
Abstract
Two classes of new materials possessing ion conductivity have been developed: a lithium ion conductor and a hydride ion conductor. Conventional perovskite and ordered rock-salt structures were adopted as frameworks for lithium migration, and electrochemically stable elements such as Al, Ga, Ta, and Sc were used in the materials to facilitate their use as low-potential negative electrodes. New compositions of (Li0.25Sr0.625V(Li,Sr)0.125)(Ga0.25Ta0.75)O3, and Li0.9Sc0.9Zr0.1O2 were found to be novel oxide-based lithium ion conductors. Oxyhydrides with K2NiF4-type structures were synthesized via a high-pressure synthesis method and their use in pure hydride ion conduction was demonstrated. The La2–x–y Sr x+y LiH1–x+y O3–y oxyhydrides showed wide composition ranges of solid solution formation and the conductivity increased with anion vacancies or the introduction of interstitial hydride ions. The performance of an all-solid-state TiH2/o-La2LiHO3 (x = y = 0, o: orthorhombic)/Ti cell provided conclusive evidence of pure H– conduction.
You have full access to this open access chapter, Download chapter PDF
Similar content being viewed by others


Keywords
1 Novel Solid-State Electrolytes
Solid materials exhibiting purely ionic conduction are used as solid-state electrolytes in a wide variety of electrochemical devices and chemical sensors, with the corresponding charge carriers being specific ions such as H+, Cu+, Ag+, Na+, Li+, F–, and O2–. The resulting charged ion flow in electrolytes creates an electric current that drives the device, the characteristics and performance of which are thus influenced by the nature of the charge carriers. Generally, in view of their small ionic radii, cations migrate easily in solid electrolytes, showing facile diffusion. For example, silver and copper ion solid electrolytes, such as RbAg4I5 and Rb4Cu16I7Cl13, show extremely high ionic conductivities of > 100 mS cm−1 at room temperature [1,2,3]. Moreover, the recently developed lithium ion conductors (Li10GeP2S12, LGPS) have achieved room temperature conductivities of >10 mS cm−1 [4, 5], with Li-based all-solid-state batteries reported to exhibit exceptionally good power characteristics. On the other hand, newly developed materials such as hydride ion conductors have expanded the research field and the scope of available energy devices [6, 7]. In this section, we focus on Li+ and H– as charge carriers and describe the structural characteristics of the corresponding newly developed materials.
2 Lithium Ion Conductors
Lithium ion conductors continue to attract much attention owing to their practical applications in all-solid-state lithium batteries [5, 8]. A wide variety of such conductors exists (e.g., LISICON, perovskite, garnet, glass, glass ceramics, thio-LISICON, and LGPS), some of which were developed in the 1970s [4, 9,10,11,12,13,14]. For instance, LGPS-based materials (σ > 10 mS cm−1 at 25 °C) enable high-power operation of solid-state lithium batteries; this is an intrinsic merit of solid-state systems, in addition to their safety and reliability. However, sulfide-based solids are sensitive to atmospheric moisture. As a result, most current research focuses on oxide-based materials, in order to satisfy the requirements of practical applications and engineering processes.
Novel ion conductors are typically developed using three methods: (i) element substitution-based, (ii) structure-based, and (iii) composition-based material searches. Approach (i) relies on existing materials with ionic conductivity of the target charge carrier [15], which are amenable to tuning of their physical and electrochemical properties [16]. Therefore, although it is relatively easy to find new materials using this method, remarkable performance improvements are difficult to achieve. Approach (ii) is initiated by selecting a suitable crystal structure candidate for ion diffusion [6, 11], which can be complicated by the fact that the diffusion of the target ion in the selected structure has usually not been demonstrated.
Finally, approach (iii) is the most challenging, but also has the greatest potential to afford new materials with unique structures and properties. This approach starts with the selection of a suitable phase diagram [17]. Subsequently, materials corresponding to the chosen region in this diagram are synthesized and characterized; in certain cases, they exhibit unique structures and properties [4, 18]. In this chapter, some examples of material searches are introduced.
