
Abstract
The knowledge of an inverse relationship between plasma high-density lipoprotein cholesterol (HDL-C) concentrations and rates of cardiovascular disease has led to the concept that increasing plasma HDL-C levels would be protective against cardiovascular events. Therapeutic interventions presently available to correct the plasma lipid profile have not been designed to specifically act on HDL, but have modest to moderate effects on plasma HDL-C concentrations. Statins, the first-line lipid-lowering drug therapy in primary and secondary cardiovascular prevention, have quite modest effects on plasma HDL-C concentrations (2–10 %). Fibrates, primarily used to reduce plasma triglyceride levels, also moderately increase HDL-C levels (5–15 %). Niacin is the most potent available drug in increasing HDL-C levels (up to 30 %), but its use is limited by side effects, especially flushing.
The present chapter reviews the effects of established hypolipidemic drugs (statins, fibrates, and niacin) on plasma HDL-C levels and HDL subclass distribution, and on HDL functions, including cholesterol efflux capacity, endothelial protection, and antioxidant properties.
You have full access to this open access chapter, Download chapter PDF
Similar content being viewed by others

Keywords
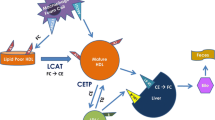
1 Effects on Plasma HDL-C Concentration
1.1 Statins
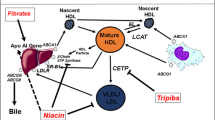
Statins are inhibitors of the hydroxymethylglutaryl coenzyme A (HMG-CoA) reductase enzyme, a key enzyme in cholesterol synthesis. In addition to effectively reducing low-density lipoprotein cholesterol (LDL-C), all statins modestly increase HDL-C levels across the range of doses used, with important differences between different statins (Jones et al. 2003). The increase in HDL-C is dose dependent with some statins, e.g., simvastatin and rosuvastatin, but not with others. In the case of atorvastatin, the highest increase in HDL-C levels is observed at the lowest dose used. Statin-induced changes in HDL-C are normally paralleled by an increase in apoA-I levels (Barter et al. 2010). Interestingly, variations in LDL-C and in HDL-C levels are independent from each other (Barter et al. 2010). The mechanism by which statins increase plasma HDL-C and apoA-I levels remains unknown, and it is apparently unrelated to the inhibition of the HMG-CoA reductase enzyme. The ability of statins to upregulate hepatic ABCA1 gene expression (Tamehiro et al. 2007), as well as the described statin-induced inhibition of cholesteryl ester transfer protein (CETP) (Guerin et al. 2000; Kassai et al. 2007) can contribute to explain the effect of statin on HDL-C levels.
1.2 Fibrates
Fibrates (fibric acid derivates), synthetic ligands for PPAR-α, significantly reduce plasma triglyceride levels (30–50 %) and moderately increase HDL-C concentrations (5–15 %), with no major differences among available molecules (Khoury and Goldberg 2011). Activation of PPAR-α leads to β-oxidation of free fatty acids in the liver reducing VLDL synthesis and secretion. Furthermore, the expression of the gene coding for lipoprotein lipase is increased and apolipoprotein C-III expression in the liver is decreased, resulting in increased hydrolysis of triglyceride-rich lipoproteins. HDL-C raises results from the increased expression of apoA-II and, at a much lesser extent, apoA-I (Yetukuri et al. 2011), as well as to the enhanced cholesterol efflux via the induction of ABCA1 in the liver (Berger et al. 2005). In addition, animal studies have shown that fenofibrate reduces CETP hepatic expression (van der Hoogt et al. 2007).
1.3 Niacin
Niacin (nicotinic acid), a GPR109a agonist used clinically for more than 50 years to lower cholesterol and triglycerides, increases HDL-C levels up to 30 % (Vega and Grundy 1994). All the different niacin formulations (regular and extended release, ER) are equally effective in increasing HDL-C levels, whereas acipimox (a nicotinic acid analog) only induces a 7 % increase (Birjmohun et al. 2005). Niacin side-effect profile, including flushing, gastrointestinal upset, and liver function testing abnormalities, has, however, limited its use, especially in warmer Mediterranean countries (Birjmohun et al. 2005). Coadministration of the prostaglandin receptor antagonist laropiprant significantly reduces but does not eliminate niacin-induced skin symptoms (Yadav et al. 2012). It is not quite clear how niacin leads to an increase in HDL-C levels, and various theories have been proposed (Soudijn et al. 2007). These include the ability of niacin to inhibit CETP (Hernandez et al. 2007), to induce ABCA1 expression (Rubic et al. 2004), and to reduce HDL catabolism (Bodor and Offermanns 2008).
2 Effects on HDL Subclass Distribution
Plasma HDL is highly heterogeneous and can be separated in several subclasses according to density, size, shape, and lipid and protein composition (Calabresi et al. 2010). According to density, two HDL subclasses can be identified, the less dense HDL2 and the more dense HDL3 (De Lalla and Gofman 1954). HDL2 and HDL3 could be further fractionated in five distinct subclasses on the basis of particle size (from large to small: HDL2b, HDL2a, HDL3a, HDL3b, and HDL3c) (Nichols et al. 1986). According to surface charge, HDL could be separated into α-HDL and preβ-HDL. The separation by charge and size, using a two-dimensional (2-D) gel electrophoresis, allow the identification of up to 12 distinct apoA-I-containing HDL subclasses (Asztalos and Schaefer 2003). Finally, according to the protein component, HDL can be separated into particles containing only apoA-I (LpA-I) or containing also apoA-II (LpA-I:A-II).
2.1 Statins
The reported effects of statins on HDL subclass distribution are not consistent, likely depending on the technique used for the analysis. Some studies showed no effect of statins on HDL subclasses (Alaupovic et al. 1994; Bard et al. 1989; Franceschini et al. 2007; Tomas et al. 2000), and on HDL particle size distribution (Franceschini et al. 1989; Homma et al. 1995). On the contrary, studies evaluating HDL subclasses by 2-D gel electrophoresis showed that all statins, although with different potency, increase the levels of the large HDL particles and decrease the levels of small particles (Asztalos et al. 2002a, b, 2007).
2.2 Fibrates
A number of studies have shown that treatment with fibrates causes a shift of HDL from large to small particles in low HDL (Franceschini et al. 2007; Otvos et al. 2006), hypertriglyceridemic (Miida et al. 2000; Sasaki et al. 2002), and hypercholesterolemic (Franceschini et al. 1989) patients. These effects could be explained by the fibrate-induced increase in hepatic lipase activity (Miida et al. 2000; Patsch et al. 1984). In addition, bezafibrate has been shown to increase small discoidal preβ1-HDL particles in hypertriglyceridemic patients (Miida et al. 2000). The analysis of HDL subclasses by 2-D electrophoresis in the large cohort of the VA-HIT trial confirmed that gemfibrozil treatment is associated with a decrease in large (α-1 and α-2) HDL and with an increase in the small, lipid-poor HDL particles (Asztalos et al. 2008).
2.3 Niacin
Sheperd et al. were the first to demonstrate in the late 1970s that niacin (3 g/day) in healthy volunteers significantly increases the large HDL2 particles, associated with a parallel reduction of the small HDL3 (Shepherd et al. 1979). Several studies have confirmed this observation in patients with different types of dyslipidemia treated with 2, 3, or 4 g/day of regular or ER niacin (Johansson and Carlson 1990; Knopp et al. 1998; Morgan et al. 2003; Wahlberg et al. 1990) and in patients with coronary artery disease (Kuvin et al. 2006). A very recent study showed that also 1 g/day of ER niacin increases large HDL particles in dyslipidemic patients, with a concomitant significant increase in mean HDL size (Franceschini et al. 2013). The analysis of HDL subclasses by 2-D gel electrophoresis recently confirmed the observation that niacin promotes the maturation of HDL into large particles (Lamon-Fava et al. 2008), likely explained by a reduction in CETP activity. In addition, Sakai et al. reported that niacin selectively increases LpA-I particles in patients with low HDL-C, due to a prolongation of apoA-I residence time (Sakai et al. 2001).
3 Effects on Cholesterol Efflux Capacity
The most relevant antiatherogenic function of HDL is the ability to promote reverse cholesterol transport, the physiological process by which excess cholesterol is removed from peripheral tissues and transported to the liver for excretion (Cuchel and Rader 2006). The first and limiting step of this process is the efflux of cellular cholesterol from lipid-laden macrophages of the arterial wall (Lewis and Rader 2005). Multiple mechanisms for efflux of cholesterol exist in macrophages (Adorni et al. 2007). The release of free cholesterol via passive diffusion occurs in all cell types and is accelerated when the scavenger receptor class B type I (SR-BI) is present on the cell plasma membrane. Both passive diffusion and SR-BI-mediated efflux occur to phospholipid-containing acceptors (i.e., HDL and lipidated apolipoproteins). ATP-binding cassette A1 (ABCA1) belongs to the ATP-binding cassette transporter family and promotes unidirectional efflux of membrane cholesterol and phospholipids to lipid-poor apolipoproteins. Macrophages may also release cholesterol via the ABCG1 pathway, another ABC transporter that mediates net mass efflux of cellular cholesterol to mature HDL (Favari et al. 2009). HDL cholesterol efflux capacity (CEC) from macrophages can be measured in vitro and provides a reliable measure of HDL functionality (Khera et al. 2011).
