in its construction. Specifically, rigid fixed-geometry structures of conductive materials (e.g., aluminium or steel), rigid fixed-geometry structures of insulating materials (e.g., glass) and flexible variable-geometry structures of insulating materials (e.g., Teflon) will each require and be best suited to particular approaches.
Whilst the methodology may vary, a common requirement for each approach is the availability of well-characterized and calibrated particle sizing and counting instrumentation and an appropriate source of particles.
The instrumentation normally comprises:
-
a mobility sizing instrument coupled to a particle counter(s) (either a differential or scanning particle mobility sizer, SMPS or DMPS, coupled to a condensation particle counter with the appropriate size cut-off), and/or
-
an optical particle spectrometer with the appropriate configuration (normally backscatter of coherent or broadband white light, but forward scatter may be more appropriate for cloud droplets).
The particle source is usually a nebulizer of some sort, capable of generating salt solution aerosol from a quantified stock. Selection of the source and stock will depend on the desired particle size, breadth of distribution and number, and of course on composition (e.g., brush generator or fluidised bed may be more appropriate for soot or dust characterisations respectively.
Maintenance of chamber facilities and transport of smaller chambers for field campaigns can induce electrostatic charges on chamber walls and increase particle wall-losses. This generates a “disturbed” chamber that can introduce significant uncertainty in the particle wall-loss rates. The recovery time can even be months after the disturbance if natural charge dissipation is the only action that reduces the charges on the walls (Wang et al. 2018a, b).
An electrostatic eliminator device (fan or air gun) can reduce the induced charges on the chamber walls significantly faster that the natural charge dissipation process. Such an electrostatic fan was used in the chambers of FORTH laboratories to reduce the induced charges after maintenance and handling of several different chambers.
In order to determine size-dependent wall loss rates, two approaches for particle generation were for example employed in the CESAM chamber. Polydisperse (NH4)2SO4 particles were nebulized from an aqueous saline solution to provide sub-micron particles and test dusts were mechanically generated for super-micron particles. Total number concentration was held below 104 cm−3 to minimize the collision probability and so as not to require a correction for coagulation. The number size distribution was measured as a function of time (with a SMPS for sub-micron particles and OPC for super-micron particles). A first order decay fit following Eq. 2.7.1 was fitted to the time evolution of each size-bin.
Wang et al. (2011) reported that particle lifetime in the stainless steel CESAM chamber ranges from 10 h to 4 days depending on particle size distribution, enabling the chamber to provide satisfactorily high-quality data on aerosol aging processes and their effects. More recently Lamkaddam (2017) has studied the physical wall loss rate as a function of particle size. Submicron ammonium sulphate particles were generated in small number to minimize coagulation and mineral dust were used for supermicron particles. The vertical air velocity was experimentally measured in the chamber and its value was used as the u* parameter in the Lai and Nazarof parameterisation (Lai and Nazaroff 2000). Plotting the particles wall loss frequency as a function of size in a log–log plot will yield a typical V-shape curve when electrostatic charges are not significant (Lai and Nazaroff 2000). Owing to its stainless steel construction, this is expected and is found to be the case for CESAM and, as shown in Fig. 2.5, the size-dependent wall loss compares satisfactorily with previous literature (Crump et al. 1983; Lai and Nazaroff 2000). Above all, even if developed for parallelepiped volumes, the Lai and Nazarrof parameterization has shown excellent agreement by just introducing the correct CESAM chamber dimensions, the measured u* and the correct density for particle material without any further adjustment or fitting of the model to the data points. The same approach was successfully adopted at the ChAMBRe facility (Massabò et al. 2018).
A range of approaches have been used to conduct an extensive characterization of the wall losses in the participating chambers. Pragmatic approaches to the characterization of each infrastructure has led to a variety of techniques according to the chamber and experimental type, instrumental availability and application-specific requirements. This diversity across the infrastructures has led to considerable expertise across the scientific applications and continuous contribution to the state-of-the-science in characterization of wall losses in simulation chambers. Best practice and model code have been shared, though design and adoption of a standardized protocol is still challenging. The recommendation is that particle wall losses are characterized as far as possible, and standardization is adopted as soon as the state-of-the-science allows. A more straightforward approach appears possible for rigid chambers constructed of the conducting material. Whilst novel mitigation approaches show promise, electrostatically enhanced particle loss in flexible plastic film chambers requires further investigation.
2.6 Characterization of the Chamber State by Gas-Phase Reference Experiments
Chamber-specific properties need to be characterized, in order to take into account the chamber background reactivity in any experimental evaluation procedure. This allows the separation of the chamber-specific chemical processes from the underlying chemistry that is being studied in experiments. They can be put into auxiliary mechanisms that complement chemical mechanisms to perform chemical modelling of chamber experiments. These auxiliary mechanisms are essential to make results from experiments carried out in different chambers comparable and transferable to the atmosphere.
Chamber auxiliary mechanisms contain a number of specific features to account for chamber properties that often arise from effects of the chamber walls. Primarily, these features consider (Fig. 2.10):

Illustration of interaction of trace gases with the wall of simulation chambers
-
Adsorption/desorption of nitrogen oxide species (NOy, including HONO, N2O5 and HNO3) and reactive organic species to/from the chamber walls.
-
Deposition of aerosol to the chamber walls.
-
Dilution of chamber trace constituents through leaks and gas removal by instruments.
Many of the chamber-specific processes can change over time due to memory effects from previous experiments carried out in the chamber. Hence experiments to characterize the processes should be performed regularly, for example at the beginning/end of intensive experimental campaigns. Characterization of wall loss is discussed in detail in Sects. 2.4 and 2.5.
