
Abstract
Nucleotide variation, whether induced or natural, is responsible for a vast majority of heritable phenotypic variation. Evaluation of genomic DNA sequence is therefore fundamental for both functional genomics and marker-assisted breeding. The starting point for this is the extraction of high-quality DNA of a suitable quantity for planned downstream applications. A myriad of kits and techniques exists, but not all are suitable for laboratories with limiting infrastructure due to high costs and reliance on toxic chemicals. We describe here a protocol for extraction of high-quality genomic DNA from leaves of a variety of plant species that uses self-prepared buffers and reagents and obviates the need for toxic organic chemicals such as chloroform. We also provide a protocol for using free image analysis software to quantify isolated DNA.
You have full access to this open access chapter, Download chapter PDF
Similar content being viewed by others

Keywords
1 Introduction
While deoxyribonucleic acid (DNA) has been known to be a component of cells since the late 1800s, it wasn’t until the pioneering work of Oswald Avery that it was understood that the molecule is the genetic material (Avery et al. 1944; Dahm 2005). Today, DNA is at the heart of many biological analyses. Molecular markers allow facile and rapid introgression of alleles into elite crop varieties, high-density variant analysis enables genomic selection, whole genome sequencing is revealing evolutionary dynamics in populations and Targeting Induced Local Lesions IN Genomes (TILLING) approaches allow reverse-genetics using induced mutations in plant and animal species (Jankowicz-Cieslak et al. 2011; Varshney et al. 2014). Many applications and methods for the molecular investigation of plants require extraction of genomic DNA. The ideal method balances throughput, per sample cost, yield per milligram of tissue and the use of toxic chemicals according to the priorities of a specific laboratory.
Commercial kits have been developed for DNA isolation from numerous tissues of plants and animals and also microorganisms. These kits are suitable for rapid and reliable DNA extractions of high purity and high quality and produce high yields. They have become the standard for sensitive molecular assays, but the cost per sample remains high and therefore DNA extraction is still a limitation for large-scale projects in some laboratories. Many commercial methods are based on the binding of DNA to silica in the presence of chaotropic salts. The silica in such kits is usually provided packed in ready-to-use spin columns. Lower-cost alternatives utilising self-made buffers exist but often have disadvantages such as the use of toxic organic phase separation. We present a protocol that utilises the DNA binding capacity of silica in the presence of chaotropic salts. Costs are reduced through the use of self-made buffers and avoiding pre-made spin columns. The principle of the silica-based DNA isolation method is the same as in commercially available kits. In the presence of high concentrations of chaotropic salt, the interaction of water with the DNA backbone is disrupted, and charged phosphate on the DNA can form a cationic bridge with silica. DNA-bound silica is separated from the other soluble cellular components by centrifugation. Washing the DNA-bound silica in the presence of a high percentage of alcohol removes excess salt. The subsequent addition of an aqueous solvent (water or buffer) drives the hydration of the DNA and its release from silica. The now soluble DNA can be separated from silica through a quick centrifugation step. The methods provided here include improvements and further cost reductions from our previously published protocol utilising chaotropic salts and silica binding (Till et al. 2015). Following DNA extraction it is necessary to assess the quality and quantity of DNA. There are many methods that can be used such as spectroscopy, fluorimetry and gel-based methods. Gel-based methods are quite common and have the advantage that only standard electrophoresis equipment common to most molecular biology labs is required. Further, gel-based methods allow the direct assessment of DNA degradation which is not easily measured using other approaches. While quantification of DNA on gels can be done visually, the precision of measurements is improved through the use of image analysis software. We describe a method utilising quantitative markers and the free image analysis software ImageJ (Schneider et al. 2012). This procedure includes the use of spreadsheet software (e.g. Microsoft Excel, LibreOffice Calc or Apache OpenOffice Calc) for rapid estimations of DNA concentrations of many samples.
2 Materials
2.1 DNA Extraction
-
1.