3.1 Statins
The impact of statin therapy on HDL ability to promote cholesterol efflux has been investigated in a number of studies. Khera et al. measured HDL CEC in patients treated with 10 and 80 mg atorvastatin or 40 mg pravastatin for 16 weeks and reported no effects of statin therapy on HDL CEC from macrophages expressing all the known cholesterol efflux pathways (ABCA1, ABCG1, SR-BI, and passive diffusion) (Khera et al. 2011). As opposed to these results, Guerin et al. have shown that atorvastatin therapy for 12 weeks (10 and 40 mg daily) could increase SR-BI-dependent CEC of plasma from patients with type IIB hyperlipidemia, in a dose-dependent manner (Guerin et al. 2002). The effect of simvastatin on HDL’s ability to promote cholesterol efflux was assessed in a randomized, double-blind, parallel group trial carried out in dyslipidemic patients with low HDL-C levels receiving either fenofibrate (160 mg/day) or simvastatin (40 mg/day) for 8 weeks (Franceschini et al. 2007). Simvastatin led to a small, significant increase in the capacity of plasma to promote SR-BI-mediated cholesterol efflux, likely related to the slight increase in HDL-C plasma levels that occurred after simvastatin treatment. The latter results are in contrast with data obtained by de Vries et al. who demonstrated that HDL from moderately hypercholesterolemic type 1 diabetic patients after 10, 20, and 40 mg simvastatin treatment does not increase their ability to promote cellular cholesterol efflux via SR-BI (De Vries et al. 2005). The effect of simvastatin on HDL functionality was further investigated recently in a small number of diabetic patients treated with simvastatin 40 mg/day or bezafibrate 400 mg/day, alone or in combination. CEC of apoB-depleted plasma samples from cholesterol-loaded macrophages, a cellular model which expresses simultaneously all the pathways of known relevance in cholesterol efflux, was increased by 14 % in response to simvastatin compared to placebo (Triolo et al. 2013a). Sviridov et al. showed that treatment with 40 mg/day rosuvastatin in overweight subjects with defined metabolic syndrome did not significantly modify HDL capacity to promote cholesterol efflux from LXR-activated human macrophages (Sviridov et al. 2008). Finally, a recent study showed a moderate ability of pitavastatin to enhance HDL CEC from macrophage foam cells in dyslipidemic subjects (Miyamoto-Sasaki et al. 2013). Overall, the impact of statin therapy on HDL CEC does not appear to be substantial, in agreement with the modest effect of statins on HDL-C concentration and on HDL subclass distribution.
3.2 Fibrates
The effect of fibrates on HDL cholesterol efflux capacity was assessed by Guerin et al. showing a significant ciprofibrate-mediated 13 % elevation in the capacity of plasma from type IIB hyperlipidemic subjects to mediate cholesterol efflux from SR-BI-expressing Fu5AH hepatoma cells (Guerin et al. 2003). The study by Franceschini et al., comparing fenofibrate and simvastatin in dyslipidemic patients (Franceschini et al. 2007), provided evidence that fenofibrate increases the plasma capacity to promote ABCA1-mediated efflux with no changes in SR-BI efflux. These observed results might be explained by a shift of HDL from large to small particles, the preferential cholesterol acceptors for ABCA1 (Adorni et al. 2007). This result was not confirmed in a more recent paper by the same group (Franceschini et al. 2013), likely because of the different HDL particle profile in the two investigated populations of patients. In the first study (Guerin et al. 2002), patients displayed a greater content of large HDL particles, whereas in patients with dyslipidemia of the last study (Franceschini et al. 2013), the small, ABCA1-interacting HDL was the predominant fraction, and no further increase in this HDL population was observed after fenofibrate therapy, consistent with the lack of changes in ABCA1-mediated CEC. Whether fenofibrate therapy modulate the ability of plasma or HDL to facilitate cholesterol efflux from macrophages has been investigated also in a subset of the FIELD study showing that cholesterol efflux values from macrophage foam cells to HDL and plasma in the fenofibrate group (200 mg/day for 5 years) were comparable to those of the placebo group (Maranghi et al. 2011). Finally, in a small study carried out in diabetic patients, it was shown that bezafibrate increases CEC of apoB-depleted plasma samples from cholesterol-loaded THP-1 macrophages (Triolo et al. 2013a). Overall, studies on the ability of fibrates to modulate HDL cholesterol efflux capacity seem not consistent, likely due to the different cell models used in the different studies and also to the different types of patients.
3.3 Niacin
The impact of niacin on HDL cholesterol efflux capacity was investigated in a placebo-controlled study carried out by Khera et al. who measured HDL CEC before and after ER niacin therapy in a small number of patients with carotid atherosclerosis (Khera et al. 2013). The study showed that niacin treatment led to favorable changes in patients’ lipid profiles without significantly improving CEC of HDL from macrophages. In the study by Franceschini et al., the effect of ER niacin therapy on plasma CEC was evaluated in a population of dyslipidemic subjects (Franceschini et al. 2013). The tendency of ER niacin in enhancing passive diffusion, SR-BI-, and ABCG1-mediated CEC observed in this study is in line with the results of a previous investigation, which demonstrated that niacin increased both HDL-C levels and CEC from human macrophages (Yvan-Charvet et al. 2010). In this study the increase in CEC achieved statistical significance, possibly because of a greater increase in plasma HDL-C levels compared with that observed in study by Franceschini et al., because passive diffusion, SR-BI-, and partially ABCG1-mediated CECs are known to be dependent on HDL-C concentrations. Consistently, a previous investigation by Morgan et al. reported that ER niacin therapy in patients with a history of primary dyslipoproteinemia had a beneficial effect on SR-BI-mediated efflux and that it was related to the change in level of HDL-C (Morgan et al. 2007). Another study evaluated specific cholesterol efflux pathway-mediated CECs (ABCA1, SR-BI, and ABCG1) of serum from men with HDL deficiency before and after treatment with niacin, but no changes were reported in all the considered efflux pathways (Alrasadi et al. 2008). Taken together, available data on the effect of niacin on the HDL-mediated ability to promote cell cholesterol efflux show modest effects on the pathways involving large HDL, in agreement with the changes observed on HDL particles.
4 Effect on HDL Ability to Preserve Endothelial Cell Homeostasis
Several in vitro and in vivo evidences indicate that the anti-atherosclerotic activity of HDL is also due to their ability to preserve endothelial cell homeostasis (Calabresi et al. 2003). HDL is able to promote vasorelaxation, to inhibit the production of cell adhesion and proinflammatory molecules, and to favor the integrity and repair of the endothelial layer, thus preventing and correcting the main features of endothelial dysfunction typical of the atherosclerotic process. HDL can modulate vascular tone by promoting the release of vasoactive molecules like nitric oxide (NO) and prostacyclin (PGI2) which also exert powerful antithrombotic effects. HDL is able to increase the protein abundance and to induce the activation of endothelial nitric oxide synthase (eNOS) (Kuvin et al. 2002; Yuhanna et al. 2001). This latter effect is driven by the interaction of HDL with the scavenger receptor SR-BI and by the subsequent activation of the PI3K/Akt signaling pathway leading to eNOS phosphorylation; in addition, the sphingolipid sphingosine-1-phosphate (S1P) carried by HDL can also mediate eNOS activation by binding with its receptor S1P3 (Nofer et al. 2004). PGI2 is a metabolite of arachidonate and HDL was shown to increase its production by different mechanisms. HDL can provide endothelial cells with the substrate arachidonate or promote its release from cellular phospholipids by activating calcium-sensitive membrane-bound phospholipases (Calabresi et al. 2003). Another feature of endothelial dysfunction is the increased production of molecules favoring blood cell adhesion to the endothelial layer and their consequent extravasation and activation. Studies on endothelial cells have shown that HDL are able to inhibit the cytokine-induced expression of cell adhesion molecules (Calabresi et al. 1997; Cockerill et al. 1995), but the precise mechanism responsible for this effect has not been fully elucidated to date (Barter et al. 2002). Different studies have shown an HDL-mediated inhibition of sphingosine kinase activity and NF-kB nuclear translocation and an increased expression of heme-oxygenase 1 mediated by SR-BI (McGrath et al. 2009; Xia et al. 1999). HDL was also shown to inhibit the production of proinflammatory cytokines, as interleukin-6 and chemokines (Gomaraschi et al. 2005). Finally, HDL was shown to promote endothelial cell migration and proliferation, to inhibit cell apoptosis through different mechanisms (Mineo and Shaul 2012), and to promote endothelial progenitor cells (EPC) differentiation and survival (Mineo and Shaul 2012; Petoumenos et al. 2009).
In certain pathological conditions HDL may lose their ability to protect the endothelium and even become dysfunctional. In particular, conditions associated with both acute and chronic inflammatory processes, such as ischemic vascular events, infections, type 2 diabetes (T2DM), obesity, metabolic syndrome, autoimmune disorders, and chronic kidney disease, have been associated with HDL displaying impaired endothelial protective activities (Riwanto and Landmesser 2013). Indeed, HDL isolated from these patients failed to promote NO production, to exert antioxidant and anti-inflammatory effects, and to favor endothelial integrity in vitro (Riwanto and Landmesser 2013).