2.6.1 Chamber Blank Experiments
Chamber blank experiments are used to assess impurities in the background air matrix as well as degassing of species from the chamber walls. The chamber is prepared as is typically done in most experiments, starting by cleaning the chamber and filling with pure air (Chap. 3). Concentrations of trace gasses are observed throughout the experiments, so that their release from the chamber walls can be parameterized. Wall sources of compounds in a chamber are often photolytic and can also be affected by the amount of water vapour present. Hence experiments are performed under light and dark conditions, and at the upper and lower limits of the typical operating range for relative humidity in the chamber (e.g., Rohrer et al. 2005, Zador et al. 2006). Figure 2.11 shows examples of reference blank experiments carried out to determine the wall sources of formaldehyde (HCHO) in the EUPHORE chamber. Chamber blank experiments can also be used for the determination of wall loss processes (see Sects. 2.6 and 2.7).

© EUPHORE
Chamber blank experiments carried out in the EUPHORE chamber in order to determine formaldehyde (HCHO) wall sources at different relative humidity values (RH) and under light and dark conditions. This behaviour is generally interpreted as a photochemical O3 production from background reactivity (unmeasured species that are formed in the sunlit chamber and could be released from the chamber wall).
2.6.2 Reference Experiments Using Well Known Chemical Systems
Reference experiments with well-known chemical systems can be used to regularly evaluate if the chemistry of the system under investigation can be separated from chamber effects. The measured time series of trace gases and radicals can be compared and contrasted to chamber simulations performed by chemical box modelling. The model must include the chemistry of trace gases present in the reference experiment and the auxiliary mechanism that describe chamber-specific processes. Several types of experiments are described below.
2.6.3 Experiments with Mixtures of NOx in Air
Because of the importance of NOx for atmospheric chemistry, the behaviour of nitrogen oxides in the chamber is often characterized in the blank experiment. Nitrogen oxide species (NOy) are known to be emitted into the gas phase from photolytic production on chamber walls, including NO, NO2, HONO, HNO3 (Rohrer et al. 2005; Zador et al. 2006). These species can also be inter-converted between each other, both in the gas phase and on the walls. For example, nitrogen dioxide may convert to nitrous acid and nitric acid (NO2 → aHONO + bHNO3). This heterogeneous chemistry can be affected by both light and relative humidity. Experiments in which NOx is added to a clean chamber can be used to explore the rates of interconversion of NOy species driven by the walls.
Chamber wall materials are typically chosen to be chemically inert. Effects from the wall material are often related to the chemical nature of adsorbed compounds arising from previous experiments. Because most of the chemical systems studied in simulation chambers lead to the formation of oxidized species, chambers walls generally exhibit an oxidative potential (Bloss et al. 2005; Hynes et al. 2005; Metzger et al. 2008).
Some the studies (Bloss et al. 2005; Metzger et al. 2008) show that the consumption of NO2 is coupled with the formation of small quantities of HONO. In Metzger et al. (2008) the wall loss rate of NO2 was (1.05 ± 0.35) × 10–6 s−1. In some cases, however, the chamber walls can be reductive as shown for metal chambers (Wang et al. 2011).
It is recommended to follow a protocol for chamber blank experiments similar to that described in Wang et al. (2011):
-
Injection of 50–200 ppbv of NO2.
-
Monitor in the dark for 1 h.
-
Irradiate the chamber air for 1 h.
-
Monitor in the dark for 30 min.
-
Monitor NO, NO2, O3, HCHO, HONO and radicals (if available).
-
Systematic studies with changing RH are recommended.
An example of this type of reference experiment from the CESAM chamber is given in Fig. 2.12 After NO2 had been injected into the chamber, a continuous loss of NO2 was observed in the dark that was accompanied by a slow production of NO. When the lights were turned, NO2 is photolyzed reaching a photo-stationary state that is established between NO2, NO and O3 concentrations within approximately 5 min. During the phase, when the chamber air was irradiated chamber wall effects lead to slow production of NO that is interpreted as NO2 conversion on the wall. Ozone concentrations decreased due to the increase of NO, but also wall loss played a role. Consequently, when the lights were turned off the concentration of NO remained high, because the available ozone concentration was not sufficient to convert all NO back to NO2. In addition, the sum of NO and NO2 was lower compared to the initially injected NO2 concentration due to chamber wall loss.

(Reused with permission from Wang et al. (2011) Open access under a CC BY 3.0 license, https://creativecommons.org/licenses/by/3.0/)
Examples of a NOx-air experiment carried out in the CESAM chamber, with initial injection of ca. 200 ppbv NO2.
2.6.4 Photochemical Oxidation of CO/Methane
Radical concentrations in chambers are often impacted by chamber processes. For example, a major source for hydroxyl radicals (OH) in Teflon chambers is often the photolysis of nitrous acid (HONO) that is emitted from the chamber walls. In addition, radicals may be lost on the chamber walls or react with organic species that are released from the chamber wall but may not be quantified. Reference experiments are useful to test if radical sources and sinks are understood.
The experimental procedure is similar to that of a blank experiment. The chamber is exposed to light, in order to trigger photolytic processes. The radical source can be the photolysis of nitrous acid released from the chamber walls. Ozone can also be injected to produce radicals from its photolysis. The chamber air would typically be humidified in the experiment, because water vapour is often needed to produce radicals. Due to the presence of sources of unknown OH reactants in the chamber, it is recommended to add an OH reactant. CO converts OH radical to hydroperoxyl radicals (HO2) and methane converts OH to methylperoxyl radicals (CH3O2). In the presence of nitric oxide (NO) a radical reaction chain is initiated in which ozone is produced in the chamber. Steady-state equilibrium concentrations of radicals are rapidly established owing to the short chemical lifetime of radicals. OH reactant concentrations are chosen such that OH radical equilibrium concentrations are above the limit of detection of instruments detecting radicals. Results from chemical box models can be compared to observed radical concentrations to test if chamber processes are appropriately taken into account. In addition, the ozone concentration increase can also be compared, because ozone is chemically produced in the reaction of peroxyl radicals with NO.
Figure 2.13 shows an example for a reference experiment with CO and CH4 injections in the SAPHIR chamber with the specific aim to test if OH radical concentrations can be described and understood. Results from a chemical box model gives excellent agreement between measured and modelled radical concentrations, if chamber-specific processes such as sources for nitrous acid (HONO) and formaldehyde (HCHO) are included and adequately described.

