
Abstract
Core activities of genebank operations include the preservation of germplasm identity and maintenance of genetic integrity. Some organisms such as banana are maintained by tissue culture that can foster accumulation of somatic mutations and loss of genetic integrity. Such changes can be reflected in their genome structure and thus be revealed by sequencing methods. Here, we propose a protocol for the detection of large chromosomal gains and/or losses that was applied to in vitro banana accessions with different levels of ploidy. Mixoploidy was detected in triploid (3x) accessions with chromosomal regions being diploid (2x) and tetraploid (4x) and in diploid accessions (2x) where large deletions resulted in partial haploidy (1x). Such abnormal molecular karyotypes can potentially explain phenotypic aberrations observed in off type material. With the affordable cost of Next Generation Sequencing (NGS) technologies and the release of the presented bioinformatic pipeline, we aim to promote the application of this methodology as a routine operation for genebank management as an important step to monitor the genetic integrity of distributed material. Moreover, genebank users can be also empowered to apply the methodology and check the molecular karyotype of the ordered material.
You have full access to this open access chapter, Download chapter PDF
Similar content being viewed by others


Keywords
1 Introduction
Somaclonal variation describes random cellular changes in plants regenerated through tissue culture. It occurs in certain crops that undergo micropropagation, and has been recorded in different explant sources, from leaves and shoots, to meristems and embryos. Banana (Musa spp.) is a clonal crop that can be conserved and multiplied in vitro. Somaclonal variations have been observed in banana after prolonged periods of in vitro culture and after intensive multiplication phases, both resulting in increased rates of subculturing for a given clone. Although somaclonal variation can result in advantageous mutations that can be useful for the genetic improvement of banana, it is undesirable in the context of micropropagation and plant conservation. This type of variation indeed is a problem for genebank managers, whose objectives are to maintain the genetic integrity of their collections for subsequent research and breeding purposes, thus preserving genetic resources for future generations.
The International Musa Germplasm Transit Centre (ITC), managed by the Alliance of Bioversity-CIAT and hosted at the Katholieke Universiteit Leuven in Belgium, is the world’s largest collection of banana germplasm with more than 1600 accessions of cultivated and wild species of banana (Ruas et al. 2017; de Langhe et al. 2018). It ensures the long-term conservation of a wide banana genepool and supports germplasm distribution all over the world. Due to the vegetative mode of propagation, banana accessions are kept in vitro under slow-growth conditions and regenerated through tissue culture. Although stress during the in vitro process is minimized by optimized multiplication and growing conditions, somaclonal variations have been observed. To avoid the conservation, and distribution, of material that holds such variations, and therefore ensure the maintenance of the genetic integrity of the germplasm, the ITC has developed the Field Verification exercise. This exercise aims at monitoring the genetic integrity of its banana accessions and combines evidence from morphological and molecular characterization to determine genetic integrity. To do so, plantlets that were maintained in vitro for more than 10 years are sent to the Musa Genotyping Centre (MGC) and to a partner field collection, USDA-ARS. In the lab, leaves are analyzed by flow cytometry and SSR markers, while in the field, plants are grown, characterized and photo documented. Once all results are obtained after a year or two, a panel of taxonomists check the morphological and molecular data and compare them to known reference information for each accession. Given that plants that have undergone somaclonal variations are expected to change in morphology, from changes in color to obvious growth issues, the panel of experts then assesses whether the accession is True-To-Type (TTT) or Off-Type (OT) (Chase et al. 2014). A major limitation of this process is the amount of time that is necessary to grow the plants and document them. It is also cumbersome to request the availability and expertise of key experts on a voluntary basis.
To fasten the process, the development of early screening methods is therefore of great interest for the community. To overcome the plant phenotyping bottleneck, investigation of modifications at the genome level has been targeted (Sahijram et al. 2003; Oh et al. 2007). The molecular basis of somaclonal variation is not precisely known, but both genetic and epigenetic mechanisms have been proposed (Kaeppler et al. 2000). Somaclonal variants in plants can be the result of various types of mutations such point mutations, gene duplication, transposable elements activation or large chromosomal rearrangements (in number and structure) (Bairu et al. 2011).
Current advances in Next Generation Sequencing (NGS) technologies and associated Genotyping-by-Sequencing methods allow the generation of high-throughput genetic markers for a large number of samples in a fast and cost-effective way. These datasets can therefore be used to study many events, including large chromosomal variations as recently reported in wheat and barley (Keilwagen et al. 2019). Such chromosome changes (e.g. aneuploidy) have also recently been reported in bananas (Cenci et al. 2019, 2021; Baurens et al. 2019, 2020). Although not all these variations may be the cause of off-type phenotypes, likely due to polyploidy that can mitigate them, it remains a change in DNA integrity of the plant that should be flagged, as well as a good reason for accession regeneration from backup or reintroduction with new material. These large chromosomal variations will be the focus of this chapter.