Vortex mixer (e.g. Vortex-Genie 2, Scientific Industries, Inc., Cat. No. SI-0236).
-
2.
Benchtop scale (e.g. Mettler Toledo Cat. No. XS2002S).
-
3.
Microcentrifuge.
-
4.
Micropipettes (1000 μl, 200 μl, 20 μl).
-
5.
Microcentrifuge tubes (1.5 ml, 2.0 ml).
-
6.
50 ml conical tube with cap.
-
7.
Metal beads (e.g. tungsten carbide beads, 3 mm, Qiagen 69997, see Note 1).
-
8.
Distilled or deionised water (dH2O).
-
9.
Celite 545 silica powder (e.g. Celite 545 AW, Sigma-Aldrich, 20,199-U).
-
10.
Sea sand (optional, see Note 2).
-
11.
RNase A (10 μg/ml).
-
12.
6 M KI (potassium iodide) or alternative buffers (see Notes 3 and 4).
-
13.
SDS (sodium dodecyl sulphate).
-
14.
Sodium acetate anhydrous (prepare 3 M sodium acetate (pH 5.2) solution).
-
15.
NaCl (sodium chloride).
-
16.
Absolute ethanol.
-
17.
Wash buffer (0.05 M NaCl in 95 % EtOH) (see Note 5).
-
18.
10× TE buffer (100 mM Tris–HCl, 10 mM EDTA, pH 8.0).
2.2 DNA Quantification
-
1.
Equipment for horizontal gel electrophoresis (combs, casting trays, gel tank, power supply)
-
2.
Agarose (for gel electrophoresis)
-
3.
0.5× Tris/Borate/EDTA (TBE) buffer (for gel electrophoresis)
-
4.
Ethidium bromide, SYBR or equivalent double-stranded DNA staining dye
-
5.
Lambda DNA (e.g. Invitrogen 25250–010) for the preparation of concentration standards (see Note 6)
-
6.
DNA gel loading dye (containing Orange G or bromophenol blue) (see Note 7)
-
7.
Gel photography system (digital camera, light box)
-
8.
Computer and monitor (see Note 8)
-
9.
ImageJ software (http://rsbweb.nih.gov/ij/) (see Note 9)
-
10.
Spreadsheet software (e.g. Microsoft Excel, LibreOffice Calc or Apache OpenOffice Calc)
3 Methods
3.1 DNA Extraction
3.1.1 Preparation of Silica Powder DNA Binding Solution
-
1.
Transfer 800 mg Celite 545 silica into a 50 ml conical tube.
-
2.
Add 30 ml dH2O.
-
3.
Vortex and invert 15 times or until a hydrated slurry forms.
-
4.
Let stand for 15 min until all solid material has settled.
-
5.
Remove (pipette off) the liquid. It is okay to leave a small amount of liquid in the tube.
-
6.
Repeat steps 2–5 to a total of three washes.
-
7.
Suspend the hydrated silica in a volume of dH2O equal to the volume of silica. This is the liquid silica stock (LSS) and can be stored at room temperature (RT) for up to 1 month (see Note 10).
-
8.
Suspend LSS by vortexing for 30 s or until a homogenised slurry is formed. For each tissue sample, transfer 50 μl into a fresh 2 ml tube.
-
9.
Add 1 ml dH2O per tube to perform a final wash step.
-
10.
Mix by vortexing for 15 s or until silica is fully suspended.
-
11.
Centrifuge at the full speed (16,000 × g) for 20 s.
-
12.
Pipette off liquid.
-
13.
Add 700 μl 6 M KI (see Note 3).
-
14.
Suspend the silica in 6 M KI by vortexing 15 s. This is the silica binding solution (SBS, see Note 11).
3.1.2 Low-Cost Extraction of Genomic DNA from Plant Tissues
-
1.
Prepare an ice bath.
-
2.
Label 2 ml tubes containing three metal beads (see Note 1) and optional sea sand with sample names. Grinding is facilitated by addition of purified sea sand (see Note 2).