Reference experiment in SAPHIR with injections of CO and CH4 indicated by the increase in measured OH reactivity (kOH). Measurements (blue dots) are compared to results of a chamber chemical box model (red lines) that include chamber-specific properties such as chamber sources of nitrous acid (HONO) and formaldehyde (HCHO). Grey areas indicate times when the chamber was kept in the dark and vertical lines give times of injections
2.6.5 Photo-Oxidation of Propene in the Presence of NOx
Experiments using more complex organic compounds with a well understood chemical oxidation mechanism, such as ethene (C2H4) or propene (C3H6), can be used to test the efficacy of the chamber auxiliary mechanism and can also be used to optimize/tune them (Bloss et al. 2005; Wang et al. 2011). Disagreements between the measured and modelled mixing ratios of precursor and product compounds are therefore assumed to be caused by chemistry driven by the chamber walls. A particularly clear chamber effect is the timing of the onset of removal of the VOC following the initial addition of the VOC and NOx. The experiment begins with no addition of a radical source, with much of the initial reactivity in a chamber being driven by HONO coming off the chamber walls and being photolyzed to produce radicals which can react with the VOC. Hence this timing is a good indication of the rate of HONO production.
It is recommended to follow a similar protocol to that used in the work of Hynes et al. (2005) and Wang et al. (2011, 2014):
-
Experiments carried out over a range of VOC–NOx concentration ratio of 0.6–17, e.g., injection of 500 ppbv C2H4 and 50–300 ppbv NO.
-
Observation of trace gases concentrations including propene, NO, NO2, ozone, HONO, HCHO, CH3CHO, HCOOH, PAN, radicals, if available, during the photo-oxidation of C2H4 for 5 h.
-
Observation of trace gas concentrations for 1 h in the dark.
-
Studies with systematic changes of the relative humidity are recommended.
An example of a propene-NOx experiment carried out in the CESAM chamber is shown in Fig. 2.14.

(Reused with permission from Wang et al. (2011) Open access under a CC BY 3.0 license, https://creativecommons.org/licenses/by/3.0/)
The overall examples of a propene-NOx experiment carried out in the CESAM chamber, with an initial injection of ~250 ppbv propene, 50 ppbv NO2, 120 ppbv NO 200 ppbv NO2, Wang et al. (2011). Comparison of simulated (solid lines) and experimental concentrations (symbols) for NO, NO2, ozone, propene, formaldehyde, acetaldehyde, formic acid and peroxyacetyl nitrate (PAN) in a propene-NOx-Air system. Solid lines are the results of modelling with an initial concentration of nitrous acid [HONO]0 = 8 ppbv.
2.7 Characterization of the Chamber State by Aerosol-Phase Reference Experiments
Reference experiments with well-known SOA precursors can be used to regularly check the overall status and conditions of a reaction chamber. Characteristic SOA formation behaviour can be established for one or more precursors and checked at regular intervals to confirm the reproducibility of the SOA production behaviour, understand changes to chamber conditions or identify potential problems and address them. In this context, the primary purpose of aerosol-phase reference experiments is to provide a method to monitor SOA production behaviour in a specific reaction chamber over time and to understand how it is influenced by any changes to the chamber infrastructure conditions (e.g., wall material, lights) or operating procedures. Reference aerosol-phase experiments could potentially also be used to compare SOA formation behaviour in different reaction chambers assuming that fundamental differences in chemical and physical factors can be accounted for.
It is not possible for one specific aerosol-phase reference experiment to fulfil this purpose for all existing chambers. This is because chambers vary in many ways including their size, temperature and relative humidity range, mode of operation (e.g., batch, continuous-stirred tank or flow reactors), light source (e.g., natural, artificial, dark), oxidant environment (e.g., OH, O3, NO3) and suitability for the use of seed particles. These and other potential factors need to be considered when deciding the required number of aerosol-phase reference experiments for each reaction chamber. The remaining part of this section will provide a brief overview of the main types of SOA formation experiments and recommend aerosol-phase reference experiments for chamber operators to select from as deemed suitable for their needs.
SOA formation occurs when one or more VOCs are oxidized to produce products of adequately low volatility to condense into the particulate phase. This can be a result of progressive oxidation steps in multiple reactions leading to multi-generation products of sufficiently low volatility, or fast auto-oxidation products such as HOMs that condense in the particulate phase quickly. This route is typically dominated by gas-to-particle conversion processes and is widely investigated in the chamber studies. Alternatively, SOA formation may occur in the condensed phase, when water soluble VOCs dissolve in droplets or particles and are subjected to aqueous phase oxidation leading to SOA products that remain dissolved. The latter route is typically investigated in bulk studies, but recent developments in analytical capabilities have enabled studies on a single particle scale with moist aerosol.
SOA formation experiments are conducted either in the presence or absence of pre-existing particles. These are types of experiments are referred to as nucleation or seeded experiments, respectively. Nucleation experiments often require a sufficiently high initial concentration of the SOA precursor(s) so that adequate amounts of low volatility oxidation products accumulate up to the threshold of homogeneous nucleation to be reached. This threshold is dependent on the parent VOC and the volatility distribution of its oxidation products. This type of experiment is useful for studies investigating properties of pure SOA particles, in addition to those focusing on nucleation rates.
Seeded experiments, on the other hand, are frequently used to avoid limitations of or lack of ability to measure aerosol size distribution or mass associated with particles of small sizes. In this type of experiments, particles of know composition are introduced into chambers with controlled amount and know size distribution. Ammonium sulphate, ammonium bisulphate and sodium chloride particles are some of the examples commonly used in this type of experiments, where their aqueous solutions are nebulized into chambers with or without the use of a classifier (e.g., DMA) producing mono- or poly-dispersed particles with known size or distribution often around 80–100 nm. This ensures that the produced SOA materials are condensed onto particles within the measurement capability of most deployed aerosol instrumentation. Seeded experiments are also often used with VOC precursors of relatively low reactivity or those which require multiple oxidation steps or aqueous phase reactions to generate SOA. The use of seed particles reduces the loss of condensable vapours to the chamber walls by offering a competing condensation sink and facilitates SOA production. The ability to clearly distinguish between and quantify the mass of seed particles and SOA material is possible using online mass spectrometry techniques. This can also offer a direct method of mass wall loss decay rate of seed particles.