The protocol described here provides an early detection method for large chromosomal indels applied to material which can be obtained at ITC. This method could be used as a routine operation to check the genetic integrity of germplasm conserved in vitro in genebanks, and also in tissue culture laboratories. It can also be used with other methods for the quick testing of variations in mutants.
2 Materials
2.1 Plant Material
Passport data of accessions held at the ITC are published on the Musa Germplasm Information System (MGIS; www.crop-diversity.org). Available material from the ITC collection can be ordered at no cost for research, education and breeding purposes using a straightforward workflow (Fig. 9.1).

Workflow to order banana samples from the International Musa Germplasm Transit Centre (ITC) via the Musa Germplasm Information System (MGIS)
The MGIS website offers several options to search for germplasm that presents specific criteria such as taxonomy, country of origin, and it is also possible to identify accessions that have been evaluated for pest and diseases resistance, biotic or abiotic stresses and genome composition. To do so:
- 1.
-
2.
First refine your search by selecting “Yes” with the exchange availability filter.
-
3.
Use the other filters to refine your search based on other criteria.
-
4.
Click on “Add to my list icon”.
MGIS permits users to create a list of accessions that, once registered and if the material is available for distribution, can be ordered online.
-
5.
Go to “My list” on the top right menu and double check the list selected accession(s).
-
6.
Click on the “order Germplasm material” button.
-
7.
If this is your first time on the website, create an account and indicate an accurate address to ensure the proper delivery of the plants.
-
8.
Fill in the order form.
The Musa Online Ordering System (MOOS) is a three-step process which generates at the end of the process a Standard Material Transfer Agreement (SMTA) in PDF format that is automatically sent to the curator of the ITC collection for preparation of the material.
-
9.
Select the appropriate SMTA acceptance method:
-
(a)
Signature: the document should be printed and signed by a person authorized to sign on behalf of the organization then sent to ITC for counter signature.
-
(b)
Shrink-wrap: by accepting the parcel, the Recipient is accepting the terms of the SMTA attached.
-
(c)
Clickwrap: you accept the SMTA online by clicking. It works the same way as when you order goods or services from other internet web sites.
-
(a)
-
10.
Please indicate under what form you want to receive the materials.
Here we recommend selecting lyophilized leaf tissue which is the fastest way to receive it if your interest is DNA extraction for GBS or RADseq. Fresh leaves, that can be obtained by growing rooted plantlets, are recommended for whole genome sequencing.
-
11.
Select the level of payment (article 6.7 or 6.11).
Although the material is distributed free of charge, several options exist to Enhance Benefit-Sharing if done for commercial purposes.
-
12.
Indicate the purpose of the use.
-
13.
Select your affiliation, professional and shipping address.
-
14.
Indicate whether an import permit is needed.
-
15.
Submit.
2.2 Data Generation
To detect changes in chromosome ploidy, SNP ratio changes need to be monitored along the chromosomes. It is therefore necessary to choose a technology that will generate high-throughput SNP-based markers for genome-wide marker discovery. Technologies based on restriction enzyme-mediated genome complexity reduction such as Genotyping by Sequencing (GBS) (Elshire et al. 2011), Restriction Site Associated DNA Sequencing (RADseq) (Davey et al. 2010), Diversity Arrays Technology Sequencing (DArTseq™) (Kilian et al. 2012) have advantages and disadvantages (Table 9.1) and are all appropriate to detect aneuploidy events. The choice of the technology can be influenced by existing datasets, in-house facilities or affordable solutions offered by service providers. Therefore, we don’t provide a protocol for data generation but list the main points related to the main methods previously tested for such analyses.
3 Methods
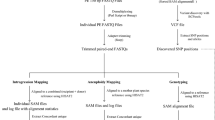
The workflow described in Figs. 9.2 and 9.3 shows the different steps involved in the analysis. It is composed of two main processes. The aim of the first part is to map data DNA from DArTseq, GBS, RADseq and RNA from RNAseq onto a reference genome sequence (Musa acuminata genome (D’Hont et al. 2012; Martin et al. 2016), or M. balbisiana genome (Wang et al. 2019)), and perform a SNP calling. The second part of the pipeline uses the VCF file obtained from SNP calling to determine the genomic structure of the accessions and define their molecular karyotype in order to reveal possible ploidy change.
-
Step 1: Read cleaning, mapping, variant calling
-
Read quality check step: Fastqc – Cutadapt
-
Mapping reads on the reference: BWA – STAR
-
Variant discovery: GATK
-
-
Step 2: Merge data, Use VcfHunter to establish the molecular karyotype
-
Combine VCF sample
-
Filtering
-
Split VCF by chromosome
-
Generate figure and assignation to each genome
-
The preprocessing pipeline presented in Fig. 9.2 shows the different steps that are compatible with four different sequencing technologies as listed in the previous section. The pipeline has to be done sample per sample according to the best practice of the software GATK (McKenna et al. 2010) used for the SNP calling part.