-
3.
Add tissue to the appropriate tube (see Note 12).
-
4.
Tape tubes onto a vortex mixer (Fig. 14.1) and vortex on high setting for 30 s or until material is ground to a fine powder.
-
5.
Add 800 μl of lysis buffer (0.5 % SDS (w/v) in 10× TE) and 4 μl RNAse A (10 μg/ml) to each tube (see Note 13).
-
6.
Vortex 2 min until ground tissue is fully hydrated and mixed with buffer.
-
7.
Incubate for 10 min at RT.
-
8.
Add 200 μl 3 M sodium acetate (pH 5.2). Mix by inversion of tubes and incubate on ice for 5 min.
-
9.
Centrifuge at 16,000 × g for 5 min at RT to pellet the leaf material.
-
10.
Label the tubes with aliquots of silica binding solution (SBS, 700 μl) with the sample name.
-
11.
Transfer liquid into appropriately labelled SBS-containing tubes (see Note 14).
-
12.
Completely suspend the silica powder by vortexing and inversion of tubes (approximately 20 s).
-
13.
Incubate 15 min at RT (on a shaker at 400 rpm or invert tubes every 3 min by hand).
-
14.
Centrifuge at 16,000 × g for 3 min at RT to pellet the silica.
-
15.
Remove the supernatant with a pipette and discard (the DNA is bound to the silica at this stage).
-
16.
Add 500 μl of freshly prepared wash buffer to each tube (see Note 5).
-
17.
Completely suspend the silica powder by vortexing or inverting tubes (approximately 20 s).
-
18.
Centrifuge at 16,000 × g for 3 min at RT to pellet the silica. Remove supernatant and keep the pellet.
-
19.
Repeat steps 16–18.
-
20.
Centrifuge pellet for 30 s and remove any residual wash buffer with a pipette.
-
21.
Open the lid on tubes containing silica pellet and place in fume hood to fully dry pellets for 30 min (see Note 15).
-
22.
Add 200 μl TE buffer to each tube to elute the DNA. The DNA is now in the liquid buffer (see Notes 16 and 17).
-
23.
Completely suspend the silica powder by vortexing and inversion of tubes (approximately 20 s).
-
24.
Incubate at RT for 5 min.
-
25.
Centrifuge at 16,000 × g for 5 min at RT to pellet the silica.
-
26.
Label new 1.5 ml tubes with sample information.
-
27.
Collect liquid containing genomic DNA and place into new tubes.
-
28.
Store DNA temporarily at 4 °C before checking quality and quantity. The approximate cost per sample is between 42 and 64 cents depending on the chaotropic buffer used (see Table 14.1). Modifications can be made to the protocol to increase throughput (see Note 18).

Grinding of plant tissues. Leaf tissue is placed in a 2 ml tube in the presence of metal balls and optional sea sand (left panel). Use of desiccated tissue often results in higher-quality DNA compared to fresh tissue. Multiple tubes are then attached to a vortex mixer using tape (middle panel). Grinding is complete when a fine powder is produced (right panel). The presence of unground tissue with the powder does not affect the quality of extracted DNA
3.2 DNA Quantification
3.2.1 Preparation of DNA Concentration Standards and Running the Gel
-
1.
Prepare DNA concentration standards. Dilute lambda DNA to a set of DNA concentrations covering the expected range of the genomic DNA samples being assayed (see Note 6).
-
2.
Prepare a 1.5 % agarose gel in 0.5× TBE buffer with 0.2 μg/ml ethidium bromide or alternative staining dye (see Note 19).
-
3.
Add 3 μl of DNA sample plus 2 μl DNA loading dye. Use the same volumes for the DNA concentrations standards.
-
4.
Load samples and concentration standards on the gel.
-
5.
Run gel at 5–6 V/cm for 30–60 min. The DNA sample (genomic DNA band) should be completely out of the well and into the gel at least 0.2 cm. Do not run the gel too long as the genomic DNA band will become diffuse and hard to quantify. You may be able to accurately quantify degraded samples by altering electrophoresis conditions. Modifications can be made to increase throughput (see Notes 18 and 20).