The choice for which reference experiment should be conducted in a chamber at regular interval should take into consideration the factors discussed so far in addition to the specific nature of the chamber and the types of experiments it is used for. The oxidant environment of a chamber plays fundamental role in its SOA formation characteristics as it influences both of its gas as well as the particle-phase chemistry. Therefore, both dark and photo-oxidation experiments should be considered when deciding on which reference experiment to conduct. These experiments could be designed to investigate SOA formation from a specific oxidant (e.g., OH, O3) or to mimic atmospheric oxidation conditions such as day- or night-time chemistry, which involves more than one oxidant at a time (e.g., OH/O3 or O3/NO3 etc.). Metrics such as SOA mass production and VOC decay should be regularly checked in dark experiments. Additional metrics should be included in the case of photo-oxidation experiments such as ozone formation behaviour. Establishing and tracking the behaviour of such metrics on a regular basis would provide useful reference knowledge to understand the overall chamber behaviour in terms of particle-phase formation.
The choice of oxidant is an important part of determining the SOA formation conditions and it is determined by the objectives of the undertaken research. Oxidation by one or a combination of hydroxyl radical, ozone and nitrate radical account for the majority of SOA formation studies in most of the existing chambers.
2.7.1 Reference Photo-Oxidation Experiments
Chamber experiments aim to mimic the degree of atmospheric oxidant exposure, which is the integral of the oxidant concentration and the experiment duration time. The latter is often limited by the residence time of the reactor. The choice of light type and characteristics are key components of each chamber’s ability to achieve its oxidant concentration target. Most atmospheric chambers operate at OH concentration levels in the range of 106–107 molecules cm−3, which are representative of daytime concentrations in most ambient environments. Depending on residence time and vapour wall loss rates, chambers typically simulate SOA formation corresponding to an oxidant exposure from a few hours to about a day or two.
In the atmosphere, OH radicals are produced from the reaction of H2O with singlet oxygen atoms (O(1D)) generated from O3 photolysis at wavelengths <320 nm. In atmospheric chambers, the viability of this source is clearly dependent on the spectrum and wavelength-dependent intensities of the light source. This method of OH generation may be suitable in chambers equipped with light sources such as xenon-arc lamps. In this case, ozone is typically produced as a secondary product of the VOC and NOx chemistry.
Other methods of OH production are needed in the case of chambers using blacklights with a spectrum peak at round 350 nm due to the lack of sufficient photon intensity required for a sustainable ozone photolysis. These methods include the use of sources such as HONO as, for example, formed from the chamber wall (Sect. 2.6) or H2O2.
The photolysis of HONO generates one OH radical and one NO radical. This means that the use of HONO as an OH source cannot be considered for experiments where NOx-free or very low NOx conditions are needed. This is because NO is produced even if no additional NOx is added to the system.
A continuous source or multiple injections of HONO are typically required in this type of experiments in order to produce sufficiently high levels of OH radicals. This is due to the substantial photolysis rate of HONO.
Alternatively, H2O2 may be as a source of OH radicals. The photolysis of H2O2 produces two OH radicals, which subsequently react with H2O2, producing HO2 radicals:
In this system, it is possible to maintain a steady OH concentration over a long period of time due to a combination of relatively slow H2O2 photolysis rate and suppression of OH propagation by reaction with H2O2 itself. Unlike HONO, H2O2 provides a NOx-free source of OH.
Several VOCs could be used to conduct a reference photo-oxidation experiment using any of the oxidant sources mentioned so far. These include toluene, 1,3,5-trimethylbenzene (TMB) or α-pinene. However, in the case of chambers relying on the photolysis of O3 for the production of OH, α-pinene experiments characterise SOA formed by a combination of ozone and OH oxidation. It is recommended that ammonium sulphate seed should be used in these explements for reasons discussed earlier in this section.
2.7.2 Reference Ozonolysis Experiments
For dark chambers without natural or artificial light sources, a reference VOC ozonolysis experiment should be conducted on a regular basis to check the overall conditions of the chamber. This serves to either confirm the reproducibility of SOA formation characteristics or provide insights into any chamber changes affecting its SOA production behaviour. Ozone reacts with VOCs containing unsaturated carbon bonds to form SOA. The gas-phase reaction of ozone with VOCs proceeds by the addition of ozone cross the C=C double bond to form an energy-rich primary ozonide, followed by decomposition of the primary ozonide to produce an energized carbonyl oxide species, known as the Criegee intermediate, and an aldehyde or ketone product. This is followed by unimolecular decay of the Criegee intermediate producing OH radicals. The OH yield from ozonolysis varies depending on the VOC molecular structure. Chamber experiments investigating the role of ozone chemistry in SOA formation often use compounds to react with the resulting OH radicals. These are known as OH scavengers and include CO, cyclohexane and 2-butanol. The use of such compounds is not needed for the purpose the reference ozonolysis experiment discussed here. This is because OH yield from ozonolysis of VOC is part of simulating night-time chemistry in the reaction chambers.
The choice of VOC precursors for carrying out a reference ozonolysis experiment is broad and includes atmospherically relevant compounds containing carbon double bonds and known to form SOA. Of these, α-pinene is commonly used in most chambers and is an obvious candidate. It is recommended that a dark ozonolysis of α-pinene is carried out in the absence of an OH scavenger. The use of ammonium sulphate seed is also recommended as discussed earlier in this section.