Schematic overview of the bioinformatics workflow for SNP calling

SNP ratio calculation and visualization. Reads are mapped to the reference genome. For a given DNA base position, multiple reads will be aligned for a global coverage. SNP detected are assigned to a different color (corresponding to different genomes for hybrid species). SNP Frequency is calculated at each site (e.g. 0.5 = half of the reads display this allele) and then plotted on a graph according to their physical position along the chromosome. Variation of SNP frequency combined with SNP coverage along the chromosome indicate chromosome segment with ploidy change
-
Step 1: Read cleaning, mapping, variant calling
This pre-processing step performs the cleaning data to the SNP calling via the read mapping and should be performed on each individual in order to produce a single VCF by sample (to be combined in Step 2).
-
Read Quality Check
-
Quality reads: Control the quality of the raw Fastq file with FASTQC
Description: In order to verify the quality of data reads, FastQC allows to check the quality score of each base and the presence or absence of the adaptor used to build the library. Adaptor depends on the sequencing technology used. https://www.bioinformatics.babraham.ac.uk/projects/fastqc/
-
Trim low-quality base with Cutadapt
Description: According to the result of FastQC, Cutadapt (Martin 2011) trims low quality ends and removes adapters (Illumina). Website: http://cutadapt.readthedocs.io/en/stable/guide.html
-
Fixed Parameters
-
-b AGATCGGAAGAGC (universal sequence for Illumina). Sequence of an adapter that was ligated to the 5′ or 3′ end. The adapter itself is trimmed and anything that follows too if located at 3′ end.
-
-O 7: Minimum overlap length. If the overlap between the read and the adapter is shorter than LENGTH, the read is not modified. This reduces the no. of bases trimmed purely due to short random adapter matches.
-
-m 30: Discard trimmed reads that are shorter than 30.
-
--quality-base = 64: Assume that quality values are encoded as ascii (quality + QUALITY_BASE). The default (33) is usually correct, except for reads produced by some versions of the Illumina pipeline, where this should be set to 64. (Default: 33).
-
-q 20,20: Trim low-quality bases from 5′ and/or 3′ ends of reads before adapter removal. If one value is given, only the 3′ end is trimmed. If two comma-separated cutoffs are given, the 5′ end is trimmed with the first cutoff, the 3′ end with the second. The algorithm is the same as the one used by BWA (see documentation).
The tool generates a trimmed fastq file (*_cutadapt.fastq.gz) files for each accession.
Mapping Reads on the Reference
Description: Align reads on a reference genome (e.g. Musa acuminata ‘DH Pahang’), with BWA (Li and Durbin 2010) for DNA, and STAR (Dobin et al. 2013) for RNA.
DNA Data: Map with BWA with default parameters with BWA-MEM.
Different types of genomic data such as DArTSeq, GBS and RADseq can be used (Fig. 9.2).
The tool generates a sam (*_.sam) files for each accession.
Website: http://bio-bwa.sourceforge.net/
RNA Data: Mapping with STAR in 2-pass mode.
Description: In the 2-pass mapping job, STAR will map the reads twice. In the first pass, the novel junctions will be detected and inserted into the genome indices. In the second pass, all reads will be re-mapped using annotated (from the GTF file given by the user) and novel (detected in the first pass) junctions. While this procedure doubles the run-time, it significantly increases sensitivity to novel splice junctions. In the absence of annotations, this option is strongly recommended.
The tool generates a folder for each accession, (names filled in column 3 “genome_name”) filled in the configuration file, which contained the SAM files (converted in BAM file) of aligned reads and a .final.out file of mapping statistics for each library. In addition, a (--prefix) folder containing a mapping statistics file (--prefix + mapping.tab) for all accession is generated.
Website: https://github.com/alexdobin/STAR
Variant Discovery
-
Add read group and (accession name from the fq.gz filename) sort BAM with Picard Tools
Description: This step replaces the reads Groups which describe the reads mapped on the reference, the sequencing technology, samples names, and library number are added.
-
ID = Read Group identifier (e.g. FLOWCELL1.LANE1).
-
PU = Platform Unit (e.g. FLOWCELL1.LANE1.UNIT1).
-
SM = Sample (e.g. DAD).
-
PL = Platform technology used to produce the read (e.g. ILLUMINA).
-
LB = DNA library identifier (e.g. LIB-DAD-1).
Website: https://broadinstitute.github.io/picard/
The tool generates a bam (*_rmdup.bam) file for each accession with the RG (Read Group) modified.