The measurement of vasodilation in response to changes in forearm blood flow (flow-mediated vasodilation, FMD) is currently considered the best method to evaluate endothelial function in vivo (Deanfield et al. 2007) and is commonly used to assess in vivo vascular condition and to evaluate the effects of pharmacological treatments. In addition, plasma levels of the soluble forms of adhesion molecules and of proinflammatory cytokines can be used as in vivo markers of endothelial inflammatory activation. Recently, circulating EPC number and migratory capacity emerged as novel biomarkers of endothelial repair (Petoumenos et al. 2009).
4.1 Statins
Several studies have shown that statins are able to improve FMD, to reduce the plasma levels of inflammatory markers, and to increase EPC number in dyslipidemic subjects (Antonopoulos et al. 2012; Liu et al. 2012; Reriani et al. 2011). These effects are likely not related to statin-induced changes in HDL. In vitro experiments have indeed shown that statin-mediated improvement of endothelial function is due to a direct effect of the drug on endothelial cells. Incubation of endothelial cells with different statins promotes a significant increase expression of SR-BI and S1P receptors and of eNOS, with a consequent increased production of NO after incubation with HDL (Igarashi et al. 2007; Kimura et al. 2008). A recent study performed on subjects with isolated low HDL-C showed that FMD was significantly improved after 4 weeks treatment with pravastatin, and multiple regression analysis revealed that changes in HDL-C and EPC number were independent predictors of FMD increase (Higashi et al. 2010). Despite the limitations due to the small number of analyzed subjects, this latter study suggests a potential role of statins on HDL capacity to maintain endothelial homeostasis.
4.2 Fibrates
Different fibrates have been tested for their ability to modulate endothelial function in dyslipidemic patients. Fenofibrate was shown to improve FMD and to decrease inflammatory biomarkers, as C-reactive protein (CRP), tumor necrosis factor alpha (TNFα), interleukin-1, and soluble CD40 ligand, in patients with combined hyperlipidemia (Malik et al. 2001; Wang et al. 2003). FMD improvement was inversely related to baseline HDL-C levels and indeed fenofibrate significantly increased FMD only in the subgroup with low HDL-C and not in subjects with normal HDL-C, in which a statin was instead effective (Wang et al. 2003). Fenofibrate, alone or in combination with a statin, was also effective in modulating FMD and decreasing plasma levels of CRP and TNFα in hypertriglyceridemic subjects (Koh et al. 2004, 2005), and a significant decrease of plasma levels of sCAMs was observed in subjects with low HDL-C levels (Calabresi et al. 2002); in this latter study, the decrease of plasma sCAMs was inversely related to the fenofibrate-induced increase of HDL-C, while no correlation was found with triglyceride reduction (Calabresi et al. 2002).
Recently, particular attention has been devoted to the effects of fibrates in subjects with T2DM. Four studies consistently demonstrated that different fibrates, i.e., ciprofibrate, gemfibrozil, fenofibrate, and bezafibrate, were all able to significantly improve FMD in T2DM, in a way that was not dependent on changes in lipid parameters, including HDL-C (Avogaro et al. 2001; Evans et al. 2000; Ghani et al. 2013; Triolo et al. 2013b). Interestingly, after 12 weeks of treatment with ciprofibrate, T2DM subjects displayed improved FMD measured in fasting conditions and in the postprandial phase (Evans et al. 2000). Ciprofibrate treatment also markedly modified lipid metabolism: the fasting triglyceride content of all lipoproteins was reduced and the increase during the postprandial phase blunted; the improvement of fasting FMD was correlated with the decreased triglyceride content in VLDL and HDL (but not with changes of HDL-C), while in the postprandial phase changes of FMD were correlated with the reduced lipoprotein TG content (Evans et al. 2000).
4.3 Niacin
The effect of niacin treatment on endothelial function has been assessed in a variety of clinical conditions, including established coronary artery disease, T2DM, metabolic syndrome, and low HDL-C. Overall, when niacin was used as monotherapy, the studies showed an increase in FMD and a reduction in inflammatory biomarkers but failed in demonstrating a correlation with the niacin-mediated increase of HDL-C levels. In 2002, Kuvin et al. demonstrated that a 3-month treatment with niacin improved FMD in patients with CAD and low HDL-C and that this improvement could be related to the HDL ability to increase eNOS protein abundance in vitro (Kuvin et al. 2002). Some years later, the same authors also showed that in statin treated patients with established CAD, the addition of niacin caused a reduction of the plasma levels of inflammatory biomarkers as CRP and lipoprotein-associated phospholipase A2 (Kuvin et al. 2006); unfortunately, likely due to the small number of participants, correlation analyses between changes of endothelial function parameters and the increase of HDL-C or other lipid variables were not performed (Kuvin et al. 2006). Later on, the ability of niacin to improve FMD and reduce inflammatory biomarkers as CRP was confirmed in subjects with isolated low HDL-C (Benjo et al. 2006), with the metabolic syndrome (Thoenes et al. 2007), and in patients after myocardial infarction (Bregar et al. 2013); the relationship between increase of HDL-C and changes of FMD was analyzed only in the latter case, and no correlation was found with any lipid parameter. The absence of correlation between niacin-mediated changes of HDL-C and endothelial function could be explained by the direct antioxidant, anti-inflammatory, and anti-adhesive activities of niacin which are independent from its role on lipid metabolism (Yadav et al. 2012). However, a recent study clearly showed that HDL isolated from subjects with T2DM after 3 months of treatment with niacin displayed an improved ability to stimulate NO production, to reduce reactive oxygen species, and to promote EPC-mediated endothelial repair in vitro when compared to HDL isolated at baseline (Sorrentino et al. 2010), further strengthening the concept that plasma HDL-C and its change may not be a reliable indicator of HDL function. Recent studies analyzed the effect of niacin treatment in patients with established CAD on top of the existing statin therapy; two of them failed to detect any significant improvement of FMD, while in one case FMD was significantly increased only in the subgroup of patients with a low baseline HDL-C level (Lee et al. 2009; Philpott et al. 2013; Warnholtz et al. 2009).
5 Effect on HDL Antioxidant Properties
ApoA-I plays a major role in HDL-mediated protection from oxidative damage (Rosenson et al. 2013). Many studies have indicated that the antioxidant function of HDL also depends upon such enzymes as lipoprotein-associated phospholipase A2 (Lp-PLA2), paraoxonase 1 (PON1), lecithin-cholesterol acyltransferase (LCAT), and glutathione selenoperoxidase (GSPx) (Florentin et al. 2008; Otocka-Kmiecik et al. 2012; Podrez 2010). GSPx can reduce lipoprotein lipid hydroperoxides (LOOHs) to the corresponding hydroxides and thereby detoxify them (Rosenson et al. 2013). Besides apoA-I, other apolipoproteins such as apoE, apoJ, apoA-II, and apoA-IV also display antioxidant properties but to a less degree than apoA-I (Otocka-Kmiecik et al. 2012). An antioxidant mechanism of HDL may also result from their ability to accept phospholipid-containing hydroperoxides (PLOOHs) and other lipid peroxidation products from oxidized LDL (Stremler et al. 1991) and their subsequent reduction by redox-active methionine residues of apoA-I, with the formation of redox-inactive phospholipid hydroxides (Kontush and Chapman 2010). In addition, HDL functions as an antioxidant enzyme by hydrolyzing oxidized phospholipids, such as F2-isoprostanes, formed during the oxidative modification of LDL. Antioxidant properties differ for the different HDL subclasses (Kontush and Chapman 2010). Activities of HDL-associated enzymes—LCAT, PON1, and platelet-activating factor acetyl hydrolase (PAF-AH)—are elevated in small, dense HDL3c (Davidson et al. 2009), which thus seem to have a potent role in protecting LDL from oxidation (Podrez 2010). It is worth emphasizing that small, dense HDL3 particles are also more resistant to oxidative modification compared with large, light HDL2 (Shuhei et al. 2010).
5.1 Statins
The role of statins in improving plasma oxidative status has been reported in a number of studies, although this effect was not always related to drug-induced changes in HDL. Different statins have been shown to significantly enhance the antioxidant activity of PON1. Atorvastatin has a positive impact on plasma total antioxidant status and PON1 activity by reducing plasma susceptibility to lipid peroxidation induced by free radicals (Fuhrman et al. 2002; Harangi et al. 2004, 2009; Nagila et al. 2009). Pitavastatin was also shown to increase the promoter activity and protein expression of PON1 in vitro via p44/42 mitogen-activated protein kinase-mediated phosphorylation of sterol-regulatory-element-binding protein-2 and binding of Sp1 to PON1 DNA (Arii et al. 2009; Yamashita et al. 2010). The improvement of antioxidant properties of HDL, via increased PON1 activity, was also observed after simvastatin and fluvastatin therapy (Bergheanu et al. 2007; Mirdamadi et al. 2008; Muacevic-Katanec et al. 2007). Atorvastatin also suppresses the enhanced cellular uptake of oxidized LDL by macrophages and reduces expression of cellular scavenger receptors (Fuhrman et al. 2002; Otocka-Kmiecik et al. 2012). Similar results were observed in the study by Kassai et al. which showed a significant increase in serum PON1 specific activity, PON/HDL ratio, and LCAT activity, as well as significant reduction of oxidized LDL and CETP activities after atorvastatin therapy (Kassai et al. 2007). Finally, statin treatment enhances the defective antioxidative activity of HDL3 in patients with acute coronary syndrome by increasing the activity of HDL-associated enzymes (Otocka-Kmiecik et al. 2012).