2.1 Novel Lithium Ion-Conducting Perovskite Oxides [15]
Lithium ion-conducting solids are key materials for all-solid-state lithium batteries, which, compared with conventional liquid electrolyte-based lithium batteries, exhibit improved energy density, stability, safety, and reliability. Among the solid electrolytes that have been developed, the oxide-based ones are among the most promising candidates, owing to their high ionic conductivities and good chemical stabilities over a wide range of operating temperatures [11, 19]. Lithium ion-conducting perovskites such as La(2/3)–x Li3x TiO3 (which exhibits an ionic conductivity above 10−3 S cm−1 at room temperature) are considered to be particularly attractive [19]. However, the interfacial reduction of Ti+4 to Ti+3 during the electrochemical process or upon contact with lithium metal gives rise to undesirable electronic conduction. On this basis, novel perovskite-structured materials were examined, in typical example of an element substitution-based material search. As a result, the Li-Sr-Ta-M-O system (M = Al, Ga) was postulated to be ideal for achieving high ionic conductivity, with (Li x Sr1–x )(M (1–x)/2Ta(1+x)/2)O3 mixed oxides expected to exhibit superior characteristics owing to (i) the presence of largely non-reducible metals (Ta and Al/Ga) in their structures, (ii) the presence of a large cation (Sr) at the A-site, (iii) the limited distortion of BO6 octahedra exhibited by Ta at the B-site, (iv) the presence of a small B-site cation (Al, Ga), and (v) the availability of controlled vacancies introduced by adjusting the concentration of the B-site cation. Thus, (Li x Sr1–x )(M (1–x)/2Ta(1+x)/2)O3 (M = Al, Ga) and (Li x Sr1–x–y V(Li,Sr)y )(Ga[(1–x)/2]–y Ta[(1+x)/2]+y )O3 systems were synthesized by solid-state reactions involving Li-rich starting materials to obtain a single phase. These were subsequently subjected to electrochemical examination and crystallographic analysis by X-ray and neutron diffraction Rietveld analysis.
The temperature-dependent conductivities of (Li0.2Sr0.65V(Li,Sr)0.15)(Ga0.25Ta0.75)O3 (x = 0.2, y = 0.15) and (Li0.25Sr0.625V(Li,Sr)0.125)(Ga0.25Ta0.75)O3 (x = 0.25, y = 0.125) are shown in Fig. 13.1, with the corresponding activation energies (E a) calculated as 35.04 and 34.64 kJ mol−1, respectively. The comparable E a values of these systems indicate that they both feature the same Li+ conduction mechanism. The highest conductivity exhibited by the (Li0.25Sr0.625V(Li,Sr)0.125)(Ga0.25Ta0.75)O3 (x = 0.25, y = 0.125) sample equaled 1.85 × 10−3 S cm−1 at 250 °C, which was the highest value measured for Li-Sr-Ga-Ta-O perovskite materials. The structure of this sample was determined by powder neutron diffraction and the obtained data were refined using a structural model of a cubic perovskite-type material with Pm-3 m symmetry.

Arrhenius plot showing the temperature-dependent conductivities of (Li x Sr1–x–y V(Li,Sr)y )(Ga[(1–x)/2]–y Ta[(1+x)/2]+y )O3 with x = 0.2, 0.25 and y = 0.125, 0.15
Figure 13.2 shows the thermal ellipsoid structure model obtained by neutron Rietveld analysis and Table 13.1 summarizes the corresponding interatomic distances, revealing that the O–O distance of 2.79411 Å in (Li0.25Sr0.625V(Li,Sr)0.125)(Ga0.25Ta0.75)O3 was slightly larger than that determined for cubic perovskite-structured Li0.5La0.5TiO3 (2.7358 Å). This increase was most likely due to substitution by the larger Sr2+ cation at the A-site, which is responsible for widening the bottleneck for lithium ion diffusion in the structure. The A–O distance of 2.79411 Å was larger than the calculated sum of the Li and O ionic radii of 2.32 Å (Li+ (CN 8): 0.92 Å and O2– (CN 6): 1.4 Å), making the Li cation more ionic in nature and, therefore, more mobile. The tolerance factor t, calculated based on the ionic radius of Sr2+ at the A-site, equaled 0.9855, which was close to unity and indicated an ideal cubic perovskite-type structure.

Crystal structure model of (Li0.25Sr0.625V(Li,Sr)0.125)(Ga0.25Ta0.75)O3 based on neutron Rietveld analysis
The increased ionic conduction was confirmed to result from the introduction of vacancies at A-sites. Average bond valence sum (BVS) values were calculated for each site of the perovskite structure using refined structural data, with the average BVS for A-sites equaling 1.98. The larger average BVS of (Li0.25Sr0.625V(Li,Sr)0.125)(Ga0.25Ta0.75)O3 compared with that of La(2/3)-x Li3x TiO3 (0.95–1.57) [20] corresponded to the greater activation energy required for ion conduction in the former. Figure 13.2 shows the perovskite structure with thermal ellipsoids for each site, revealing their approximately isotropic thermal nature. The presence of vacancies in conjunction with Li ions at the A-sites suggests that Li diffusion proceeds according to the vacancy mechanism.
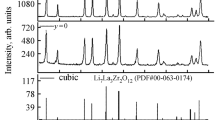
2.2 M-Doped LiScO2 (M = Zr, Nb, Ta) [21] as New Lithium Ion Conductors
No material has yet been discovered that satisfies all requirements imposed on lithium ion conductors as solid electrolytes for battery applications (i.e., high ionic conductivity at room temperature, chemical stability, electrochemical stability, thermal stability, and low cost). This clearly indicates the need for further research efforts in this direction. Herein, we focus on LiScO2, which has an ionic conductivity of 4 × 10−9 S cm−1 at 573 K. Although this value is not overly high, the above material is still attractive in view of its enhanced thermodynamic stability in contact with lithium metal [22].