3.2.2 Photographing the Gel and Quantification
-
1.
Adjust the image and take the longest possible exposure that does not saturate the image of any of the samples being assayed (see Note 21).
-
2.
Save this image in TIFF format (see Note 22 and Fig. 14.2).
-
3.
Start the ImageJ software.
-
4.
Open the TIFF image to be analysed (File > Open) (see Note 23 for an example image).
-
5.
Open the rotate dialogue box to straighten the image (Image> Transform> Rotate).
-
6.
In the rotate dialogue box, select ‘preview’, set the grid lines number to ‘30’, select one of the interpolation features (either ‘bilinear’ or ‘bicubic’) and adjust the angle in degrees until the bands are in line with the grid lines (see Note 24).
-
7.
When finished, click OK.
-
8.
Subtract background noise (Process> Subtract Background). Deselect ‘light background’. Ensure that rolling ball radius is set properly. A default of five pixels works for most mini gels (see Note 25).
-
9.
Select the magnifying glass tool in the ImageJ toolkit dialogue box and make the gel image as large as reasonable.
-
10.
Select the rectangle selection tool in the ImageJ toolkit dialogue box.
-
11.
Find the highest intensity or largest band on the gel to be analysed and draw a box around it. Partially degraded samples can still be quantified (see Fig.14.3 and Notes 20, 26 and 27).
-
12.
Left click and hold the mouse over the box and move it so that it is positioned around lane 1.
-
13.
Measure the box by hitting the ‘m’ key. A results table should appear with columns for sample number, area, mean, min and max values (see Note 28).
-
14.
Minimise the table so that you can again view the gel image.
-
15.
Move the box to lane 2 and hit the ‘m’ key (see Note 29).
-
16.
Continue to move the box to the next lane and hit the ‘m’ key until all the lanes in a gel tier are measured, including the standards.
-
17.
Evaluate the table to ensure every sample has the same area value. If not, redo the analysis.
-
18.
Check that the number of samples equals the number of lanes on the gel. If not, you either missed a lane or counted a lane more than once. If so, you need to start over.
-
19.
When you are satisfied that the table is correct, select the entire contents of the table (Ctrl + A), copy and paste into a spreadsheet program (e.g. Microsoft Excel, OpenOffice or LibreOffice Calc) (see Note 23).
-
20.
Prepare a standard curve (Insert> Charts> Scatter) using the ‘Mean’ values of the lambda standards from the table.
-
21.
Add a trend line (Chart> Add Trendline> Polynomial). Under type, click polynomial and select second order. Typically, the second-order polynomial is suitable (Fig. 14.4) (see Note 30 for the use of higher-order polynomial).
-
22.
Click Options and select Display equation on chart and Display r-squared value on chart.
-
23.
Click OK.
-
24.
Inspect the graph: are there any points that are clearly off of the trend? If so, consider removing this data point and redrawing the graph (Fig. 14.5).
-
25.
Use the equation in the graph to solve for the y value (DNA concentration). The X-axis is your density data collected from ImageJ.
-
26.
Use the spreadsheet formula tools to calculate the DNA concentrations in the samples (see Note 31). The results will be the DNA concentrations in ng/μl. Use the autofill function for the other samples.
-
27.
Visually compare the band intensities on the image with the concentrations estimated from the standard curve. Bands with lower intensity by visual inspection should have lower concentrations listed on the worksheet. If not, consider repeating the measurement.