-
Mark duplicate reads and index BAM with MarkDuplicates from PicardTools
Description: PCR duplicate removal, where PCR duplicates arise from multiple PCR products from the same template molecule binding on the flow cell. These are removed because they can lead to false positive variant calls. Sort the BAM file and mark the duplicated reads.
Website: https://broadinstitute.github.io/picard/
The tool generate a bam (*_rmdup.bam) files for each accession with duplicated reads removed. In addition, a file named (--prefix + rmdup*stat.tab) file containing duplicate statistics for each accession was generated in the (--prefix) folder.
-
Index BAM with Samtools
Description: This step reorders the bam file according to the genome index position.
Website: http://samtools.sourceforge.net/
The tool generates a reordered (*_reorder.bam) and bai (*_reorder.bai) files for each accession.
-
Split ‘N CIGAR’ reads with SplitNCigarReads from GATK
Description: Splits reads that contain Ns in their cigar string (e.g. spanning splicing events in RNAseq data). Identifies all N cigar elements and creates k+1 new reads (where k is the number of N cigar elements). The first read includes the bases that are to the left of the first N element, while the part of the read that is to the right of the N (including the Ns) is hard clipped and so on for the rest of the new reads. Used for post-processing RNA reads aligned against the full reference.
Website: https://gatk.broadinstitute.org/hc/en-us/articles/360036727811-SplitNCigarReads
The tool generates a split and trimmed (on splicing sites) bam (*_trim.bam) and bai (*_trim.bai) files for each accession.
-
Realign indels with IndelRealigners from GATK (2 steps)
Description: The mapper BWA has some difficulties to manage the alignment close to the Indel. The step is not necessary with HaplotypeCaller but is necessary with UnifiedGenotyper. The tool generates a bam (*_realigned.bam) and bai (*_realigned.bai) files realigned around indel for each accession. This step is done with the GATK version 3.8.
The tool generates a bam (*_realigned.bam) and bai (*_realigned.bai) files realigned around indel for each accession.
-
Create a VCF file with HaplotypeCaller from GATK
Description: The HaplotypeCaller is able to call SNPs and indels simultaneously via local de-novo assembly of haplotypes in an active region. Whenever the program encounters a region showing variation, it discards the existing mapping information and completely reassembles the reads in that region. This allows the HaplotypeCaller to be more accurate when calling regions that are traditionally difficult to call.
Parameters:
-
--genotyping_mode DISCOVERY.
-
--variant_index_type LINEAR.
-
--variant_index_parameter 128,000.
https://gatk.broadinstitute.org/hc/en-us/articles/360036712151-HaplotypeCaller
Note: All these steps can be performed separately or with workflows reported in the literature (e.g. Toggle(Monat et al. 2015; Tranchant-Dubreuil et al. 2018)) or using the scripts we made available on GitHub at https://github.com/CathyBreton/Genomic_Evolution, which follow all the steps.
The tools are developed in Perl, bash, Python3, Java and work on the Linux system and require:
-
Bamtools v2.4, https://github.com/pezmaster31/bamtools
-
BWA v0.7.12, http://bio-bwa.sourceforge.net
-
STAR v2.5, https://github.com/alexdobin/STAR
-
Picard Tools v2.7, https://broadinstitute.github.io/picard/
-
Samtools v1.2, https://github.com/samtools/samtools
-
VCFHunter v1, https://github.com/SouthGreenPlatform/VcfHunter
Description: Get one FASTQ file ready for SNP calling per accession from raw sequence data (fastq.gz files).
USAGE: <Technique>_<Type_of_data>_fastq_to_vcf_job_array_Total_GATK4.pl -r ref.fasta -x fq.gz -cu accession
Parameters:
-
-r (string): reference FASTA filename.
-
-x (string): file extension (fq.gz).
-
-cu (string): cultivar.
-
Step 2: Molecular Karyotyping and Ploidy Change Detection
Based on known sequence variability, SNP variants can be assigned to the ancestral genomes in order to plot the genome allele coverage ratio and to calculate the normalized site coverage along chromosomes as described in (Baurens et al. 2019). This method can detect chromosome changes such as homoeologous exchanges (Cenci et al. 2021) but also is powerful enough to detect ploidy variation along the chromosomes as illustrated in Fig. 9.3.
The method has been developed within the VCFHunter software (Baurens et al. 2019) and can be used with the following procedure (Fig. 9.4).

Pipeline to determine the genome structure with VCFHunter
-
Merge datasets
Script name: CombineVcf.pl.
Description: The script merges and prepares the final VCF file, this step combines multiple VCF and performs pre-filtering using GATK. The samples to analyze are combined to the reference samples to allow allele assignation. Reference samples are representative genotypes that are relevant to the identification of your samples (i.e. acuminata or balbisiana without admixture). Whenever necessary, such SNP datasets can be downloaded on MGIS (https://www.crop-diversity.org/mgis/gigwa with RADseq_ABB_AB datasets) via GIGWA (Cenci et al. 2021; Sempéré et al. 2019).