5.2 Fibrates
Tsimihodimos et al. reported that fenofibrate induces redistribution of Lp-PLA2 from apoB-containing lipoproteins to HDL in dyslipidemic patients (Tsimihodimos et al. 2003). This effect enhanced the anti-inflammatory and antioxidant potential of the enzyme. In a recent study, Vazanna et al. examined whether HDL levels are related to in vivo oxidative stress and platelet activation in patients with coronary heart disease treated with fenofibrate (Vazzana et al. 2013). The authors measured urinary 8-iso-prostaglandin (PG) F2α and 11-dehydrothromboxane (TX) B2—in vivo markers of oxidative stress and platelet activation, respectively. They observed significantly higher urinary 8-iso-PGF2α and 11-dehydro-TXB2 in patients with low HDL than in individuals with higher HDL levels, as well as an inverse relation between the markers of oxidation and HDL concentrations. Fenofibrate treatment significantly reduced the 2 eicosanoids in healthy subjects, in parallel with an HDL increase (Vazzana et al. 2013). Tkac et al. showed a significant reduction of circulating conjugated dienes, a nonsignificant decrease in the production of malondialdehyde, and an increase in the GPx activity in patients with combined hyperlipidemia treated with fenofibrate (Tkac et al. 2006). Similar results were obtained in patients with visceral obesity and dyslipidemia treated with micronized fenofibrate (Broncel et al. 2006). Few studies have evaluated the effect of fibrates on PON1 activity. Paragh et al. evaluated this effect in a group of coronary patients with type IIb hyperlipidemia; after 3 months of treatment, HDL-C and apoA-I levels increased significantly, and PON1 specific activity also increased significantly (Paragh et al. 2003). Very similar results were obtained by Phuntuwate et al., who evaluated the effect of fenofibrate on PON1 levels, as well as the effects of the PON1 polymorphisms on lipid and PON1 responses to the drug, in dyslipidemic patients with low HDL (Phuntuwate et al. 2008). A significant increases in PON1 concentration and activity was observed after treatment, and a significant positive correlation between changes in HDL-C and changes in PON1 concentration/activity was detected (Phuntuwate et al. 2008). The response of lipid parameters to fenofibrate was independent of PON1 polymorphisms; however, PON1 Q192R and T-108C polymorphisms significantly affected the increase in PON1 activity and concentration (Phuntuwate et al. 2008). An increase of PON1 activity, which was significantly correlated with the changes in HDL-C, was also observed in patients with combined hyperlipidemia (Yesilbursa et al. 2005). On the contrary, Dullart et al. did not observe such increase after short-term (8 weeks) administration of simvastatin and bezafibrate, even when combined in diabetic patients (Dullaart et al. 2009). It needs to be also emphasized that besides the above mentioned positive HDL associated antioxidant properties of fibrates, they also elevate plasma total homocysteine, which is known to induce oxidative stress and endothelial dysfunction. Among others, a subgroup analysis of the FIELD study showed that fenofibrate treatment was associated with an increase of homocysteine levels, with no changes of serum PON-1 mass (Maranghi et al. 2011).
5.3 Niacin
There is still very limited data on the potential role of niacin therapy on HDL-related antioxidant properties. Recent studies indicate that niacin increases vascular endothelial cell redox state, resulting in the inhibition of oxidative stress (Kamanna and Kashyap 2008). These effects were investigated by Ganji et al. in vitro, who showed that in cultured endothelial cells niacin increases NADPH levels and reduces GSH (Ganji et al. 2009). In the same study, niacin also inhibited angiotensin II-induced reactive oxygen species (ROS) production (Ganji et al. 2009). These findings indicate for the first time that niacin inhibits vascular inflammation by decreasing endothelial ROS production and subsequent LDL oxidation (Ganji et al. 2009). Kaplon et al. have recently tested the hypothesis that vascular endothelial function and oxidative stress are related to dietary niacin intake among healthy middle-aged and older adults (Kaplon et al. 2014). They observed that plasma ox-LDL was inversely related to niacin intake. Moreover, in endothelial cells sampled from the brachial artery of a subgroup patients, dietary niacin intake was inversely related to nitrotyrosine, a marker of oxidative damage, leading to the hypothesis that higher dietary niacin intake is associated with greater vascular endothelial function related to lower systemic and vascular oxidative stress (Kaplon et al. 2014). The antioxidant properties of niacin were also observed patients with hypercholesterolemia and low HDL-C treated with niacin for 12 weeks (Hamoud et al. 2013). Subjects with low HDL-C levels exhibited higher oxidative stress compared with subjects with normal HDL-C levels. Niacin treatment in hypercholesterolemic patients caused a significant increase in HDL-C and apoA-I levels and a decrease in TG levels. Niacin also significantly reduced oxidative stress, as measured by a significant decrease in the serum content of thiobarbituric acid reactive substances and lipid peroxides and increase of PON activity, compared with the levels before treatment (Hamoud et al. 2013).
Conclusions and Perspectives
Currently, optimal statin treatment manages to achieve a risk reduction for cardiovascular mortality of approximately 25–35 %, mainly due to a reduction of LDL-C. This leaves an important residual risk that needs to be considered for the optimal management of patients at risk of cardiovascular disease. Raising HDL-C was seen as a viable and promising way to further reduce the risk of cardiovascular mortality. However, things look to be difficult with HDL (Table 1).
Although all fibrates have been shown to increase HDL-C significantly, their beneficial effect on cardiovascular and all-cause mortality remains controversial (Saha et al. 2007). The early VA-HIT and BIP secondary prevention trials have shown that treating CHD patients with low plasma HDL-C levels and especially those with moderate triglyceride elevations and some degree of insulin resistance (Rubins et al. 2002; The Bezafibrate Infarction Prevention (BIP) Study Group 2000) with a fibrate leads to significant reductions in the risk of a recurrent cardiovascular event, part of the effect being mediated by the fibrate-induced increase in HDL-C (Robins et al. 2001). In the FIELD study in people with type 2 diabetes mellitus, fenofibrate induced a highly significant reduction of cardiovascular events in patients with low HDL-C levels, versus a minimal effectiveness of the drug in patients with higher HDL-C (The FIELD study investigators 2005). The more recent ACCORD trial shows no benefits of adding fibrate therapy to statin on cardiovascular events except possibly in patients with residual atherogenic dyslipidemia (Ginsberg et al. 2010).
Earlier studies with niacin suggested a favorable effect of the drug on cardiovascular mortality (Canner et al. 1986), but the AIM-HIGH trial, which compared the combination niacin/simvastatin with simvastatin alone, was stopped for lack of efficacy, as niacin on the top of statin, despite increasing HDL-C levels, failed to determine an incremental clinical benefit in patients with LDL-C values at target (Boden et al. 2011). The more recent and much larger HPS2-THRIVE trial, also designed to assess the effect of adding niacin to a statin therapy in patients with established cardiovascular disease, also gave disappointing results (HPS2-THRIVE Collaborative Group 2013).
Novel HDL-raising drugs targeting new players in the HDL system, and more potent than the available drugs, are urgently needed. The promising CETP inhibitors, despite substantially increasing HDL-C levels, did not give so far positive results on cardiovascular risk (Barter et al. 2007; Schwartz et al. 2013). Different is the case of the HDL mimetics, which appears to be more promising, at least from results on surrogate end points (Nissen et al. 2003; Tardif et al. 2007), awaiting morbidity and mortality studies.