As shown in Fig. 13.3, LiScO2 has a fractional cationic ordered rock-salt structure exhibiting tetragonal I41/amd symmetry [23], which has the potential to partially rearrange depending on the synthesis conditions and the doped element [24]. Although element doping is an effective method of increasing the ionic conductivities of solid lithium ion conductors such as LiScO2, no corresponding investigations have been reported. Thus, in an attempt to improve the ionic conductivity of LiScO2 by introducing lithium vacancies into its structure, this material was doped by M = Zr4+, Nb5+, and Ta5+, and the crystal structures and ionic conductivities of the thus prepared Li1−y Sc1−x M x O2 were evaluated in detail.

Crystal structure of LiScO2 [23], with blue octahedra and green spheres indicating ScO6 and Li, respectively
Li1−y Sc1−x M x O2 (M = Zr4+, Nb5+, or Ta5+; x = 0.1) were obtained via a solid-state reaction (sintering at 1073–1623 K for 1–12 h in air). Their impedance spectra and temperature-dependent conductivities are presented in Fig. 13.4. The conductivities were calculated from the corresponding impedance spectra, which comprised semicircles and spikes corresponding to contributions of the bulk and grain boundary and the electrode, respectively. The bulk and grain boundary contributions could not be separated and were therefore calculated together.

a Representative impedance plots at 623 K and b Arrhenius plots showing the temperature-dependent conductivities of doped Li1−y Sc1−x M x O2 (M = Zr4+, Nb5+, or Ta5+)
Resistances were calculated from the diameter of the aforementioned semicircles and used to determine conductivities. The diameters of these semicircles decreased upon doping, indicating the suitability of impedance spectroscopy to survey and evaluate ionic conductivities, with capacitance values corresponding to the observed semicircles being in the range of 10−10–10−12 F. Table 13.2 summarizes the ionic conductivities and activation energies of Li1−y Sc1−x M x O2 (x = 0.1) at 573 K, along with the values previously reported for LiScO2. All doped samples showed higher conductivities than the parent compound, owing to the formation of solid solutions upon aliovalent cation doping. Furthermore, this doping decreased the activation energies by more than 10%, indicating that the formation of lithium vacancies in the LiScO2 lattice reduced the energy barrier of lithium diffusion. Zr4+-doped samples showed the highest ionic conductivities, with a maximum value of 9.73 × 10−7 S cm−1 observed at 573 K. In order to verify the changes in ionic conductivities caused by Zr4+ doping, the corresponding crystal structures were evaluated in detail.
Tables 13.3 and 13.4 summarize the refinement-determined structural parameters for x = 0.0 and x = 0.1, respectively. All diffraction peaks were indexed to the I41/amd(141) space group with tetragonal symmetry, with the exception of reflections ascribed to impurities. The lattice parameters of LiScO2 were determined as a = b = 4.1791(18) Å and c = 9.3610(4) Å, making them nearly identical to the reported values of a = b = 4.182 Å and c = 9.318 Å [23]. The lattice parameters calculated for x = 0.1 (a = b = 4.1804(16) Å and c = 9.4186(3) Å) were increased by doping with Zr4+, with refinement results showing that 10% Zr4+ was doped at Sc3+ sites in the above structure, in agreement with the ratio of utilized reactants. Concomitantly, lithium vacancies were probably formed to maintain the charge balance in LiScO2, since the doped Zr4+ ion has a higher charge than Sc3+. These results demonstrate that the ionic conductivity of LiScO2 was markedly improved by substitution with certain aliovalent cations, owing to the resulting lattice expansion and formation of lithium vacancies.
3 Development of Hydride Ion Conductors
Hydride ion conduction is particularly attractive, as H– is similar in size to fast ionic conduction-suitable oxide and fluoride ions, while exhibiting strong reducing properties (standard H−/H2 redox potential = − 2.3 V), comparable to those of Mg/Mg2+ (−2.4 V) (Fig. 13.5). Thus, hydride ion conductors may be applied in energy storage/conversion devices with high energy densities. To indicate a new direction for next-generation battery systems beyond lithium ion batteries and fuel cells, we herein focus on hydride ion conduction in solids.