USAGE: perl CombineVcf.pl -r reference_fasta -p output_prefix -x extension_file_to_treat.
Parameters:
-
-r (string): reference FASTA filename.
-
-p (string): VCF output prefix.
-
-x (string): file extension (VCF).
Website: https://github.com/CathyBreton/Genomic_Evolution
-
Filter SNP dataset
Script name: vcfFilter.1.0.py
Description: Filter VCF file based on most common parameters such as the coverage, missing data, MAF (minor allele frequency). The tool keeps bi-allelic sites and removes mono-allelic, tri-allelic, tetra-allelic sites.
USAGE: python3 vcfFilter.1.0.py --vcf file.prefiltered.vcf --prefix file.filtered --MinCov 8 --MaxCov 200 --MinAl 3 --MinFreq 0.05 --nMiss 50 --names All_names.tab --RmAlAlt 1:3:4:5:6:7:8:9:10 --RmType SnpCluster.
Website: https://github.com/SouthGreenPlatform/VcfHunter/blob/master/tutorial_DnaSeqVariantCalling.md
Parameters:
-
--MinCov, --MaxCov (int): min and max coverage for each genotype. If not, converted into missing data.
-
--MinAl (int): min coverage for each allele. If not, converted into missing data.
-
--MinFreq (float): minor allele frequency.
-
--nMiss (int): max number of accession with missing data--RmAlAlt : keeping diallelic sites.
-
--RmType SnpCluster (string): remove SNP clusters (define as 2 adjacent SNPs).
-
--names (string): A file containing accession names (one per line) in the output file.
-
Split VCF by chromosome
Description: Generate a VCF file for each chromosome with VcfTools (Danecek et al. 2011), in order to obtain a representation (chromosome Painting) of the SNP position along each chromosome.
USAGE: vcftools --vcf file_filtered.vcf --chr chr01 --recode --out batchall_filt_chr01.
Parameters:
-
--chr (string): generate a VCF from a given chromosome.
-
--recode: generate a new VCF file.
-
--out (string): output file name.
-
Generate molecular karyotype
Description: The program allows to perform a chromosome painting for all chromosomes of a given accession.
Script name: vcf2allPropAndCov.py.
USAGE: python3 vcf2allPropAndCov.py --conf <chromosomes.conf> --origin <origin.conf> --acc <sample_name> --ploidy 2
Parameters:
-
--acc: name of the sample to be analyzed (as in the VCF file).
-
--conf chromosomes.conf: list of VCF files for the chromosomes to be processed.
-
--origin origin.conf: name of the sample to be used for allele attribution.
-
--ploidy: expected ploidy of the sample (e.g. 2, 3 or 4).
The tool generates the 4 following files:
-
_AlleleOriginAndRatio.tab is a file describing each grouped allele, its origin and the proportion of reads having this allele at the studied position in the accession.
-
_stats.tab is a file reporting statistics on SNP sites used, sites where an allele is attributed to each group and alleles number attributed to each group in the accession.
-
_Cov.png is a figure showing read coverage along chromosomes (see figure below for interpretation).
-
_Ratio.png: is a figure showing grouped allele read ratio along chromosomes (see figure below for interpretation).
Website: https://github.com/SouthGreenPlatform/VcfHunter/blob/master/tutorial_ChromosomePainting.md
-
Use Cases: Application to Accessions at ITC
This section describes several examples of samples across the banana taxonomy that were processed by our method and allowed the detection of large chromosome variations.
-
Chromosomal changes in allotriploids (AAB, ABB).
Comprehensive characterization of the ABB samples has been conducted on ITC materials, revealing the genome structure or molecular karyotypes of most of the existing taxonomic subgroups (Cenci et al. 2021). Using VCFHunter on RADseq data, we were able to uncover patterns of chromosome segment recombinations between A and B genomes for most of the accessions. Among them, one displayed a clear case of chromosome number change as illustrated on Fig. 9.5. For this genotype belonging to the Bluggoe subgroup, no SNP variant was assigned to the A subgenome (in green) along the whole chromosome 8. Most of the B variants (in red) were located at the top of the diagram (value = 1) with an unimodal distribution for residual B genome heterozygosity around 0.5 (instead of 0.33/0.67 expected in the presence of three B chromosome 8). Moreover, the diagram on the right shows a lower SNP density coverage in comparison to all the other chromosomes, showing that chromosome 8 was diploid (2x) for this accession. This pattern observed here is due to the loss of the A genome version of chromosome 8. For comparison, chromosome 4 exhibits two regions with irregular patterns compared to expectation for an ABB. One is located on the first arm of the chromosome and the second one is placed on the distal part of the second arm. However, in both regions, allelic frequencies for heterozygous sites are consistent with triploidy (0.33/0.67) and no distortion of the coverage density is observed.