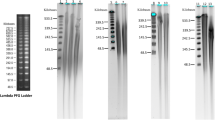
Example of genomic DNA with lambda DNA standards

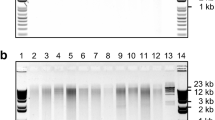
ImageJ analysis of cassava genomic DNA samples assayed on a high-throughput E-Gel 96 2 % agarose gel. Seven of 96 samples assayed are shown. Well positions on the 96-well sample plate are indicated for each sample along with the location of lambda DNA standards. Sample H09 shows extensive smearing indicating DNA degradation. This sample can be quantified by enlarging the size of the analysis box. Care should be taken when quantifying degraded DNA samples (see Notes 18 and 20). This gel image was produced in the Plant Breeding and Genetics Laboratory by Daniel Tello and Lina Kafuri

Example of standard curve with second-order polynomial trend line and equation

Example of correcting a standard curve when there is one erroneous data point. (a) The intensity value of the 10 ng/μl data point (red arrow) is considered to be wrong because the density value is much higher than expected (based on the trend line in Fig. 14.4, the 10 ng/μl should have a value closer to 23). (b) A correction is made by removing this data point and redrawing the standard curve. A curve more similar to that shown in Fig. 14.4 is achieved and concentration accuracy is improved
4 Notes
-
1.
While costly, tungsten carbide beads are durable and can be washed and reused many times, making the actual cost per extraction extremely low. An alternative is to use 3-mm steel ball bearings. Ball bearings can be purchased in bulk at a low cost allowing for disposal of bearings after a single use (cost per sample is less than 1 cent).
-
2.
Adding approximately 0.1 g (a pinch) of purified sea sand (e.g. Sigma-Aldrich Cat. No. 274,739) per sample can greatly improve tissue grinding.
-
3.
It takes several hours to prepare 6 M KI. Prepare in advance and avoid exposure to light by storing in brown bottle or foil-covered bottle. The buffer will eventually turn yellow but is still suitable for use.
-
4.
Alternative chaotropic buffers can be used at the same molarity. Examples include guanidine thiocyanate and sodium iodide. We have not observed any difference in DNA quantity or quality recovered using different buffers. The buffer represents the most expensive part of the assay (Table 14.1). We therefore recommend testing each buffer and choosing the one that can be purchased at lowest cost.
-
5.
Prepare fresh because salt precipitates during storage.
-
6.
Lambda DNA is a high molecular weight DNA at a precise concentration. Dilute this lambda stock in 1× TE to about ten different concentrations in the range between 2.5 and 150 ng/μl to serve as DNA concentration standards. The choice of the optimal DNA concentration standards depends on the concentrations of the sample DNAs. Calculate dilutions based on the information printed on the stock tube of the lambda DNA (note that the concentration of lambda DNA may vary from batch to batch). Store stocks at 4 °C.
-
7.
Prepare gel loading dye containing 30 % glycerol and colour dye. Do not use xylene cyanol as it migrates near the genomic DNA fragment and can interfere with quantification.
-
8.
Most computer operating systems are suitable (Windows, Macintosh, Linux).
-
9.
Download and install ImageJ software on your computer. Follow provided instructions for your operating system. The version used for this chapter was 1.49 v (Java version). The software is a free public domain program developed by Wayne Rasband of the National Institutes of Health, USA. Full documentation can be obtained from the website.
-
10.
Long-term storage of LSS is to be avoided to reduce proliferation of microorganisms that may lead to degradation of genomic DNA. Longevity may be prolonged by addition of an antimicrobial agent such as sodium azide. However, due to the low cost of silica and the ease of preparation, we suggest making fresh preparations monthly and avoiding the addition of toxic chemicals.
-
11.
Tubes containing SBS are typically prepared within 24 h of use. The effect of long-term storage has not been tested.
-
12.
The type and condition of starting tissue is a key component of the quality and quantity of extracted genomic DNA. We suggest tissue desiccation of leaf material using silica gel as outlined in Till et al. (2015).
-
13.
Alternative lysis buffers can be used if yields are low or sample degradation is observed. A description of alternative lysis buffers is provided in Till et al. (2015).
-
14.
Transfer liquid only. Avoid transferring solid material. We have observed that transfer of a minor amount of particulate from leaf preparations does not negatively affect quality or quantity of resulting genomic DNA.