Abbreviations
- ABCA1:
-
ATP-binding cassette A1
- CETP:
-
Cholesteryl ester transfer protein
- CEC:
-
Cholesterol efflux capacity
- CRP:
-
Reactive protein
- eNOS:
-
Endothelial nitric oxide synthase
- EPC:
-
Endothelial progenitor cells
- FMD:
-
Flow-mediated vasodilation
- GSPx:
-
Glutathione selenoperoxidase
- HDL-C:
-
HDL cholesterol
- LDL-C:
-
LDL cholesterol
- LCAT:
-
Lecithin:cholesterol acyltransferase
- LOOHs:
-
Lipoprotein lipid hydroperoxides
- LpA-I:
-
HDL containing apoA-I only
- LpA-I:A-II:
-
HDL containing apoA-I and apoA-II
- Lp-PLA2:
-
Lipoprotein-associated phospholipase A2
- NO:
-
Nitric oxide
- PGI2 :
-
Prostacyclin
- PON1:
-
Paraoxonase 1
- ROS:
-
Reactive oxygen species
- SR-BI:
-
Scavenger receptor class B type I
- S1P:
-
Sphingolipid sphingosine-1-phosphate
- T2DM:
-
Type 2 diabetes
- TNFα:
-
Tumor necrosis factor alpha
References
Adorni MP, Zimetti F, Billheimer JT, Wang N, Rader DJ, Phillips MC, Rothblat GH (2007) The roles of different pathways in the release of cholesterol from macrophages. J Lipid Res 48:2453–2462
Alaupovic P, Hodis HN, Knight Gibson C, Mack WJ, LaBree L, Cashin Hemphill L, Corder CN, Kramsch DM, Blankenhorn DH (1994) Effects of lovastatin on ApoA- and ApoB-containing lipoproteins families in a subpopulation of patients participating in the monitored atherosclerosis regression study (MARS). Arterioscler Thromb 14:1906–1913
Alrasadi K, Awan Z, Alwaili K, Ruel I, Hafiane A, Krimbou L, Genest J (2008) Comparison of treatment of severe high-density lipoprotein cholesterol deficiency in men with daily atorvastatin (20 mg) versus fenofibrate (200 mg) versus extended-release niacin (2 g). Am J Cardiol 102:1341–1347
Antonopoulos AS, Margaritis M, Lee R, Channon K, Antoniades C (2012) Statins as anti-inflammatory agents in atherogenesis: molecular mechanisms and lessons from the recent clinical trials. Curr Pharm Des 18:1519–1530
Arii K, Suehiro T, Ota K, Ikeda Y, Kumon Y, Osaki F, Inoue M, Inada S, Ogami N, Takata H, Hashimoto K, Terada Y (2009) Pitavastatin induces PON1 expression through p44/42 mitogen-activated protein kinase signaling cascade in Huh7 cells. Atherosclerosis 202:439–445
Asztalos BF, Schaefer EJ (2003) High-density lipoprotein subpopulations in pathologic conditions. Am J Cardiol 91:12E–17E
Asztalos BF, Horvath K, McNamara J, Roheim P, Rubinstein J, Schaefer E (2002a) Comparing the effects of five different statins on the HDL subpopulation profiles of coronary heart disease patients. Atherosclerosis 164:361–369
Asztalos BF, Horvath KV, McNamara JR, Roheim PS, Rubinstein JJ, Schaefer EJ (2002b) Effects of atorvastatin on the HDL subpopulation profile of coronary heart disease patients. J Lipid Res 43:1701–1707
Asztalos BF, Le MF, Dallal GE, Stein E, Jones PH, Horvath KV, McTaggart F, Schaefer EJ (2007) Comparison of the effects of high doses of rosuvastatin versus atorvastatin on the subpopulations of high-density lipoproteins. Am J Cardiol 99:681–685
Asztalos BF, Collins D, Horvath KV, Bloomfield HE, Robins SJ, Schaefer EJ (2008) Relation of gemfibrozil treatment and high-density lipoprotein subpopulation profile with cardiovascular events in the veterans affairs high-density lipoprotein intervention trial. Metabolism 57:77–83
Avogaro A, Miola M, Favaro A, Gottardo L, Pacini G, Manzato E, Zambon S, Sacerdoti D, de Kreutzenberg S, Piliego T, Tiengo A, Del Prato S (2001) Gemfibrozil improves insulin sensitivity and flow-mediated vasodilatation in type 2 diabetic patients. Eur J Clin Invest 31:603–609
Bard JM, Luc G, Douste-Blazy P, Drouin P, Ziegler O, Jacotot B, Dachet C, de Gennes JL, Fruchart JC (1989) Effect of simvastatin on plasma lipids, apolipoproteins and lipoprotein particles in patients with primary hypercholesterolaemia. Eur J Clin Pharmacol 37:545–550
Barter PJ, Baker PW, Rye KA (2002) Effect of high-density lipoproteins on the expression of adhesion molecules in endothelial cells. Curr Opin Lipidol 13:285–288
Barter PJ, Caulfield M, Eriksson M, Grundy SM, Kastelein JJ, Komajda M, Lopez-Sendon J, Mosca L, Tardif JC, Waters DD, Shear CL, Revkin JH, Buhr KA, Fisher MR, Tall AR, Brewer B (2007) Effects of torcetrapib in patients at high risk for coronary events. N Engl J Med 357:2109–2122
Barter PJ, Brandrup-Wognsen G, Palmer MK, Nicholls SJ (2010) Effect of statins on HDL-C: a complex process unrelated to changes in LDL-C: analysis of the VOYAGER Database. J Lipid Res 51:1546–1553
Benjo AM, Maranhao RC, Coimbra SR, Andrade AC, Favarato D, Molina MS, Brandizzi LI, da Luz PL (2006) Accumulation of chylomicron remnants and impaired vascular reactivity occur in subjects with isolated low HDL cholesterol: effects of niacin treatment. Atherosclerosis 187:116–122
Berger JP, Akiyama TE, Meinke PT (2005) PPARs: therapeutic targets for metabolic disease. Trends Pharmacol Sci 26:244–251
Bergheanu SC, Van Tol A, Dallinga-Thie GM, Liem A, Dunselman PH, Van der Bom JG, Jukema JW (2007) Effect of rosuvastatin versus atorvastatin treatment on paraoxonase-1 activity in men with established cardiovascular disease and a low HDL-cholesterol. Curr Med Res Opin 23:2235–2240
Birjmohun RS, Hutten BA, Kastelein JJ, Stroes ES (2005) Efficacy and safety of high-density lipoprotein cholesterol-increasing compounds. A meta-analysis of randomized controlled trials. J Am Coll Cardiol 45:185–197
Boden WE, Probstfield JL, Anderson T, Chaitman BR, Desvignes-Nickens P, Koprowicz K, McBride R, Teo K, Weintraub W (2011) Niacin in patients with low HDL cholesterol levels receiving intensive statin therapy. N Engl J Med 365:2255–2267
Bodor ET, Offermanns S (2008) Nicotinic acid: an old drug with a promising future. Br J Pharmacol 153(Suppl 1):S68–S75
Bregar U, Jug B, Keber I, Cevc M, Sebestjen M (2013) Extended-release niacin/laropiprant improves endothelial function in patients after myocardial infarction. Heart Vessel 29(3):313–319
Broncel M, Cieslak D, Koter-Michalak M, Duchnowicz P, Mackiewicz K, Chojnowska-Jezierska J (2006) The anti-inflammatory and antioxidants effects of micronized fenofibrate in patients with visceral obesity and dyslipidemia. Pol Merkur Lekarski 20:547–550
Calabresi L, Franceschini G, Sirtori CR, de Palma A, Saresella M, Ferrante P, Taramelli D (1997) Inhibition of VCAM-1 expression in endothelial cells by reconstituted high density lipoproteins. Biochem Biophys Res Commun 238:61–65
Calabresi L, Gomaraschi M, Villa B, Omoboni L, Dmitrieff C, Franceschini G (2002) Elevated soluble cellular adhesion molecules in subjects with low HDL-cholesterol. Arterioscler Thromb Vasc Biol 22:656–661
Calabresi L, Gomaraschi M, Franceschini G (2003) Endothelial protection by high-density lipoproteins: from bench to bedside. Arterioscler Thromb Vasc Biol 23:1724–1731
Calabresi L, Gomaraschi M, Franceschini G (2010) High-density lipoprotein quantity or quality for cardiovascular prevention? Curr Pharm Des 16:1494–1503
Canner PL, Berge KG, Wenger NK, Stamler J, Friedman L, Prineas RJ, Friedewald WT (1986) Fifteen year mortality in coronary drug project patients: longterm benefit with niacin. J Am Coll Cardiol 8:1245–1255
Cockerill GW, Rye KA, Gamble JR, Vadas MA, Barter PJ (1995) High-density lipoproteins inhibit cytokine-induced expression of endothelial cell adhesion molecules. Arterioscler Thromb Vasc Biol 15:1987–1994
Cuchel M, Rader DJ (2006) Macrophage reverse cholesterol transport: key to the regression of atherosclerosis? Circulation 113:2548–2555
Davidson WS, Silva RA, Chantepie S, Lagor WR, Chapman MJ, Kontush A (2009) Proteomic analysis of defined HDL subpopulations reveals particle-specific protein clusters: relevance to antioxidative function. Arterioscler Thromb Vasc Biol 29:870–876
De Lalla OF, Gofman JW (1954) Ultracentrifugal analysis of serum lipoproteins. Methods Biochem Anal 1:459–478
De Vries R, Kerstens MN, Sluiter WJ, Groen AK, Van TA, Dullaart RP (2005) Cellular cholesterol efflux to plasma from moderately hypercholesterolaemic type 1 diabetic patients is enhanced, and is unaffected by simvastatin treatment. Diabetologia 48:1105–1113
Deanfield JE, Halcox JP, Rabelink TJ (2007) Endothelial function and dysfunction: testing and clinical relevance. Circulation 115:1285–1295