References
Atkinson, R., Baulch, D.L., Cox, R.A., Crowley, J.N., Hampson, R.F., Hynes, R.G., Jenkin, M.E., Rossi, M.J., Troe, J.: Evaluated kinetic and photochemical data for atmospheric chemistry: Volume I—gas phase reactions of Ox, HOx, NOx and SOx species. Atmos. Chem. Phys. 6, 1461–1738 (2004). https://doi.org/10.5194/acp-4-1461-2004
Avnet Inc.: https://www.avnet.com/wps/portal/abacus/solutions/technologies/sensors/pressure-sensors/core-technologies/. Accessed 01 June 2020
Baratron capacitance manometers: https://www.mksinst.com/n/baratron-capacitance-manometers. Accessed 01 June 2020
Becker, K.H.: The European photoreactor “EUPHORE”. European Community, Final report (1996)
Bloss, C., Wagner, V., Bonzanini, A., Jenkin, M.E., Wirtz, K., Martin-Reviejo, M., Pilling, M.J.: Evaluation of detailed aromatic mechanisms (MCMv3 and MCMv3.1) against environmental chamber data. Atmos. Chem. Phys. 5, 623–639 (2005). https://doi.org/10.5194/acp-5-623-2005
Bohn, B., Zilken, H.: Model-aided radiometric determination of photolysis frequencies in a sunlit atmosphere simulation chamber. Atmos. Chem. Phys. 5, 191–206 (2005). https://doi.org/10.5194/acp-5-191-2005
Bohn, B., Rohrer, F., Brauers, T., Wahner, A.: Actinometric measurements of NO2 photolysis frequencies in the atmosphere simulation chamber SAPHIR. Atmos. Chem. Phys. 5, 493–503 (2005). https://doi.org/10.5194/acp-5-493-2005
Burkholder, J.B., Sander, S.P, Abbatt, J.P.D., Barker, J.R., Cappa, C., Crounse, J.D., Dibble, T.S., Hule, R.E., Kolb, C.E., Kurylo, M.J., Orkin, V.L., Percival, C.J., Wilmouth, D.M., Wine, P.H.: Chemical kinetics and photochemical data for use in atmospheric studies. Evaluation Numbber 19, Jet Proopulsion Laboratory, California Institute of Technology (2020)
Burrows, J.P., Dehn, A., Deters, B., Himmelmann, S., Richter, A., Voigt, S., Orphal, J.: Atmospheric remote-sensing reference data from GOME: Part 1. Temperature-dependent absorption cross-sections of NO2 in the 231–794 nm range. J. Quant. Spectrosc. Radiat. Transfer 60, 1025–1031 (1998). https://doi.org/10.1016/S0022-4073(97)00197-0
Carter, W.P.L., Cocker, D.R., III., Fitz, D.R., Malkina, I.L., Bumiller, K., Sauer, C.G., Pisano, J.T., Bufalino, C., Song, C.: A new environmental chamber for evaluation of gas-phase chemical mechanisms and secondary aerosol formation. Atmos. Environ. 39, 7768–7788 (2005)
Crump, J.G., Flagan, R.C., Seinfeld, J.H.: Particle wall loss rates in vessels. Aerosol Sci. Techn. 2, 303–309 (1983)
Curtis, A.R.: The FACSIMILE numerical integrator for stiff initial value problems. Computer science and systems division AERE Harwell, Harwell, Oxfordshire, Unclassified AERE—R 9352 (1979)
Deming, B., Pagonis, D., Liu, X., Day, D., Talukdar, R., Krechmer, J., de Gouw, J.A., Jimenez, J.L., Ziemann, P.J.: Measurements of delays of gas-phase compounds in a wide variety of tubing materials due to gas–wall interaction. Atmos. Meas. Tech. 12, 3453–3461 (2019). https://doi.org/10.5194/amt-12-3453-2019
Dunlea, E.J., Herndon, S.C., Nelson, D.D., Volkamer, R.M., San Martini, F., Sheehy, P.M., Zahniser, M.S., Shorter, J.H., Wormhoudt, J.C., Lamb, B.K., Allwine, E.J., Gaffney, J.S., Marley, N.A., Grutter, M., Marquez, C., Blanco, S., Cardenas, B., Retama, A., Ramos Villegas, C.R., Kolb, C.E., Molina, L.T., Molina, M.J.: Evaluation of nitrogen dioxide chemiluminescence monitors in a polluted urban environment. Atmos. Chem. Phys. ACP 7, 2691–2704 (2007)
Fahey, D.W., Gao, R.S., Möhler, O., Saathoff, H., Schiller, C., Ebert, V., Krämer, M., Peter, T., Amarouche, N., Avallone, L.M., Bauer, R., Bozóki, Z., Christensen, L.E., Davis, S.M., Durry, G., Dyroff, C., Herman, R.L., Hunsmann, S., Khaykin, S.M., Mackrodt, P., Meyer, J., Smith, J.B., Spelten, N., Troy, R.F., Vömel, H., Wagner, S., Wienhold, F.G.: The AquaVIT-1 intercomparison of atmospheric water vapor measurement techniques. Atmos. Meas. Tech. 7, 3177–3213 (2014). https://doi.org/10.5194/amt-7-3177-2014
Fry, J.L., Kiendler-Scharr, A., Rollins, A.W., Brauers, T., Brown, S.S., Dorn, H.P., Dubé, W.P., Fuchs, H., Mensah, A., Rohrer, F., Tillmann, R., Wahner, A., Wooldridge, P.J., Cohen, R.C.: SOA from limonene: role of NO3 in its generation and degradation. Atmos. Chem. Phys. 11, 3879–3894 (2011). https://doi.org/10.5194/acp-11-3879-2011
Grosjean, D.: Wall loss of gaseous pollutants in outdoor simulation chambers. Environ. Sci. Tech. 19, 1059–1065 (1985)
Hofzumahaus, A.: Measurement of photolysis frequencies in the atmosphere. In: Heard, D.E. (ed.) Analytical Techniques for Atmospheric Measurement. Blackwell Publishing, pp. 406–500 (2006)
Hofzumahaus, A., Kraus, A., Kylling, A., Zerefos, C.S.: Solar actinic radiation (280–420 nm) in the cloud-free troposphere between ground and 12 km altitude: measurements and model results. J. Geophys. Res. 107, PAU 6-1–PAU 6-11 (2002). https://doi.org/10.1029/2001JD900142