-
15.
The colour of pellets turns from grey to white when dried. If a fume hood is not available, pellets can be dried for a longer period of time on the benchtop.
-
16.
A buffered solution is preferred over water to prevent sample degradation, e.g. TE buffer (Tris buffer maintains pH, while presence of EDTA chelates divalent cations and inactivates nucleases that could damage DNA).
-
17.
The amount of TE buffer used in elution can be adjusted if a higher sample concentration is desired. This should be determined empirically.
-
18.
Various modifications can be made to increase the throughput of sample processing. Reactions can be scaled to a 96-well format. Specialised equipment exists for grinding tissue in 96-well plates (e.g. the TissueLyser from Qiagen). In addition, modular 96-well rapid agarose gel electrophoresis such as the E-Gel 96 system (Thermo Fisher Scientific) can be loaded with multichannel pipettors and be used to electrophorese hundreds of samples in less than 10 min (see Fig. 14.3). Images from these gels are suitable for the quantification method described here. It is important to note that scaling reactions to a 96-well plate format will likely result in lower yields of DNA due to physical limitations on the weight of tissue that will fit into a well of a plate.
-
19.
Caution: Ethidium bromide is mutagenic. Wear gloves, lab coat and goggles. Dispose of gloves in toxic trash when through. Avoid contaminating other lab items (equipment, phones, door handles, light switches) with ethidium bromide. SYBR Safe DNA gel stain can be used instead of ethidium bromide.
-
20.
The genomic DNA band should be clear of the gel well for accurate quantification. If a sample is still partially in the gel well, quantification will be less accurate due to fluorescence that can occur at the well interface. Partially degraded samples can be quantified by extending the size of the analysis box (Fig. 14.3). However, it is often best to improve gel images by running samples again on a 3 % MetaPhor agarose gel (~10.5 g MetaPhor (Cambrex) in 350 ml 0.5× TBE) following manufacturer’s instructions. This allows for a sharper band to be produced for more accurate quantification. The aim is to quantify the entire DNA that can be used productively in PCR reactions. It is advisable to test PCR performance of degraded DNA prior to use as excessively nicked DNA may inhibit PCR amplification. If a high percentage of samples are degraded, consider changing lysis buffer (see Note 13) and make sure tissue is properly handled prior to extraction (Till et al. 2015).
-
21.
It is important to get a proper exposure of the gel that shows a difference in ethidium bromide staining in the concentration ranges you are assaying. The image of a reference sample of a higher concentration than any of the samples being assayed can be saturated. For example, if all of your samples are at 20 ng/μl, you should be able to observe a noticeable difference in the concentration standards around 20 ng/μl.
-
22.
Caution: Do not use compressed file formats such as JPEG.
-
23.
Go to http://www-naweb.iaea.org/nafa/pbg/public/manuals-pbg.html to download an example of DNA gel image and an example of DNA concentration workbook with data added. The example of DNA concentration workbook shows how a spreadsheet can be used to semiautomate the task of concentration determination. The second worksheet in this workbook shows how formula can be applied to group (‘bin’) samples of similar concentration together. This is especially useful when normalising DNA samples to a common concentration prior to sample pooling (such as in TILLING assays). Binning samples can dramatically reduce the number of pipetting volumes to adjust samples to a common concentration.
-
24.
You can use positive values for clockwise rotation and negative values (by placing a minus ‘−’ sign) for counterclockwise rotation. You may have to use a decimal setting to get the lanes to line up.
-
25.
Information on rolling ball radius settings can be found at http://imagej.nih.gov/ij/docs/menus/process.html#background. In general, your image should be completely black with the exception of DNA-dye-stained bands. Try several different pixel radii to see how your gel looks. Notice that when the number is very high, you can see the outline of your gel. If you do not fully subtract these pixels, they will be counted and your DNA concentration calculations will be less accurate.
-
26.
ImageJ reports the pixels in the box. Variation from counting background pixels is controlled for by keeping the box size constant.