Dullaart RP, De VR, Voorbij HA, Sluiter WJ, Van TA (2009) Serum paraoxonase-I activity is unaffected by short-term administration of simvastatin, bezafibrate, and their combination in type 2 diabetes mellitus. Eur J Clin Invest 39:200–203
Evans M, Anderson RA, Graham J, Ellis GR, Morris K, Davies S, Jackson SK, Lewis MJ, Frenneaux MP, Rees A (2000) Ciprofibrate therapy improves endothelial function and reduces postprandial lipemia and oxidative stress in type 2 diabetes mellitus. Circulation 101:1773–1779
Favari E, Calabresi L, Adorni MP, Jessup W, Simonelli S, Franceschini G, Bernini F (2009) Small discoidal pre-beta1 HDL particles are efficient acceptors of cell cholesterol via ABCA1 and ABCG1. Biochemistry 48:11067–11074
Florentin M, Liberopoulos EN, Wierzbicki AS, Mikhailidis DP (2008) Multiple actions of high-density lipoprotein. Curr Opin Cardiol 23:370–378
Franceschini G, Sirtori M, Vaccarino V, Gianfranceschi G, Chiesa G, Sirtori CR (1989) Plasma lipoprotein changes after treatment with pravastatin and gemfibrozil in patients with familial hypercholesterolemia. J Lab Clin Med 114:250–259
Franceschini G, Calabresi L, Colombo C, Favari E, Bernini F, Sirtori CR (2007) Effects of fenofibrate and simvastatin on HDL-related biomarkers in low-HDL patients. Atherosclerosis 195:385–391
Franceschini G, Favari E, Calabresi L, Simonelli S, Bondioli A, Adorni MP, Zimetti F, Gomaraschi M, Coutant K, Rossomanno S, Niesor EJ, Bernini F, Benghozi R (2013) Differential effects of fenofibrate and extended-release niacin on high-density lipoprotein particle size distribution and cholesterol efflux capacity in dyslipidemic patients. J Clin Lipidol 7:414–422
Fuhrman B, Koren L, Volkova N, Keidar S, Hayek T, Aviram M (2002) Atorvastatin therapy in hypercholesterolemic patients suppresses cellular uptake of oxidized-LDL by differentiating monocytes. Atherosclerosis 164:179–185
Ganji SH, Qin S, Zhang L, Kamanna VS, Kashyap ML (2009) Niacin inhibits vascular oxidative stress, redox-sensitive genes, and monocyte adhesion to human aortic endothelial cells. Atherosclerosis 202:68–75
Ghani RA, Bin YI, Wahab NA, Zainudin S, Mustafa N, Sukor N, Wan Mohamud WN, Kadir KA, Kamaruddin NA (2013) The influence of fenofibrate on lipid profile, endothelial dysfunction, and inflammatory markers in type 2 diabetes mellitus patients with typical and mixed dyslipidemia. J Clin Lipidol 7:446–453
Ginsberg HN, Elam MB, Lovato LC, Crouse JR III, Leiter LA, Linz P, Friedewald WT, Buse JB, Gerstein HC, Probstfield J, Grimm RH, Ismail-Beigi F, Bigger JT, Goff DC Jr, Cushman WC, Simons-Morton DG, Byington RP (2010) Effects of combination lipid therapy in type 2 diabetes mellitus. N Engl J Med 362:1563–1574
Gomaraschi M, Basilico N, Sisto F, Taramelli D, Eligini S, Colli S, Sirtori CR, Franceschini G, Calabresi L (2005) High-density lipoproteins attenuate interleukin-6 production in endothelial cells exposed to pro-inflammatory stimuli. Biochim Biophys Acta 1736:136–143
Guerin M, Lassel TS, Le Goff W, Farnier M, Chapman MJ (2000) Action of atorvastatin in combined hyperlipidemia : preferential reduction of cholesteryl ester transfer from HDL to VLDL1 particles. Arterioscler Thromb Vasc Biol 20:189–197
Guerin M, Egger P, Soudant C, Le Goff W, van Tol A, Dupuis R, Chapman MJ (2002) Dose-dependent action of atorvastatin in type IIB hyperlipidemia: preferential and progressive reduction of atherogenic apoB-containing lipoprotein subclasses (VLDL-2, IDL, small dense LDL) and stimulation of cellular cholesterol efflux. Atherosclerosis 163:287–296
Guerin M, Le Goff W, Frisdal E, Schneider S, Milosavljevic D, Bruckert E, Chapman MJ (2003) Action of ciprofibrate in type IIb hyperlipoproteinemia: modulation of the atherogenic lipoprotein phenotype and stimulation of high-density lipoprotein-mediated cellular cholesterol efflux. J Clin Endocrinol Metab 88:3738–3746
Hamoud S, Kaplan M, Meilin E, Hassan A, Torgovicky R, Cohen R, Hayek T (2013) Niacin administration significantly reduces oxidative stress in patients with hypercholesterolemia and low levels of high-density lipoprotein cholesterol. Am J Med Sci 345:195–199
Harangi M, Seres I, Varga Z, Emri G, Szilvassy Z, Paragh G, Remenyik E (2004) Atorvastatin effect on high-density lipoprotein-associated paraoxonase activity and oxidative DNA damage. Eur J Clin Pharmacol 60:685–691
Harangi M, Mirdamadi HZ, Seres I, Sztanek F, Molnar M, Kassai A, Derdak Z, Illyes L, Paragh G (2009) Atorvastatin effect on the distribution of high-density lipoprotein subfractions and human paraoxonase activity. Transl Res 153:190–198
Hernandez M, Wright SD, Cai TQ (2007) Critical role of cholesterol ester transfer protein in nicotinic acid-mediated HDL elevation in mice. Biochem Biophys Res Commun 355:1075–1080
Higashi Y, Matsuoka H, Umei H, Sugano R, Fujii Y, Soga J, Kihara Y, Chayama K, Imaizumi T (2010) Endothelial function in subjects with isolated low HDL cholesterol: role of nitric oxide and circulating progenitor cells. Am J Physiol Endocrinol Metab 298:E202–E209
Homma Y, Ozawa H, Kobayashi T, Yamaguchi H, Sakane H, Nakamura H (1995) Effects of simvastatin on plasma lipoprotein subfractions, cholesterol esterification rate, and cholesteryl ester transfer protein in type II hyperlipoproteinemia. Atherosclerosis 114:223–234
HPS2-THRIVE Collaborative Group (2013) HPS2-THRIVE randomized placebo-controlled trial in 25 673 high-risk patients of ER niacin/laropiprant: trial design, pre-specified muscle and liver outcomes, and reasons for stopping study treatment. Eur Heart J 34:1279–1291
Igarashi J, Miyoshi M, Hashimoto T, Kubota Y, Kosaka H (2007) Statins induce S1P(1) receptors and enhance endothelial nitric oxide production in response to high-density lipoproteins. Br J Pharmacol 150:470–479
Johansson J, Carlson LA (1990) The effects of nicotinic acid treatment on high density lipoprotein particle size subclass levels in hyperlipidaemic subjects. Atherosclerosis 83:207–216
Jones PH, Davidson MH, Stein EA, Bays HE, McKenney JM, Miller E, Cain VA, Blasetto JW (2003) Comparison of the efficacy and safety of rosuvastatin versus atorvastatin, simvastatin, and pravastatin across doses (STELLAR* Trial). Am J Cardiol 92:152–160
Kamanna VS, Kashyap ML (2008) Mechanism of action of niacin. Am J Cardiol 101:20B–26B
Kaplon RE, Gano LB, Seals DR (2014) Vascular endothelial function and oxidative stress are related to dietary niacin intake among healthy middle-aged and older adults. J Appl Physiol (1985) 116:156–163
Kassai A, Illyes L, Mirdamadi HZ, Seres I, Kalmar T, Audikovszky M, Paragh G (2007) The effect of atorvastatin therapy on lecithin:cholesterol acyltransferase, cholesteryl ester transfer protein and the antioxidant paraoxonase. Clin Biochem 40:1–5
Khera AV, Cuchel M, De LL-M, Rodrigues A, Burke MF, Jafri K, French BC, Phillips JA, Mucksavage ML, Wilensky RL, Mohler ER, Rothblat GH, Rader DJ (2011) Cholesterol efflux capacity, high-density lipoprotein function, and atherosclerosis. N Engl J Med 364:127–135
Khera AV, Patel PJ, Reilly MP, Rader DJ (2013) The addition of niacin to statin therapy improves high-density lipoprotein cholesterol levels but not metrics of functionality. J Am Coll Cardiol 62:1909–1910
Khoury N, Goldberg AC (2011) The use of fibric acid derivatives in cardiovascular prevention. Curr Treat Option Cardiovasc Med 13:335–342
Kimura T, Mogi C, Tomura H, Kuwabara A, Im DS, Sato K, Kurose H, Murakami M, Okajima F (2008) Induction of scavenger receptor class B type I is critical for simvastatin enhancement of high-density lipoprotein-induced anti-inflammatory actions in endothelial cells. J Immunol 181:7332–7340
Knopp RH, Alagona P, Davidson M, Goldberg AC, Kafonek SD, Kashyap M, Sprecher D, Superko HR, Jenkins S, Marcovina S (1998) Equivalent efficacy of a time-release form of niacin (Niaspan) given once-a-night versus plain niacin in the management of hyperlipidemia. Metabolism 47:1097–1104
Koh KK, Yeal AJ, Hwan HS, Kyu JD, Sik KH, Cheon LK, Kyun SE, Sakuma I (2004) Effects of fenofibrate on lipoproteins, vasomotor function, and serological markers of inflammation, plaque stabilization, and hemostasis. Atherosclerosis 174:379–383
Koh KK, Quon MJ, Han SH, Chung WJ, Ahn JY, Seo YH, Choi IS, Shin EK (2005) Additive beneficial effects of fenofibrate combined with atorvastatin in the treatment of combined hyperlipidemia. J Am Coll Cardiol 45:1649–1653