Hofzumahaus, A., Lefer, B.L., Monks, P.S., Hall, S.R., Kylling, A., Mayer, B., Shetter, R.E., Junkermann, W., Bais, A., Calvert, J.G., Cantrell, C.A., Madronich, S., Edwards, G.D., Kraus, A., Müller, M., Bohn, B., Schmitt, R., Johnston, P., McKenzie, R., Frost, G.J., Griffioen, E., Krol, M., Martin, T., Pfister, G., Röth, E.P., Ruggaber, A., Swartz, W.H., Lloyd, S.A., Van Weele, M.: Photolysis frequency of O3 to O(1D): Measurements and modeling during the International Photolysis Frequency Measurement and Modeling Intercomparison (IPMMI), J. Geophys. Res. 109 (2004). https://doi.org/10.1029/2003JD004333
Holmes, J.R., O’Brien, R.J., Crabtree, J.H., Hecht, T.A., Seinfeld, J.H.: Measurement of ultraviolet radiation intensity in photochemical smog studies. Environ. Sci. Tech. 7, 519–523 (1973). https://doi.org/10.1021/es60078a002
Hoyle, C.R., Fuchs, C., Järvinen, E., Saathoff, H., Dias, A., El Haddad, I., Gysel, M., Coburn, S.C., Tröstl, J., Bernhammer, A.K., Bianchi, F., Breitenlechner, M., Corbin, J.C., Craven, J., Donahue, N.M., Duplissy, J., Ehrhart, S., Frege, C., Gordon, H., Höppel, N., Heinritzi, M., Kristensen, T.B., Molteni, U., Nichman, L., Pinterich, T., Prévôt, A.S.H., Simon, M., Slowik, J.G., Steiner, G., Tomé, A., Vogel, A.L., Volkamer, R., Wagner, A.C., Wagner, R., Wexler, A.S., Williamson, C., Winkler, P.M., Yan, C., Amorim, A., Dommen, J., Curtius, J., Gallagher, M.W., Flagan, R.C., Hansel, A., Kirkby, J., Kulmala, M., Möhler, O., Stratmann, F., Worsnop, D.R., Baltensperger, U.: Aqueous phase oxidation of sulphur dioxide by ozone in cloud droplets. Atmos. Chem. Phys. 16, 1693–1712 (2016). https://doi.org/10.5194/acp-16-1693-2016
https://www.nist.gov/system/files/documents/2020/03/24/gmp-11-calibration-intervals-20190506.pdf, Accessed 16 Dec 2020
Huang, Y., Zhao, R., Charan, S.M., Kenseth, C.M., Zhang, X., Seinfeld, J.H.: Unified theory of vapor–wall mass transport in Teflon-walled environmental chambers. Environ. Sci. Tech. 52, 2134–2142 (2018). https://doi.org/10.1021/acs.est.7b05575
Hunter, J.F., Carrasquillo, A.J., Daumit, K.E., Kroll, J.H.: Secondary organic aerosol formation from acyclic, monocyclic, and polycyclic alkanes. Environ. Sci. Techol. 48, 10227–10234 (2014). https://doi.org/10.1021/es502674s
Hynes, R.G., Angove, D., Saunders, S., Haverd, V., Azzi, M.: Evaluation of two MCM v3.1 alkene mechanisms using indoor environmental chamber data. Atmos. Enviro. 39, 7251–7262 (2005). https://doi.org/10.1016/j.atmosenv.2005.09.005
Ibrahim, S.S., Bilger, R.W., Mudford, N.R.: Turbulence effects on chemical reactions in smog chamber flows. Atmos. Environ. 21, 2609–2621 (1987). https://doi.org/10.1016/0004-6981(87)90192-2
Karl, M., Brauers, T., Dorn, H.-P., Holland, F., Komenda, M., Poppe, D., Rohrer, F., Rupp, L., Schaub, A., Wahner, A.: Kinetic Study of the OH-isoprene and O3-isoprene reaction in the atmosphere simulation chamber, SAPHIR. Geophys. Res. Lett. 31, L05117 (2004)
Keller-Rudek, H., Moortgart, G.K., Sander, R., Sörensen, R.: The MPI-Mainz UV/VIS spectral atlas of gaseous molecules of atmospheric interest. Earth Syst. Sci. Data 5, 365–373 (2013). https://doi.org/10.5194/essd-5-365-2013
Kraus, A., Rohrer, F., Hofzumahaus, A.: Intercomparison of NO2 photolysis frequency measurements by actinic flux spectroradiometry and chemical actinometry during JCOM97. Geophys. Res. Lett. 27, 1115–1118 (2000). https://doi.org/10.1029/1999GL011163
Krechmer, J., Pagonis, D., Ziemann, P.J., Jimnez, J.L.: Quantification of gas-wall partitioning in Teflon environmental chambers using rapid bursts of low-volatility oxidized species generated in situ. Environ. Sci. Tech. 50, 5757–5765 (2016). https://doi.org/10.1021/acs.est.6b00606
Lai, A., Nazaroff, W.: Modeling indoor particle deposition from turbulent flow onto smooth surfaces. J. Aerosol Sci. 31, 463–476 (2000). https://doi.org/10.1016/s0021-8502(99)00536-4
Lake Shore Cryotronics, Inc.: https://www.lakeshore.com/resources/sensors (2019). Accessed 16 Dec 2020
Lamkaddam, H.: Study under simulated condition of the secondary organic aerosol from the photooxydation of n-dodecane: Impact of the physical-chemical processes, Université Paris-Est (2017)
Leighton, P.A.: Photochemistry of air pollution. Academic Press, New York (1961)
Leskinen, A., Yli-Pirilä, P., Kuuspalo, K., Sippula, O., Jalava, P., Hirvonen, M.R., Jokiniemi, J., Virtanen, A., Komppula, M., Lehtinen, K.E.J.: Characterization and testing of a new environmental chamber. Atmos. Meas. Tech. 8, 2267–2278 (2015). https://doi.org/10.5194/amt-8-2267-2015