Standard redox potentials of typical charge carriers, showing that the highly negative potential of H2/H− is similar to that of Mg2+/Mg
Hydride ion conduction in CaH2 was first described by Andresen et al. in 1977 [25], with similar reports on other materials following in later years [26,27,28,29,30,31]. However, experimental evidence of H– conduction was not obtained until Irvine et al. determined the transport number of BaH2 by electromotive force measurements in 2015 [7]. Although alkaline earth metal hydrides such as BaH2 act as pure H– conductors, they are also strong reducing agents. This complicates their use as solid electrolytes of energy devices, in which electrochemical stability to both oxidation and reduction is required. Indeed, these metal hydrides have not yet been applied to battery reactions. From the viewpoint of material design, the structural inflexibility of metal hydrides complicates the control of their lattice structure (which is required to create smooth transport pathways) and their conducting hydride ion content. Thus, little progress has been achieved in the development of H– conductors. We have considered oxyhydrides, in which hydride and oxide ions share anion sublattices, as prospective hydride conductors with flexible anion sublattices. Known oxyhydrides include A 2 BH x O4–x (K2NiF4 structure; A = La, Ce, Nd, Pr, Sr; B = Co, V, Li; 0 < x ≤ 1), Sr3Co2O4.33H0.84 (Ruddlesden-Popper structure), ATiO3–x H x (perovskite structure; A = Ba, Sr, Ca) [32,33,34,35,36,37], and [Ca24Al28O64]4+·4H– (mayenite structure) [38,39,40]. However, none of these materials display pure H– conductivity, since hydride ions have been reported to act as electron donors in oxide-based materials [38,39,40,41,42], donating electrons to their lattice and thus causing electron conduction accompanied by a characteristic change in hydrogen charge from H– to H+. Indeed, perovskite- and mayenite-type oxyhydrides predominantly exhibit electron conduction caused by the dissociation of hydride ions into electrons and protons [33, 38,39,40, 43]. Taking this into consideration, preventing the above electron donation may be important for achieving pure H– conduction in the oxide framework structure. Herein, we attempted to synthesize a series of K2NiF4-type oxyhydrides, La2–x–y Sr x+y LiH1–x+y O3–y (0 ≤ x ≤ 1, 0 ≤ y ≤ 2, 0 ≤ x + y ≤ 2) featuring cation sublattices that contain cations that are more electron-donating than H– and anion sublattices that allow flexible storage of H–, O2–, and vacancies.
3.1 Hydride-Conducting Oxyhydrides La2–X–Y Sr x+Y H1–X+Y O3–Y
Novel La2–x–y Sr x+y LiH1–x+y O3–y oxyhydrides were synthesized by a high-temperature solid-state reaction in a cubic anvil cell [6] under high pressure to prevent the loss of light elements such as hydrogen, which can easily vaporize at high temperatures. The compositions and structures of La2–y Sr y LiH1+y O3–y (y = 0, 1, 2) were determined by X-ray and neutron Rietveld analyses (Fig. 13.6). In La2LiHO3 (x = y = 0). The two apical sites of LiX 6 (X = H–, O) octahedra were occupied only by O2–, with the four in-plane apexes occupied by O2– and H–. These results indicate that highly charged cations, i.e., La3+ and Sr2+, need to be surrounded by highly charged anions. LaSrLiH2O2 (x = 0, y = 1) was composed of tetragonal (LiH2)– and (LaSrO2)+ layers alternately stacked along the c-axis, with the further increased hydride content of Sr2LiH3O resulting in the formation of (Sr2HO)+ layers. Considering the above series of compositions, it should be noted that there exists a K2NiF4-type H––free oxide, La2LiO3.5, in which the anion vacancies are randomly distributed in basal (LiO0.75)0.5– layers [44].

Crystal structures of t-La2LiHO3 and La2–y Sr y LiH1+y O3–y (y = 0, 1, 2)
Remarkably, t-La2LiHO3 contains anion vacancies (V(H,O)), which are best represented as La2Li(H0.53O1.21V(H,O)0.26)O2 and exhibit H–, O2–, and V(H,O) disorder at the axial sites of LiX 6 octahedra. By contrast, the orthorhombic phase, o-La2LiHO3, is stoichiometric, with H– and O2–located in axial anion sites. This symmetry change can be attributed to the order–disorder transition of H– and O2– in axial sites, both with and without vacancies. The crystal structures of the anion-deficient series, La2–x Sr x LiH1–x O3 and La1–x Sr1+x LiH2–x O2, were also determined by Rietveld analysis. Representative results obtained for La0.7Sr1.3LiH1.7O2 (x = 0.3, y = 1) are shown in Fig. 13.7 and Table 13.5. The H and O occupancy parameters were calculated as g H1 = 0.938(2), g H2 = 0.118(3), and g O1 = 0.882(3), leading to a composition of La0.7Sr1.3Li(H1.88V(H)0.12)H0.24O1.76. Thus, doping resulted in the generation of vacancies in the LiH4 plane and caused H–/O2– anion mixing at apical sites.

Neutron Rietveld analysis of anion-deficient La0.7Sr1.3LiH1.7O2 (x = 0.3, y = 1)
The valence states of all constituent atoms in La2LiHO3, LaSrLiH2O2, and Sr 2 LiH3O were determined by valence charge integration over the corresponding Voronoi cells (Table 13.6). The valences of hydrogen and oxygen in all materials were estimated at approximately –0.8 to –1.0 and –1.3 to –1.6, respectively, indicating that these elements were present as H– and O2–. Electronic density of states calculations corroborated the presence of hydride ions (Fig. 13.8), with their localized electrons located between approximately 0 and –5 eV below the Fermi level, confirming the ionic nature of the Li–H– bond.