Molecular karyotype of an ABB banana cultivar. On the left, SNP ratio distribution along the chromosomes. Each SNP is illustrated by a dot and assigned to a genome by a color (A = green, B = red). The red arrows with values (0.33/0.5/0.67) refer to the SNP frequency ratio. On the right, read coverage at SNP positions along the chromosomes. Heterozygous SNP frequency distribution around 0.5 and lower SNP coverage along the whole chromosome 8 indicate a BB pattern (loss of chromosome 8A) in ‘Dole’ (ITC0767)
Two additional examples of aneuploidy detection in ABB are illustrated in Fig. 9.6. In the accession ‘Simili Radjah’ (ITC0123, ABB), the loss of the second arm in one of the B chromosomes of chromosome 5 can be inferred by the SNP frequency at 0.5 for both A and B assigned SNP and by the lower SNP density coverage compared to the first arm (Fig. 9.6a). In the accession ‘INIVIT PF-2003’ (ITC1600, ABB), in the second arm of chromosome 10, an interstitial region appears to have SNP frequency at 0.5. Since the SNP density coverage in this region is higher than in the remaining chromosome having 1A and 2B chromosomes, the duplication of the interstitial region in chromosome A was deducted, being the SNP ratio in this region 2A:2B.

Patterns of chromosome loss and gain in Musa ABB (A = blue, B = red, read coverage = black). (a) Pattern of ‘Simili Radjah’ (ITC0123) chromosome 5, showing frequency of A and B variants (y axes) around 0.5 and lower SNP coverage on the second arm of one B chromosome, indicates AB pattern (loss of one B chromosome 5 second arm). (b) Pattern of ‘INIVIT PB-2003’ (ITC1600) chromosome 10, containing an interstitial region showing frequency of A and B variants (y axes) around 0.5 and higher read coverage indicates AABB pattern (duplication of A chromosome interstitial region)
Finally, other examples were also detected in AAB Plantain cultivars (Fig. 9.7). We observed in ‘Nzumoigne’ (ITC0718), a SNP frequency of A and B alleles at 0.5 on chromosome 2 that was combined to lower SNP density coverage (compared to other chromosomes as exemplified with chromosome 3) (Fig. 9.7a). In ‘Ihitisim’ (ITC0121), multiple events were detected including a chromosome gain on chromosome 3 with SNP ratio of ~0.25 and ~ 0.75, supported by a higher SNP coverage and a partial loss at the beginning of the chromosome 4 (Fig. 9.7b).

Patterns of chromosome loss and gain in Musa AAB Plantains (A = green, B = red). (a) Pattern of ‘Nzumoigne’ (ITC0718) chromosome 3 with regular pattern (variants A and B with frequency at 0.67 and 0.33 (y axis), respectively) compared to chromosome 2 having both ends with A and B variants at 0.5 frequency. The pattern indicates loss of both ends of one A chromosome 2. (b) A and B variant frequencies (y axes) in chromosome 3 (0.75 and 0.25, respectively) and read coverage higher in chromosome 3 than in chromosome 4 indicate the presence of an additional chromosome 3A in ‘Ihitisim’ (ITC0121)
-
Chromosomal changes in diploid banana accessions (AA).
The survey of more than 200 AA accessions with the same procedure revealed molecular karyotypes corresponding to chromosome arms with large interstitial or terminal deletions for a few individuals (Fig. 9.8).

Chromosome pattern of mutated chromosomes in cultivated AA diploid accessions. (a) Patterns of ‘No.110’ (AA, ITC0413) second arm terminal region of chromosome 5. Absence of heterozygous variants and lower read coverage indicates terminal deletion in one copy of second arm. (b) Patterns of ‘Pahang’ (AA, ITC0727) second arm interstitial region of chromosome 8. Absence of heterozygous variants and lower read coverage indicates interstitial deletion of a copy on second arm
4 Notes
-
1.
Lyophilized leaf tissue allows sufficient DNA quality extraction for GBS, RADseq or DArtSeq (Doyle et al. 1990).
-
2.
We recommend using the PSTI restriction enzyme for GBS, RADseq or DArTseq (Chan et al. 2014).
-
3.
From our experience, GBS will generate fewer markers than RADseq but higher read coverage (Hueber et al. 2015).
-
4.
A minimum read coverage of 10x by haplotype would be required to support SNP detection statistically (e.g. 30x for triploids).
-
5.
SNP calling by individual and then merge is more efficient for SNP calling. However, SNP calling can be performed in different way and used directly with Step 2 on molecular karyotyping (https://github.com/CathyBreton/Genomic_Evolution).
-
6.