Liu, X., Deming, B., Pagonis, D., Day, D.A., Palm, B.B., Talukdar, R., Roberts, J.M., Veres, P.R., Krechmer, J.E., Thornton, J.A., de Gouw, J.A., Ziemann, P.J., Jimenez, J.L.: Effects of gas–wall interactions on measurements of semivolatile compounds and small polar molecules. Atmos. Meas. Tech. 12, 3137–3149 (2019). https://doi.org/10.5194/amt-12-3137-2019
Loza, C.L., Chan, A.W.H., Galloway, M.M., Keutsch, F.N., Flagan, R.C., Seinfeld, J.H.: Characterization of vapor wall loss in laboratory chambers. Environ. Sci. Technol. 44, 5074–5078 (2010). https://doi.org/10.1021/es100727v
Massabò, D., Danelli, S.G., Brotto, P., Comite, A., Costa, C., Di Cesare, A., Doussin, J.F., Ferraro, F., Formenti, P., Gatta, E., Negretti, L., Oliva, M., Parodi, F., Vezzulli, L., Prati, P.: ChAMBRe: a new atmospheric simulation chamber for aerosol modelling and bio-aerosol research. Atmos. Meas. Tech. 11, 5885–5900 (2018). https://doi.org/10.5194/amt-11-5885-2018
Matsunaga, A., Ziemann, P.J.: Gas-Wall partitioning of organic compounds in a Teflon film chamber and potential effects on reaction product and aerosol yield measurements. Aerosol Sci. Technol. 44, 881–892 (2010). https://doi.org/10.1080/02786826.2010.501044
MBW Calibration Ltd.: www.mbw.ch (2020). Accessed 16 Dec 2020
Metcalf, A.R., Loza, C.L., Coggon, M.M., Craven, J.S., Jonsson, H.H., Flagan, R.C., Seinfeld, J.H.: Secondary organic aerosol coating formation and evaporation: chamber studies using black carbon seed aerosol and the single-particle soot photometer. Aerosol Sci. Technol. 47, 326–347 (2013). https://doi.org/10.1080/02786826.2012.750712
METEOMET: https://www.meteomet.org/ (2020). Accessed 16 Dec 2020
Metzger, A., Dommen, J., Gaeggeler, K., Duplissy, J., Prevot, A. S.H., Kleffmann, J., Elshorbany, Y., Wisthaler, A., Baltensperger, U.: Evaluation of 1,3,5 trimethylbenzene degradation in the detailed tropospheric chemistry mechanism, MCMv3.1, using environmental chamber data. Atmos. Chem. Phys. 8, 6453–6468 (2008). https://doi.org/10.5194/acp-8-6453-2008
Murphy, D.M., Koop, T.: Review of the vapour pressures of ice and supercooled water for atmospheric applications. Quart. J. r. Meteorol. Soc. 131, 1539–1565 (2005). https://doi.org/10.1256/qj.04.94
Nachbar, M., Duft, D., Leisner, T.: Volatility of amorphous solid water. J. Phys. Chem. B 122, 10044–10050 (2018a). https://doi.org/10.1021/acs.jpcb.8b06387
Nachbar, M., Duft, D., Leisner, T.: The vapor pressure over nano-crystalline ice. Atmos. Chem. Phys. 18, 3419–3431 (2018b). https://doi.org/10.5194/acp-18-3419-2018
Nannoolal, Y., Rarey, J., Ramjugernath, D.: Estimation of pure component properties: Part 3. Estimation of the vapor pressure of non-electrolyte organic compounds via group contributions and group interactions. Fluid Phase Equilib. 269, 117–133 (2008). https://doi.org/10.1016/j.fluid.2008.04.020
NIST 2019: See Good Measurement Practices GMP 11 & 13: https://www.nist.gov/system/files/documents/2019/06/21/gmp-13-ensuring-traceability-20190621.pdf
Pagonis, D., Krechmer, J.E., de Gouw, J., Jimenez, J.L., Ziemann, P.J.: Effects of gas–wall partitioning in Teflon tubing and instrumentation on time-resolved measurements of gas-phase organic compounds. Atmos. Meas. Tech. 10, 4687–4696 (2017). https://doi.org/10.5194/amt-10-4687-2017
Rohrer, F., Bohn, B., Brauers, T., Brüning, D., Johnen, F.J., Wahner, A., Kleffmann, J.: Characterisation of the photolytic HONO-source in the atmosphere simulation chamber SAPHIR. Atmos. Chem. Phys. 5, 2189–2201 (2005). https://doi.org/10.5194/acp-5-2189-2005
Saathoff, H., Möhler, O., Schurath, U., Kamm, S., Dippel, B., Mihelcic, D.: The AIDA soot aerosol characterisation campaign 1999. J. Aerosol Sci. 34, 1277–1296 (2003). https://doi.org/10.1016/s0021-8502(03)00363-x
Schütze, M., Stratmann, F.: Numerical simulation of cloud droplet formation in a tank. Comput. Geosci. 34, 1034–1043 (2008). https://doi.org/10.1016/j.cageo.2007.06.013
Shetter, R.E., Junkermann, W., Swartz, W.H., Frost, G.J., Crawford, J.H., Lefer, B.L., Barrick, J.D., Hall, S.R., Hofzumahaus, A., Bais, A., Calvert, J.G., Cantrell, C.A., Madronich, S., Müller, M., Kraus, A., Monks, P.S., Edwards, G.D., McKenzie, R., Johnston, P., Schmitt, R., Griffioen, E., Krol, M., Kylling, A., Dickerson, R.R., Lloyd, S.A., Martin, T., Gardiner, B., Mayer, B., Pfister, G., Röth, E.P., Koepke, P., Ruggaber, A., Schwander, H., van Weele, M.: Photolysis frequency of NO2: measurement and modeling during the International Photolysis Frequency Measurement and Modeling Intercomparison (IPMMI). J. Geophys. Res. 108 (2003). https://doi.org/10.1029/2002JD002932
Shetter, R.E., Müller, M.: Photolysis frequency measurements using actinic flux spectroradiometry during the PEM-Tropics mission: instrumentation description and some results. J. Geophys. Res. 104, 5647–5661 (1999). https://doi.org/10.1029/98JD01381