Electronic densities of states for La2–y Sr y LiH1+y O3-y (y = 0, 1, 2) determined by first principles calculations
3.2 Hydride Ion Conductivity of La2–X–Y Sr x+Y H1–X+YO3–Y
The ionic conductivities of La2–x–y Sr x+y LiH1–x+y O3–y were examined by impedance measurements. The Arrhenius plots of conductivities are shown in Fig. 13.9. In the case of La2-y Sr y LiH1+y O3-y (x = 0), conductivity increased with increasing H– content, with the highest value of 3.2 × 10−5 S cm−1 at 573 K observed for Sr2LiH3O (y = 2) (Fig. 13.9a). Thus, introduction of hydride ions into the anion sites of the K2NiF4 structure improved ionic conductivity, confirming that these ions were primary charge carriers. Conduction was further facilitated by the introduction of vacancies, indicating that structural defects can affect ionic diffusion, as can be seen for La2–x Sr x LiH1–x O3 (y = 0) and La1–x Sr1+x LiH2–x O2 (y = 1), with conductivities of up to 2.1 × 10−4 S cm−1 observed for La0.6Sr1.4LiH1.6O2 at 590 K (activation energy ~ 68.4 kJ mol−1) (Fig. 13.9b, c).

Temperature-dependent ionic conductivities of La2–x–y Sr x+y H1–x+yO3–y . a La2–y Sr y LiH1+y O3–y (x = 0, y = 0, 1, and 2) with a fixed cation/anion ratio of (A2B)/X4, where A, B, and X are La(Sr), Li, and O(H), respectively. Anion-deficient series: b La2–x Sr x LiH1–x O3 (y = 0, 0 ≤ x ≤ 0.2) and c La1–x Sr1+x LiH2–x O2 (y = 1, 0 ≤ x ≤ 0.4)
To further identify the nature of the charge carriers, the electrical conductivity of La0.6Sr1.4LiH1.6O2 (x = 0.4, y = 1.0) was evaluated by the Hebb-Wagner polarization method [45] at 480 and 590 K using an asymmetric (–) Pd/La0.6Sr1.4LiH1.6O2/Mo (+) cell, with the total electrical conductivities (electrons + holes) at the irreversible Mo-electrolyte interface (2.9 × 10−8 and 4.1 × 10−7 S cm−1, respectively) showing that La0.6Sr1.4LiH1.6O2 is a purely ionic conductor (Fig. 13.10 and Table 13.7).

Hebb-Wagner polarization curves of the (–) Pd/La0.6Sr1.4LiH1.6O2/Mo (+) cell at a 590 K and b 480 K
3.3 Development of Electrochemical Devices Based on Hydride Ion Conduction
To verify the occurrence of H– conduction in La2–x–y Sr x+y LiH1–x+y O3–y , we constructed a Ti/o-La2LiHO3/TiH2 all-solid-state cell and subjected it to galvanostatic discharge, with an electrode configuration (powdered mixture of electrode and electrolyte materials) similar to that previously used in an all-solid-state lithium battery [46]. Figure 13.11a shows the discharge curve of the cell, revealing a constant discharge current of 0.5 μA at 300 °C. Moreover, the cell showed an initial open circuit voltage of 0.28 V, which was consistent with the theoretical value calculated from the standard Gibbs energy of formation of TiH2 [47]. During the electrochemical reaction, the cell voltage rapidly dropped from 0.28 to 0.06 V and then gradually decreased to 0.0 V. The initial steep drop corresponded to an increase in hydride ion content at the anode, owing to the following constant current discharge reaction:

All-solid-state cell fabricated for verification of H− conduction in La2–x–y Sr x+y LiH1–x+y O3–y . a Discharge curve for a Ti/o-La2LiHO3/TiH2 solid-state battery, with the inset showing a schematic illustration of the cell and the proposed electrochemical reaction. b X-ray diffraction patterns of electrolyte (o-La2LiHO3), cathode (TiH2 + o-La2LiHO3), and anode (Ti + o-La2LiHO3) materials after the reaction; the two right panels show expanded ranges of 13–13.8° and 15.1–15.8°
with the cathode reaction represented as:
The occurrence of these discharge reactions was confirmed by analysis of the produced phases. Figure 13.11b shows the synchrotron X-ray diffraction patterns of the cathode, electrolyte, and anode materials before and after the reaction. The absence of any variation in the diffraction patterns of the electrolyte indicates that the La2LiHO3 electrolyte was stable in contact with the Ti and TiH2 electrodes during the reaction. Conversely, phase changes were observed for the cathode and anode materials, as expected from the Ti-H phase diagram [47], where the δ-TiH2 (Fm \( \overline{3} \) m) phase releases hydrogen and is transformed into α-Ti (P63/mmc), passing through a two-phase (α-TiH b + δ-TiH2-a ) coexistence region found below ~ 573 K. In the case of the cathode, additional diffraction peaks corresponding to species with P63/mmc symmetry were detected, and the signals of TiH2 shifted to a higher angle, indicating that the release of hydrogen from TiH2 induced lattice shrinkage. In the case of the anode, peaks corresponding to species with Fm \( \bar{3} \) m symmetry were detected. Thus, the results indicate that during the electrochemical reaction, hydride ions were released from the TiH2 cathode and diffused into the Ti anode through o-La2LiHO3. The present success in the construction of an all-solid-state electrochemical cell exhibiting H– diffusion confirms not only the ability of oxyhydrides to act as H– solid electrolytes, but also the possibility of developing electrochemical solid devices based on H– conduction.