The pipeline provides graphical output for interpretation of insertion deletions by chromosome. A workflow is developed in github (https://github.com/CathyBreton/Genomic_Evolution).
-
7.
The method can detect aneuploidy. In case of partial gain on a chromosome, as this is based on allelic ratio mapped of the reference genome, the type of event such as insertion or duplication cannot be distinguished. Inversion and translocation are not identified. (Cenci et al. 2019)
-
8.
Large chromosome variations provide information on the loss of genetic integrity. However, those events in banana seems to not be systematically synonyms of somaclonal variations leading to change in observed phenotype. Further research is needed to clarify the importance of the events linked to the loss of True-To-Typeness status.
References
Bairu MW, Aremu AO, Van Staden J (2011) Somaclonal variation in plants: causes and detection methods. Plant Growth Regul 63:147–173. https://doi.org/10.1007/s10725-010-9554-x
Baurens F-C, Martin G, Hervouet C, Salmon F, Yohomé D, Ricci S et al (2019) Recombination and large structural variations shape interspecific edible bananas genomes. Mol Biol Evol 36:97–111. https://doi.org/10.1093/molbev/msy199
Busche M, Pucker B, Viehöver P, Weisshaar B, Stracke R (2020) Genome sequencing of Musa acuminata dwarf Cavendish reveals a duplication of a large segment of chromosome 2. G3: genes, genomes. Genetics 10:37–42. https://doi.org/10.1534/g3.119.400847
Cenci A, Hueber Y, Zorrilla-Fontanesi Y, van Wesemael J, Kissel E, Gislard M et al (2019) Effect of paleopolyploidy and allopolyploidy on gene expression in banana. BMC Genomics 20:244. https://doi.org/10.1186/s12864-019-5618-0
Cenci A, Sardos J, Hueber Y, Martin G, Breton C, Roux N, Swennen R, Carpentier SC, Rouard M (2021) Unravelling the complex story of intergenomic recombination in ABB allotriploid bananas, Ann Bot 127(1):7–20, https://doi.org/10.1093/aob/mcaa032
Chan A, Xavier Perrier, Christophe Jenny, Jean-Pierre Jacquemoud-Collet, Mathieu Rouard, Julie Sardos, Nicolas Roux, Christopher D Town (2014) Using genotyping-by-sequencing to understand Musa diversity. Poster 449 PAG XXIII Conference. San Diego (USA)
Chase R, Sardos J, Ruas M, Van den Houwe I, Roux N, Hribova E, et al (2014) The field verification activity: a cooperative approach to the management of the global Musa in vitro collection at the international transit Centre. In: XXIX international horticultural congress on horticulture: sustaining lives, livelihoods and landscapes (IHC2014): IX 1114, pp 61–66
D’Hont A, Denoeud F, Aury J-M, Baurens F-C, Carreel F, Garsmeur O et al (2012) The banana (Musa acuminata) genome and the evolution of monocotyledonous plants. Nature 488:213
Danecek P, Auton A, Abecasis G, Albers CA, Banks E, DePristo MA et al (2011) The variant call format and VCFtools. Bioinformatics 27:2156–2158. https://doi.org/10.1093/bioinformatics/btr330
Davey JW, Davey JL, Blaxter ML, Blaxter MW (2010) RADSeq: next-generation population genetics. Brief Funct Genomics 9:416–423. https://doi.org/10.1093/bfgp/elq031
de Langhe E, Laliberte B, Chase R, Domaingue R, Horry JP, Karamura D, et al. The 2018 The 2016 Global strategy for the conservation and use of Musa genetic resources – key strategic elements. Acta Hortic 71–78. doi:https://doi.org/10.17660/ActaHortic.2018.1196.8
Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S et al (2013) STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29:15–21. https://doi.org/10.1093/bioinformatics/bts635
Doyle JJ, Doyle JL (1990) A rapid total DNA preparation procedure for fresh plant tissue. Focus: 13–15
Elshire RJ, Glaubitz JC, Sun Q, Poland JA, Kawamoto K, Buckler ES et al (2011) A robust, simple genotyping-by-sequencing (GBS) approach for high diversity species. PLoS One 6:e19379. https://doi.org/10.1371/journal.pone.0019379
Hueber Y, Sardos J, Hribova E, Van den Houwe I, Roux N, Rouard, M (2015) Application of NGS-generated SNP data to complex crops studies: the example of Musa spp. (banana). Poster presented at Plant and Animal Genome - PAG XXIII Conference. San Diego (USA) 10–14
Kaeppler SM, Kaeppler HF, Rhee Y (2000) Epigenetic aspects of somaclonal variation in plants. Plant Mol Biol 43:179–188. https://doi.org/10.1023/A:1006423110134
Keilwagen J, Lehnert H, Berner T, Beier S, Scholz U, Himmelbach A et al (2019) Detecting large chromosomal modifications using short read data from genotyping-by-sequencing. Front Plant Sci 10. https://doi.org/10.3389/fpls.2019.01133