-
27.
Caution: Make sure that the box surrounds the entire signal but does not overlap with the signal from another band. Failure to do so will lead to an inaccurate reading.
-
28.
‘Area’ is area of selection in square pixels or in calibrated square units. ‘Mean’ is the average grey value within the selection (sum of the grey values of all the pixels in the selection divided by the number of pixels). ‘Min’ and ‘max’ values are minimum and maximum grey values within the selection.
-
29.
Caution: Do not change the size of the box. You must measure the same volume of the box for each lane. If you accidentally change the size of the box while measuring lanes, start over.
-
30.
Optional: You may try a higher-order polynomial to evaluate how differences in curve fitting can affect your concentration estimation. For example, the third-order polynomial for the graph in Fig. 14.4 is the formula:
y = − 0.0002 × x 3 + 0.0244 × x 2 − 0.2174 × x + 3.3653.
-
31.
For example, using the graph in Fig. 14.4, if the density data for your sample is in cell D5, select an empty cell in row 5 and enter the following formula:
y = 0.0133 × D5 × D5 + 0.0183 × D5 + 1.9631.
The result will be the concentration in ng/μl. Select the cell and drag the right corner down to autofill for other samples. See Note 23 for examples.
References
Avery OT, Macleod CM, McCarty M (1944) Studies on the chemical nature of the substance inducing transformation of pneumococcal types: induction of transformation by a desoxyribonucleic acid fraction isolated from pneumococcus type III. J Exp Med 79(2):137–158
Dahm R (2005) Friedrich Miescher and the discovery of DNA. Dev Biol 278(2):274–288
Jankowicz-Cieslak J, Huynh OA, Bado S, Matijevic M, Till BJ (2011) Reverse-genetics by TILLING expands through the plant kingdom. Emirates J Food Agric 23(4):290–300
Schneider CA, Rasband WS, Eliceiri KW (2012) NIH Image to ImageJ: 25 years of image analysis. Nat Methods 9(7):671–675
Till BJ, Jankowicz-Cieslak J, Huynh OA, Beshir M, Laport RG, Hofinger BJ (2015) Low-cost methods for molecular characterization of mutant plants. Springer, Switzerland
Varshney RK, Terauchi R, McCouch SR (2014) Harvesting the promising fruits of genomics: applying genome sequencing technologies to crop breeding. PLoS Biol 12(6):e1001883
Acknowledgements
Funding for this work was provided by the Food and Agriculture Organization of the United Nations and the International Atomic Energy Agency through their Joint FAO/IAEA Programme of Nuclear Techniques in Food and Agriculture. This work is part of IAEA Coordinated Research Project D24012. We thank Sana’a Khraisat for assistance in preparation of Fig. 14.1 and Daniel Tello and Lina Kafuri for the gel image used to produce Fig. 14.3.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Open Access This chapter is distributed under the terms of the Creative Commons Attribution-Noncommercial 2.5 License (http://creativecommons.org/licenses/by-nc/2.5/) which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
The images or other third party material in this chapter are included in the work’s Creative Commons license, unless indicated otherwise in the credit line; if such material is not included in the work’s Creative Commons license and the respective action is not permitted by statutory regulation, users will need to obtain permission from the license holder to duplicate, adapt or reproduce the material.
Copyright information
© 2017 International Atomic Energy Agency
About this chapter
Cite this chapter
Huynh, O.A., Jankowicz-Cieslak, J., Saraye, B., Hofinger, B., Till, B.J. (2017). Low-Cost Methods for DNA Extraction and Quantification. In: Jankowicz-Cieslak, J., Tai, T., Kumlehn, J., Till, B. (eds) Biotechnologies for Plant Mutation Breeding. Springer, Cham. https://doi.org/10.1007/978-3-319-45021-6_14
Download citation
DOI: https://doi.org/10.1007/978-3-319-45021-6_14
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-45019-3
Online ISBN: 978-3-319-45021-6
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)