3.4 Ambient-Pressure Synthesis of H–-Conductive Oxyhydrides
The abovementioned high-pressure method is efficient for synthesizing oxyhydrides, owing to its ability to inhibit hydrogen desorption from the starting materials during sintering. However, in order to apply H– conductors to electrochemical devices, a simple synthetic protocol needs to be established for oxyhydrides, in parallel with the development of highly H–-conductive novel materials. Here, we describe the synthesis of LaSrLiH2O2 by a conventional solid-state reaction under ambient pressure and characterize its electrochemical properties.
The starting materials (which were identical to those used in the high-pressure method) were pelletized and placed in a sealed sample container made of stainless steel, with subsequent sintering performed at 650 °C for 6 h under H2.
Figure 13.12 shows the X-ray diffraction patterns of LaSrLiH2O2 synthesized using different amounts of LiH (stoichiometric, 20, 50, and 100 wt% excess), with the main diffraction peaks corresponding to the space group of LaSrLiH2O2, i.e., I4/mmm. However, small diffraction peaks indexed to SrO, SrH2, and/or La2O3, which were present in the raw starting materials, were observed for samples synthesized using a small excess or no excess of LiH (stoichiometric, 20 wt%, and 50 wt%). The amount of residual starting materials decreased as the amount of LiH increased, with LaSrLiH2O2 obtained as a single phase only at a 100 wt% excess. In addition, excess LiH improved the crystallinity of LaSrLiH2O2, i.e., the magnification of the normalized 004 peaks (Fig. 13.12) showed that their full width at half maximum decreased as the amount of LiH increased. Therefore, excess LiH not only prevented the loss of lithium and hydrogen during the synthesis of LaSrLiH2O2, but also acted as a flux for reducing the synthesis temperature.

X-ray diffraction profiles of LaSrLiH2O2 synthesized using various amounts of LiH: a stoichiometric, b 20, c 50, and d 100 wt% excess
Crystal structure analysis revealed that the sample prepared under ambient pressure had almost the same structure as the high-pressure one, with the refined site occupancies of each atom indicating that the former exhibited a nearly stoichiometric composition without vacancies. However, mixing of H− and O in the 4c axial anion site (g(H1) = 0.9361(5) and g(O1) = 1 − g(H1)) in Li octahedra was detected for the ambient-pressure sample, whereas this site was exclusively occupied by H− in the high-pressure sample.
The ionic conductivity of LaSrLiH2O2 synthesized at ambient pressure was evaluated by AC impedance measurements. The corresponding impedance and Arrhenius plots are shown in Fig. 13.13, with the conductivity of the high-pressure LaSrLiH2O2 synthesized in our previous study also plotted for comparison [6]. The impedance plot exhibited a typical form, comprising a semicircle in the high-frequency range and a spike in the low-frequency range, which corresponded to contributions of the bulk and grain boundary and the electrode, respectively. The former contribution was estimated by fitting impedance spectra using an equivalent circuit, as shown in Fig. 13.13. For the ambient-pressure sample, the activation energy of ionic conduction was calculated as 80.7 kJ mol−1, which is nearly equal to that observed for the high-pressure sample [6]. The total conductivity (bulk + grain boundary) of the ambient-pressure sample was determined as 3.2 × 10−6 S cm−1 at 300 °C, slightly less than that of the high-pressure sample. Given the crystal structure of LaSrLiH2O2, in which tetragonal (LiH2)− and (LaSrO2)+ layers are alternately stacked along the c-axis, the hydride ions were expected to exhibit two-dimensional diffusion in the LiH4 plane. Hence, the movement of H− in the crystal lattice of ambient-pressure LaSrLiH2O2 may have been inhibited by the presence of oxide ions in the (LiH2)− layer.

Arrhenius and Cole-Cole plots of LaSrLiH2O2. The inset shows an equivalent circuit used to model impedance data
Thus, we successfully synthesized LaSrLiH2O2 by a conventional solid-state reaction under ambient pressure [48], with a two-fold molar excess of LiH required to obtain single-phase LaSrLiH2O2. The sample synthesized at ambient pressure exhibited a crystal structure and H− conductivity similar to those observed for the high-pressure sample, implying that the method described here should increase the applicability of H− conductors as solid electrolytes.
4 Concluding Remarks
This chapter outlined the properties of ion conductors and material search methods, introducing Li+ and H− conductors and providing examples of material search (e.g., element substitution and structure-based methods). However, broader material variability will be required to fabricate viable electrochemical devices based on solid electrolytes, necessitating the utilization of composition-based material search, which is one of the conventional material discovery methods. The approach described is significantly influenced by the experience and intuition of researchers, and it generally takes longer than element substitution and structure-based methods. However, the recent development of theoretical calculation and material informatics methods is expected to shorten the time required [49,50,51,52,53], allowing high-speed screening of prospective compositions/structures. In some cases, this approach might be misleading, since not all theoretically predicted compositions or structures can be obtained by the present synthetic techniques, as exemplified by the failure of the composition/structure-based search in the case of Li+ and H− conductors. Thus, the area of materials informatics for composition-based material search is still in its infancy, but it holds promise for the future.