Tuesday, C. S.: The atmospheric photooxidation of trans-2-butene and nitric oxide. In: Chemical Reactions in the Lower and Upper Atmosphere, pp. 1–49. Interscience, New York, S.Y. (1961)
Valorso, R., Aumont, B., Camredon, M., Raventos-Duran, T., Mouchel-Vallon, C., Ng, N.L., Seinfeld, J.H., Lee-Taylor, J., Madronich, S.: Explicit modelling of SOA formation from α-pinene photooxidation: sensitivity to vapour pressure estimation. Atmos. Chem. Phys. 11, 6895–6910 (2011). https://doi.org/10.5194/acp-11-6895-2011
Villena, G., Bejan, I., Kurtenbach, R., Wiesen, P., Kleffmann, J.: Interferences of commercial NO2 instruments in the urban atmosphere and in a smog chamber. Atmos. Chem. Tech. 5, 149–159 (2012). https://doi.org/10.5194/amt-5-149-2012
Voigtländer, J., Duplissy, J., Rondo, L., Kürten, A., Stratmann, F.: Numerical simulations of mixing conditions and aerosol dynamics in the CERN CLOUD chamber. Atmos. Chem. Phys. 12, 2205–2214 (2012). https://doi.org/10.5194/acp-12-2205-2012
Wang, J., Doussin, J.F., Perrier, S., Perraudin, E., Katrib, Y., Pangui, E., Picquet-Varrault, B.: Design of a new multi-phase experimental simulation chamber for atmospheric photosmog, aerosol and cloud chemistry research. Atmos. Meas. Tech. 4, 2465–2494 (2011). https://doi.org/10.5194/amt-4-2465-2011
Wang, X., Liu, T., Bernard, F., Ding, X., Wen, S., Zhang, Y., Zhang, Z., He, Q., Lü, S., Chen, J., Saunders, S., Yu, J.: Design and characterization of a smog chamber for studying gas-phase chemical mechanisms and aerosol formation. Atmos. Meas. Tech. 7, 301–313 (2014). https://doi.org/10.5194/amt-7-301-2014
Wang, N., Jorga, S.D., Pierce, J.R., Donahue, N.M., Pandis, S.N.: Particle wall-loss correction methods in smog chamber experiments. Atmos. Meas. Tech. 11, 6577–6588 (2018a). https://doi.org/10.5194/amt-11-6577-2018
Wang, N., Kostenidou, E., Donahue, N.M., Pandis, S.N.: Multi-generation chemical aging of α-pinene ozonolysis products by reactions with OH. Atmos. Chem. Phys. 18, 3589–3601 (2018b). https://doi.org/10.5194/acp-18-3589-2018
Wilck, M., Stratmann, F., Whitby, E.: A fine particle model for fluent: description and application. In: Sixth International Aerosol Conference, Taipei, Taiwan, pp. 1269–1270 (2002)
WMO: World meteorological organization, guide to instruments and methods of observation, WMO-No. 8, Geneva (2018). ISBN: 978-92-63-10008-5
Yeh, G.K., Ziemann, P.J.: Identification and yields of 1,4-hydroxynitrates formed from the reactions of C8–C16 n-alkanes with OH radicals in the presence of NOx. J. Phys. Chem. A 118, 8797–8806 (2014a). https://doi.org/10.1021/jp505870d
Yeh, G.K., Ziemann, P.J.: Alkyl nitrate formation from the reactions of C8–C14 n-alkanes with OH radicals in the presence of NOx: measured yields with essential corrections for gas–wall partitioning. J. Phys. Chem. A 118, 8147–8157 (2014b). https://doi.org/10.1021/jp500631v
Yeh, G.K., Ziemann, P.J.: Gas-wall partitioning of oxygenated organic compounds: measurements, structure–activity relationships, and correlation with gas chromatographic retention factor. Aerosol. Sci. Technol. 49, 727–738 (2015). https://doi.org/10.1080/02786826.2015.1068427
Zador, J., Turányi, T., Wirtz, K., Pilling, M.J.: Measurement and investigation of chamber radical sources in the European Photoreactor (EUPHORE). J. Atmos. Chem. 55, 147–166 (2006). https://doi.org/10.1007/s10874-006-9033-y
Zhang, X., Cappa, C.D., Jathar, S.H., McVay, R.C., Ensberg, J.J., Kleemann, M.J., Seinfeld, J.H.: Influence of vapor wall loss in laboratory chambers on yields on secondary organic yields. Proc. Nat. Acad. Sci. 11, 5802–5807 (2014). https://doi.org/10.1073/pnas.1404727111
Zhang, X., Schwantes, R.H., McVay, R.C., Lignell, H., Coggon, M.M., Flagan, R.C., Seinfeld, J.H.: Vapor wall deposition in Teflon chambers. Atmos. Chem. Phys. 15, 4197–4214 (2015). https://doi.org/10.5194/acp-15-4197-2015
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Open Access This chapter is licensed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license and indicate if changes were made.
The images or other third party material in this chapter are included in the chapter's Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the chapter's Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.
Copyright information
© 2023 The Author(s)
About this chapter

Cite this chapter
Alfarra, R. et al. (2023). Physical and Chemical Characterization of the Chamber. In: Doussin, JF., Fuchs, H., Kiendler-Scharr, A., Seakins, P., Wenger, J. (eds) A Practical Guide to Atmospheric Simulation Chambers. Springer, Cham. https://doi.org/10.1007/978-3-031-22277-1_2
Download citation
DOI: https://doi.org/10.1007/978-3-031-22277-1_2
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-031-22276-4
Online ISBN: 978-3-031-22277-1
eBook Packages: Chemistry and Materials ScienceChemistry and Material Science (R0)