Kontush A, Chapman MJ (2010) Antiatherogenic function of HDL particle subpopulations: focus on antioxidative activities. Curr Opin Lipidol 21:312–318
Kuvin JT, Ramet ME, Patel AR, Pandian NG, Mendelsohn ME, Karas RH (2002) A novel mechanism for the beneficial vascular effects of high-density lipoprotein cholesterol: enhanced vasorelaxation and increased endothelial nitric oxide synthase expression. Am Heart J 144:165–172
Kuvin JT, Dave DM, Sliney KA, Mooney P, Patel AR, Kimmelstiel CD, Karas RH (2006) Effects of extended-release niacin on lipoprotein particle size, distribution, and inflammatory markers in patients with coronary artery disease. Am J Cardiol 98:743–745
Lamon-Fava S, Diffenderfer MR, Barrett PH, Buchsbaum A, Nyaku M, Horvath KV, Asztalos BF, Otokozawa S, Ai M, Matthan NR, Lichtenstein AH, Dolnikowski GG, Schaefer EJ (2008) Extended-release niacin alters the metabolism of plasma apolipoprotein (Apo) A-I and ApoB-containing lipoproteins. Arterioscler Thromb Vasc Biol 28:1672–1678
Lee JM, Robson MD, Yu LM, Shirodaria CC, Cunnington C, Kylintireas I, Digby JE, Bannister T, Handa A, Wiesmann F, Durrington PN, Channon KM, Neubauer S, Choudhury RP (2009) Effects of high-dose modified-release nicotinic acid on atherosclerosis and vascular function a randomized, placebo-controlled, magnetic resonance imaging study. J Am Coll Cardiol 54:1787–1794
Lewis GF, Rader DJ (2005) New insights into the regulation of HDL metabolism and reverse cholesterol transport. Circ Res 96:1221–1232
Liu Y, Wei J, Hu S, Hu L (2012) Beneficial effects of statins on endothelial progenitor cells. Am J Med Sci 344:220–226
Malik J, Melenovsky V, Wichterle D, Haas T, Simek J, Ceska R, Hradec J (2001) Both fenofibrate and atorvastatin improve vascular reactivity in combined hyperlipidaemia (fenofibrate versus atorvastatin trial – FAT). Cardiovasc Res 52:290–298
Maranghi M, Hiukka A, Badeau R, Sundvall J, Jauhiainen M, Taskinen MR (2011) Macrophage cholesterol efflux to plasma and HDL in subjects with low and high homocysteine levels: a FIELD substudy. Atherosclerosis 219:259–265
McGrath KC, Li XH, Puranik R, Liong EC, Tan JT, Dy VM, DiBartolo BA, Barter PJ, Rye KA, Heather AK (2009) Role of 3beta-hydroxysteroid-Delta 24 reductase in mediating antiinflammatory effects of high-density lipoproteins in endothelial cells. Arterioscler Thromb Vasc Biol 29:877–882
Miida T, Sakai K, Ozaki K, Nakamura Y, Yamaguchi T, Tsuda T, Kashiwa T, Murakami T, Inano K, Okada M (2000) Bezafibrate increases Prebeta1-HDL at the expense of HDL(2b) in hypertriglyceridemia. Arterioscler Thromb Vasc Biol 20:2428–2433
Mineo C, Shaul PW (2012) Novel biological functions of high-density lipoprotein cholesterol. Circ Res 111:1079–1090
Mirdamadi HZ, Sztanek F, Derdak Z, Seres I, Harangi M, Paragh G (2008) The human paraoxonase-1 phenotype modifies the effect of statins on paraoxonase activity and lipid parameters. Br J Clin Pharmacol 66:366–374
Miyamoto-Sasaki M, Yasuda T, Monguchi T, Nakajima H, Mori K, Toh R, Ishida T, Hirata K (2013) Pitavastatin increases HDL particles functionally preserved with cholesterol efflux capacity and antioxidative actions in dyslipidemic patients. J Atheroscler Thromb 20:708–716
Morgan JM, Capuzzi DM, Baksh RI, Intenzo C, Carey CM, Reese D, Walker K (2003) Effects of extended-release niacin on lipoprotein subclass distribution. Am J Cardiol 91:1432–1436
Morgan JM, De LL-M, Capuzzi DM (2007) Effects of niacin and Niaspan on HDL lipoprotein cellular SR-BI-mediated cholesterol efflux. J Clin Lipidol 1:614–619
Muacevic-Katanec D, Bradamante V, Poljicanin T, Reiner Z, Babic Z, Simeon-Rudolf V, Katanec D (2007) Clinical study on the effect of simvastatin on paraoxonase activity. Arzneimittelforschung 57:647–653
Nagila A, Permpongpaiboon T, Tantrarongroj S, Porapakkham P, Chinwattana K, Deakin S, Porntadavity S (2009) Effect of atorvastatin on paraoxonase1 (PON1) and oxidative status. Pharmacol Rep 61:892–898
Nichols AV, Krauss RM, Musliner TA (1986) Nondenaturing polyacrylamide gradient gel electrophoresis. Methods Enzymol 128:417–431
Nissen SE, Tsunoda T, Tuzcu EM, Schoenhagen P, Cooper CJ, Yasin M, Eaton GM, Lauer MA, Sheldon WS, Grines CL, Halpern S, Crowe T, Blankenship JC, Kerensky R (2003) Effect of recombinant ApoA-I Milano on coronary atherosclerosis in patients with acute coronary syndromes: a randomized controlled trial. JAMA 290:2292–2300
Nofer JR, van der Giet M, Tolle M, Wolinska I, von Wnuck LK, Baba HA, Tietge UJ, Godecke A, Ishii I, Kleuser B, Schafers M, Fobker M, Zidek W, Assmann G, Chun J, Levkau B (2004) HDL induces NO-dependent vasorelaxation via the lysophospholipid receptor S1P3. J Clin Invest 113:569–581
Otocka-Kmiecik A, Mikhailidis DP, Nicholls SJ, Davidson M, Rysz J, Banach M (2012) Dysfunctional HDL: a novel important diagnostic and therapeutic target in cardiovascular disease? Prog Lipid Res 51:314–324
Otvos JD, Collins D, Freedman DS, Shalaurova I, Schaefer EJ, McNamara JR, Bloomfield HE, Robins SJ (2006) Low-density lipoprotein and high-density lipoprotein particle subclasses predict coronary events and are favorably changed by gemfibrozil therapy in the veterans affairs high-density lipoprotein intervention trial. Circulation 113:1556–1563
Paragh G, Seres I, Harangi M, Balogh Z, Illyes L, Boda J, Szilvassy Z, Kovacs P (2003) The effect of micronised fenofibrate on paraoxonase activity in patients with coronary heart disease. Diabetes Metab 29:613–618
Patsch JR, Prasad S, Gotto AM Jr, Bengtsson-Olivecrona G (1984) Postprandial lipemia. A key for the conversion of high density lipoprotein2 into high density lipoprotein3 by hepatic lipase. J Clin Invest 74:2017–2023
Petoumenos V, Nickenig G, Werner N (2009) High-density lipoprotein exerts vasculoprotection via endothelial progenitor cells. J Cell Mol Med 13:4623–4635
Philpott AC, Hubacek J, Sun YC, Hillard D, Anderson TJ (2013) Niacin improves lipid profile but not endothelial function in patients with coronary artery disease on high dose statin therapy. Atherosclerosis 226:453–458
Phuntuwate W, Suthisisang C, Koanantakul B, Chaloeiphap P, Mackness B, Mackness M (2008) Effect of fenofibrate therapy on paraoxonase1 status in patients with low HDL-C levels. Atherosclerosis 196:122–128
Podrez EA (2010) Anti-oxidant properties of high-density lipoprotein and atherosclerosis. Clin Exp Pharmacol Physiol 37:719–725
Reriani MK, Dunlay SM, Gupta B, West CP, Rihal CS, Lerman LO, Lerman A (2011) Effects of statins on coronary and peripheral endothelial function in humans: a systematic review and meta-analysis of randomized controlled trials. Eur J Cardiovasc Prev Rehabil 18:704–716
Riwanto M, Landmesser U (2013) High density lipoproteins and endothelial functions: mechanistic insights and alterations in cardiovascular disease. J Lipid Res 54:3227–3243
Robins SJ, Collins D, Wittes JT, Papademetriou V, Deedwania PC, Schaefer EJ, McNamara JR, Kashyap ML, Hershman JM, Wexler LF, Rubins HB (2001) Relation of gemfibrozil treatment and lipid levels with major coronary events: VA-HIT: a randomized controlled trial. JAMA 285:1585–1591
Rosenson RS, Brewer HB Jr, Ansell B, Barter P, Chapman MJ, Heinecke JW, Kontush A, Tall AR, Webb NR (2013) Translation of high-density lipoprotein function into clinical practice: current prospects and future challenges. Circulation 128:1256–1267
Rubic T, Trottmann M, Lorenz RL (2004) Stimulation of CD36 and the key effector of reverse cholesterol transport ATP-binding cassette A1 in monocytoid cells by niacin. Biochem Pharmacol 67:411–419
Rubins HB, Robins SJ, Collins D, Nelson DB, Elam MB, Schaefer EJ, Faas FH, Anderson JW (2002) Diabetes, plasma insulin, and cardiovascular disease: subgroup analysis from the department of veterans affairs high-density lipoprotein intervention trial (VA-HIT). Arch Intern Med 162:2597–2604
Saha SA, Kizhakepunnur LG, Bahekar A, Arora RR (2007) The role of fibrates in the prevention of cardiovascular disease–a pooled meta-analysis of long-term randomized placebo-controlled clinical trials. Am Heart J 154:943–953
Sakai T, Kamanna VS, Kashyap ML (2001) Niacin, but not gemfibrozil, selectively increases LP-AI, a cardioprotective subfraction of HDL, in patients with low HDL cholesterol. Arterioscler Thromb Vasc Biol 21:1783–1789