References
B.B. Owens, G.R. Argue, Science 157, 308 (1967)
T. Takahashi, O. Yamamoto, S. Yamada, S. Hayashi, J. Electrochem. Soc. 126, 1654 (1979)
T. Takahashi, R. Kanno, Y. Takeda, O. Yamamoto, Solid State Ion. 3, 283 (1981)
N. Kamaya, K. Homma, Y. Yamakawa, M. Hirayama, R. Kanno, M. Yonemura, T. Kamiyama, Y. Kato, S. Hama, K. Kawamoto, A. Mitsui, Nat. Mater. 10, 682 (2011)
Y. Kato, S. Hori, T. Saito, K. Suzuki, M. Hirayama, A. Mitsui, M. Yonemura, H. Iba, R. Kanno, Nat. Energy 1, 16030 (2016)
G. Kobayashi, Y. Hinuma, S. Matsuoka, A. Watanabe, M. Iqbal, M. Hirayama, M. Yonemura, T. Kamiyama, I. Tanaka, R. Kanno, Science 351, 1314 (2016)
M.C. Verbraeken, C. Cheung, E. Suard, J.T.S. Irvine, Nat. Mater. 14, 95 (2015)
V. Thangadurai, S. Narayanan, D. Pinzaru, Chem. Soc. Rev. 43, 4714 (2014)
U.V. Alpen, M.F. Bell, W. Wichelhaus, K.Y. Cheung, G.J. Dudley, Electrochim. Acta 23, 1395 (1978)
Y. Inaguma, L. Chen, M. Itoh, T. Nakamura, Solid State Ion. 70–71 Part 1, 196 (1994)
V. Thangadurai, H. Kaack, W.J.F. Weppner, J. Am. Ceram. Soc. 86, 437 (2003)
A. Hayashi, S. Hama, H. Morimoto, M. Tatsumisago, T. Minami, Chem. Lett. 30, 872 (2001)
R. Kanno, M. Murayama, J. Electrochem. Soc. 148, A742 (2001)
K. Minami, A. Hayashi, M. Tatsumisago, J. Am. Ceram. Soc. 94, 1779 (2011)
T. Phraewphiphat, M. Iqbal, K. Suzuki, Y. Matsuda, M. Yonemura, M. Hirayama, R. Kanno, J. Solid State Chem. 225, 431 (2015)
Y. Sun, K. Suzuki, S. Hori, M. Hirayama, R. Kanno, Chem. Mater. 29, 5858 (2017)
R. Kanno, T. Hata, Y. Kawamoto, M. Irie, Solid State Ion. 130, 97 (2000)
Y. Inoue, K. Suzuki, N. Matsui, M. Hirayama, R. Kanno, J. Solid State Chem. 246, 334 (2017)
Y. Inaguma, C. Liquan, M. Itoh, T. Nakamura, T. Uchida, H. Ikuta, M. Wakihara, Solid State Commun. 86, 689 (1993)
Y. Inaguma, T. Katsumata, M. Itoh, Y. Morii, T. Tsurui, Solid State Ion. 177, 3037 (2006)
G. Zhao, I. Muhammad, K. Suzuki, M. Hirayama, R. Kanno, Mater. Trans. 57, 1370 (2016)
E.E. Hellstrom, W. Van Gool, Solid State Ion. 2, 59 (1981)
C.J.M. Rooymans, Z. Anorg, Allg. Chem. 313, 234 (1961)
M. Tabuchi, S. Tsutsui, C. Masquelier, R. Kanno, K. Ado, I. Matsubara, S. Nasu, H. Kageyama, J. Solid State Chem. 140, 159 (1998)
A.F. Andresen, A.J. Maeland, D. Slotfeldt-Ellingsen, J. Solid State Chem. 20, 93 (1977)
B. Wegner, R. Essmann, J. Bock, H. Jacobs, P. Fischer, Eur. J. Solid State Inorg. Chem. 29, 1217 (1992)
F. Altorfer, W. Buhrer, B. Winkler, G. Coddens, R. Essmann, H. Jacobs, Solid State Ion. 70–71, 272 (1994)
B. Zhu, X. Yang, Electrochem. Commun. 1, 411 (1999)
B. Zhu, X. Liu, Electrochem. Commun. 2, 10 (2000)
F.W. Poulsen, Solid State Ion. 145, 387 (2001)
M.C. Verbraeken, E. Suard, J.T.S. Irvine, J. Mater. Chem. 19, 2766 (2009)
R.M. Helps, N.H. Rees, M.A. Hayward, Inorg. Chem. 49, 11062 (2010)
Y. Kobayashi, O.J. Hernandez, T. Sakaguchi, T. Yajima, T. Roisnel, Y. Tsujimoto, M. Morita, Y. Noda, Y. Mogami, A. Kitada, M. Ohkura, S. Hosokawa, Z. Li, K. Hayashi, Y. Kusano, J.E. Kim, N. Tsuji, A. Fujiwara, Y. Matsushita, K. Yoshimura, K. Takegoshi, M. Inoue, M. Takano, H. Kageyama, Nat. Mater. 11, 507 (2012)