Kilian A, Wenzl P, Huttner E, Carling J, Xia L, Blois H et al (2012) Diversity arrays technology: a generic genome profiling technology on open platforms. Methods Mol Biol 888:67–89. https://doi.org/10.1007/978-1-61779-870-2_5
Li H, Durbin R (2010) Fast and accurate long-read alignment with burrows-wheeler transform. Bioinformatics 26:589–595. https://doi.org/10.1093/bioinformatics/btp698
Martin M (2011) Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnetjournal 17:10. https://doi.org/10.14806/ej.17.1.200
Martin G, Baurens F-C, Droc G, Rouard M, Cenci A, Kilian A et al (2016) Improvement of the banana “Musa acuminata” reference sequence using NGS data and semi-automated bioinformatics methods. BMC Genomics 17:243. https://doi.org/10.1186/s12864-016-2579-4
McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A et al (2010) The genome analysis toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 20:1297–1303. https://doi.org/10.1101/gr.107524.110
Monat C, Tranchant-Dubreuil C, Kougbeadjo A, Farcy C, Ortega-Abboud E, Amanzougarene S et al (2015) TOGGLE: toolbox for generic NGS analyses. BMC Bioinf 16:374. https://doi.org/10.1186/s12859-015-0795-6
Oh TJ, Cullis MA, Kunert K, Engelborghs I, Swennen R, Cullis CA (2007) Genomic changes associated with somaclonal variation in banana (Musa spp.). Physiol Plant 129:766–774. https://doi.org/10.1111/j.1399-3054.2007.00858.x
Ruas M, Guignon V, Sempere G, Sardos J, Hueber Y, Duvergey H et al (2017) MGIS: managing banana (Musa spp.) genetic resources information and high-throughput genotyping data. Database (Oxford) 2017. https://doi.org/10.1093/database/bax046
Sahijram L, Soneji JR, Bollamma K (2003) Analyzing somaclonal variation in micropropagated bananas (Musa spp.). In Vitro Cell Dev Biol Plant 39:551–556
Sempéré G, Pétel A, Rouard M, Frouin J, Hueber Y, De Bellis F et al (2019) Gigwa v2—extended and improved genotype investigator. Gigascience 8. https://doi.org/10.1093/gigascience/giz051
Tranchant-Dubreuil C, Ravel S, Monat C, Sarah G, Diallo A, Helou L et al (2018) TOGGLe, a flexible framework for easily building complex workflows and performing robust large-scale NGS analyses. bioRxiv:245480. https://doi.org/10.1101/245480
Wang Z, Miao H, Liu J, Xu B, Yao X, Xu C et al (2019) Musa balbisiana genome reveals subgenome evolution and functional divergence. Nature Plants 1. https://doi.org/10.1038/s41477-019-0452-6
Acknowledgements
This work was supported by the CGIAR Fund, and in particular by the CGIAR Research Program Roots, Tubers and Bananas.
Author information
Authors and Affiliations
Corresponding authors
Editor information
Editors and Affiliations
Rights and permissions
The opinions expressed in this chapter are those of the author(s) and do not necessarily reflect the views of the [NameOfOrganization], its Board of Directors, or the countries they represent
Open Access This chapter is licensed under the terms of the Creative Commons Attribution 3.0 IGO license (http://creativecommons.org/licenses/by/3.0/igo/), which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the [NameOfOrganization], provide a link to the Creative Commons license and indicate if changes were made.
Any dispute related to the use of the works of the [NameOfOrganization] that cannot be settled amicably shall be submitted to arbitration pursuant to the UNCITRAL rules. The use of the [NameOfOrganization]'s name for any purpose other than for attribution, and the use of the [NameOfOrganization]'s logo, shall be subject to a separate written license agreement between the [NameOfOrganization] and the user and is not authorized as part of this CC-IGO license. Note that the link provided above includes additional terms and conditions of the license.
The images or other third party material in this chapter are included in the chapter's Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the chapter's Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.
Copyright information
© 2022 The Author(s)
About this chapter

Cite this chapter
Breton, C. et al. (2022). A Protocol for Detection of Large Chromosome Variations in Banana Using Next Generation Sequencing. In: Jankowicz-Cieslak, J., Ingelbrecht, I.L. (eds) Efficient Screening Techniques to Identify Mutants with TR4 Resistance in Banana. Springer, Berlin, Heidelberg. https://doi.org/10.1007/978-3-662-64915-2_9
Download citation
DOI: https://doi.org/10.1007/978-3-662-64915-2_9
Published:
Publisher Name: Springer, Berlin, Heidelberg
Print ISBN: 978-3-662-64914-5
Online ISBN: 978-3-662-64915-2
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)