Sasaki J, Yamamoto K, Ageta M (2002) Effects of fenofibrate on high-density lipoprotein particle size in patients with hyperlipidemia: a randomized, double-blind, placebo-controlled, multicenter, crossover study. Clin Ther 24:1614–1626
Schwartz GG, Olsson AG, Barter PJ (2013) Dalcetrapib in patients with an acute coronary syndrome. N Engl J Med 368:869–870
Shepherd J, Packard CJ, Patsch JR, Gotto AM Jr, Taunton OD (1979) Effects of nicotinic acid therapy on plasma high density lipoprotein subfraction distribution and composition on apolipoprotein A metabolism. J Clin Invest 63:858–867
Shuhei N, Soderlund S, Jauhiainen M, Taskinen MR (2010) Effect of HDL composition and particle size on the resistance of HDL to the oxidation. Lipids Health Dis 9:104
Sorrentino SA, Besler C, Rohrer L, Meyer M, Heinrich K, Bahlmann FH, Mueller M, Horvath T, Doerries C, Heinemann M, Flemmer S, Markowski A, Manes C, Bahr MJ, Haller H, Von EA, Drexler H, Landmesser U (2010) Endothelial-vasoprotective effects of high-density lipoprotein are impaired in patients with type 2 diabetes mellitus but are improved after extended-release niacin therapy. Circulation 121:110–122
Soudijn W, van Wijngaarden I, Ijzerman AP (2007) Nicotinic acid receptor subtypes and their ligands. Med Res Rev 27:417–433
Stremler KE, Stafforini DM, Prescott SM, McIntyre TM (1991) Human plasma platelet-activating factor acetylhydrolase. Oxidatively fragmented phospholipids as substrates. J Biol Chem 266:11095–11103
Sviridov D, Hoang A, Ooi E, Watts G, Barrett PH, Nestel P (2008) Indices of reverse cholesterol transport in subjects with metabolic syndrome after treatment with rosuvastatin. Atherosclerosis 197:732–739
Tamehiro N, Shigemoto-Mogami Y, Kakeya T, Okuhira K, Suzuki K, Sato R, Nagao T, Nishimaki-Mogami T (2007) Sterol regulatory element-binding protein-2- and liver X receptor-driven dual promoter regulation of hepatic ABC transporter A1 gene expression: mechanism underlying the unique response to cellular cholesterol status. J Biol Chem 282:21090–21099
Tardif JC, Gregoire J, L'Allier PL, Ibrahim R, Lesperance J, Heinonen TM, Kouz S, Berry C, Basser R, Lavoie MA, Guertin MC, Rodes-Cabau J (2007) Effects of reconstituted high-density lipoprotein infusions on coronary atherosclerosis: a randomized controlled trial. JAMA 297:1675–1682
The Bezafibrate Infarction Prevention (BIP) Study Group (2000) Secondary prevention by raising HDL cholesterol and reducing triglycerides in patients with coronary artery disease: the Bezafibrate Infarction Prevention (BIP) study. Circulation 102:21–27
The FIELD study investigators (2005) Effects of long-term fenofibrate therapy on cardiovascular events in 9795 people with type 2 diabetes mellitus (the FIELD study): randomised controlled trial. Lancet 366:1849–1861
Thoenes M, Oguchi A, Nagamia S, Vaccari CS, Hammoud R, Umpierrez GE, Khan BV (2007) The effects of extended-release niacin on carotid intimal media thickness, endothelial function and inflammatory markers in patients with the metabolic syndrome. Int J Clin Pract 61:1942–1948
Tkac I, Molcanyiova A, Javorsky M, Kozarova M (2006) Fenofibrate treatment reduces circulating conjugated diene level and increases glutathione peroxidase activity. Pharmacol Res 53:261–264
Tomas M, Senti M, Garcia-Faria F, Vila J, Torrents A, Covas M, Marrugat J (2000) Effect of simvastatin therapy on paraoxonase activity and related lipoproteins in familial hypercholesterolemic patients. Arterioscler Thromb Vasc Biol 20:2113–2119
Triolo M, Annema W, de Boer JF, Tietge UJ, Dullaart RP (2013a) Simvastatin and bezafibrate increase cholesterol efflux in men with type 2 diabetes. Eur J Clin Invest 44(3):240–248
Triolo M, Kwakernaak AJ, Perton FG, De Vries R, Dallinga-Thie GM, Dullaart RP (2013b) Low normal thyroid function enhances plasma cholesteryl ester transfer in type 2 diabetes mellitus. Atherosclerosis 228:466–471
Tsimihodimos V, Kakafika A, Tambaki AP, Bairaktari E, Chapman MJ, Elisaf M, Tselepis AD (2003) Fenofibrate induces HDL-associated PAF-AH but attenuates enzyme activity associated with apoB-containing lipoproteins. J Lipid Res 44:927–934
van der Hoogt CC, de Haan W, Westerterp M, Hoekstra M, Dallinga-Thie GM, Romijn JA, Princen HM, Jukema JW, Havekes LM, Rensen PC (2007) Fenofibrate increases HDL-cholesterol by reducing cholesteryl ester transfer protein expression. J Lipid Res 48:1763–1771
Vazzana N, Ganci A, Cefalu AB, Lattanzio S, Noto D, Santoro N, Saggini R, Puccetti L, Averna M, Davi G (2013) Enhanced lipid peroxidation and platelet activation as potential contributors to increased cardiovascular risk in the low-HDL phenotype. J Am Heart Assoc 2:e000063
Vega GL, Grundy SM (1994) Lipoprotein responses to treatment with lovastatin, gemfibrozil, and nicotinic acid in normolipidemic patients with hypoalphalipoproteinemia. Arch Intern Med 154:73–82
Wahlberg G, Walldius G, Olsson AG, Kirstein P (1990) Effects of nicotinic acid on serum cholesterol concentrations of high density lipoprotein subfractions HDL2 and HDL3 in hyperlipoproteinaemia. J Intern Med 228:151–157
Wang TD, Chen WJ, Lin JW, Cheng CC, Chen MF, Lee YT (2003) Efficacy of fenofibrate and simvastatin on endothelial function and inflammatory markers in patients with combined hyperlipidemia: relations with baseline lipid profiles. Atherosclerosis 170:315–323
Warnholtz A, Wild P, Ostad MA, Elsner V, Stieber F, Schinzel R, Walter U, Peetz D, Lackner K, Blankenberg S, Munzel T (2009) Effects of oral niacin on endothelial dysfunction in patients with coronary artery disease: results of the randomized, double-blind, placebo-controlled INEF study. Atherosclerosis 204:216–221
Xia P, Vadas MA, Rye KA, Barter PJ, Gamble JR (1999) High density lipoproteins (HDL) interrupt the sphingosine kinase signaling pathway. A possible mechanism for protection against atherosclerosis by HDL. J Biol Chem 274:33143–33147
Yadav R, France M, Younis N, Hama S, Ammori BJ, Kwok S, Soran H (2012) Extended-release niacin with laropiprant : a review on efficacy, clinical effectiveness and safety. Expert Opin Pharmacother 13:1345–1362
Yamashita S, Tsubakio-Yamamoto K, Ohama T, Nakagawa-Toyama Y, Nishida M (2010) Molecular mechanisms of HDL-cholesterol elevation by statins and its effects on HDL functions. J Atheroscler Thromb 17:436–451
Yesilbursa D, Serdar A, Saltan Y, Serdar Z, Heper Y, Guclu S, Cordan J (2005) The effect of fenofibrate on serum paraoxonase activity and inflammatory markers in patients with combined hyperlipidemia. Kardiol Pol 62:526–530
Yetukuri L, Huopaniemi I, Koivuniemi A, Maranghi M, Hiukka A, Nygren H, Kaski S, Taskinen MR, Vattulainen I, Jauhiainen M, Oresic M (2011) High density lipoprotein structural changes and drug response in lipidomic profiles following the long-term fenofibrate therapy in the FIELD substudy. PLoS ONE 6:e23589
Yuhanna IS, Zhu Y, Cox BE, Hahner LD, Osborne-Lawrence S, Lu P, Marcel YL, Anderson RG, Mendelsohn ME, Hobbs HH, Shaul PW (2001) High-density lipoprotein binding to scavenger receptor-BI activates endothelial nitric oxide synthase. Nat Med 7:853–857
Yvan-Charvet L, Kling J, Pagler T, Li H, Hubbard B, Fisher T, Sparrow CP, Taggart AK, Tall AR (2010) Cholesterol efflux potential and antiinflammatory properties of high-density lipoprotein after treatment with niacin or anacetrapib. Arterioscler Thromb Vasc Biol 30:1430–1438
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Open Access This chapter is distributed under the terms of the Creative Commons Attribution Noncommercial License, which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Copyright information
© 2015 The Author(s)
About this chapter
Cite this chapter
Gomaraschi, M., Adorni, M.P., Banach, M., Bernini, F., Franceschini, G., Calabresi, L. (2015). Effects of Established Hypolipidemic Drugs on HDL Concentration, Subclass Distribution, and Function. In: von Eckardstein, A., Kardassis, D. (eds) High Density Lipoproteins. Handbook of Experimental Pharmacology, vol 224. Springer, Cham. https://doi.org/10.1007/978-3-319-09665-0_19
Download citation
DOI: https://doi.org/10.1007/978-3-319-09665-0_19
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-09664-3
Online ISBN: 978-3-319-09665-0
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)