T. Sakaguchi, Y. Kobayashi, T. Yajima, M. Ohkura, C. Tassel, F. Takeiri, S. Mitsuoka, H. Ohkubo, T. Yamamoto, J.E. Kim, N. Tsuji, A. Fujihara, Y. Matsushita, J. Hester, M. Avdeev, K. Ohoyama, H. Kageyama, Inorg. Chem. 51, 11371 (2012)
H. Schwarz, Ph.D. Thesis, University of Karlsruhe, 1991
J. Bang, S. Matsuishi, H. Hiraka, F. Fujisaki, T. Otomo, S. Maki, J.-I. Yamaura, R. Kumai, Y. Murakami, H. Hosono, J. Am. Chem. Soc. 136, 7221 (2014)
M.A. Hayward, E.J. Cussen, J.B. Claridge, M. Bieringer, M.J. Rosseinsky, C.J. Kiely, S.J. Blundell, I.M. Marshall, F.L. Pratt, Science 295, 1882 (2002)
K. Hayashi, S. Matsuishi, T. Kamiya, M. Hirano, H. Hosono, Nature 419, 462 (2002)
K. Hayashi, P.V. Sushko, A.L. Shluger, M. Hirano, H. Hosono, J. Phys. Chem. B 109, 23836 (2005)
S. Matsuishi, K. Hayashi, M. Hirano, H. Hosono, J. Am. Chem. Soc. 127, 12454 (2005)
C.G. Van de Walle, Phys. Rev. Lett. 85, 1012 (2000)
C.G. Van de Walle, J. Neugebauer, Nature 423, 626 (2003)
J. Zhang, G. Gou, B. Pan, J. Phys. Chem. C 118, 17254 (2014)
J.P. Attfield, G. Férey, J. Solid State Chem. 80, 112 (1989)
B.J. Neudecker, W. Weppner, J. Electrochem. Soc. 143, 2198 (1996)
N. Kamaya, K. Homma, Y. Yamakawa, M. Hirayama, R. Kanno, M. Yonemura, T. Kamiyama, Y. Kato, S. Hama, K. Kawamoto, A. Mitsui, Nat. Mater. 10, 682 (2011)
A. San-Martin, F.D. Manchester, Bull. Alloy Phase Diagrams 8, 30 (1987)
A. Watanabe, G. Kobayashi, N. Matsui, M. Yonemura, A. Kubota, K. Suzuki, M. Hirayama, R. Kanno, Electrochemistry 85, 88 (2017)
M. Nishijima, T. Ootani, Y. Kamimura, T. Sueki, S. Esaki, S. Murai, K. Fujita, K. Tanaka, K. Ohira, Y. Koyama, I. Tanaka, Nat. Commun. 5, 4553 (2014)
Y. Hinuma, T. Hatakeyama, Y. Kumagai, L.A. Burton, H. Sato, Y. Muraba, S. Iimura, H. Hiramatsu, I. Tanaka, H. Hosono, F. Oba, Nat. Commun. 7, 11962 (2016)
A.D. Sendek, Q. Yang, E.D. Cubuk, K.-A.N. Duerloo, Y. Cui, E.J. Reed, Energy Environ. Sci. 10, 306 (2017)
W.D. Richards, Y. Wang, L.J. Miara, J.C. Kim, G. Ceder, Energy Environ. Sci. 9, 3272 (2016)
A. Seko, H. Hayashi, K. Nakayama, A. Takahashi, I. Tanaka, Phys. Rev. B 95, 144110 (2017)
Acknowledgements
This research was supported by JST, PRESTO, and Grant-in-Aid for Young Scientists (A) no. 15H05497 and (B) no. 24750209; Grant-in-Aid for Challenging Exploratory Research no. 15K13803, 23655191, and 25620180; and Grant-in-Aid for Scientific Research on Innovative Areas no. 25106005 and 25106009, from the Japan Society for the Promotion of Science. Synchrotron and neutron radiation experiments were carried out as four projects approved by the Japan Synchrotron Radiation Research Institute (JASRI) (proposals no. 2013A1704, 2015A1778, and 2015B1768), the Japan Proton Accelerator Research Complex (J-PARC) (proposal no. 2010A0058), the Spallation Neutron Source (SNS) in the Oakridge National Laboratory (proposal no. IPTS5808 and 10030), and the Neutron Scattering Program Advisory Committee of IMSS, KEK (proposal no. 2014S10). Part of the neutron experiments (proposal no. 2014S10) was performed at the BL09 Special Environment Neutron Powder Diffractometer (SPICA) developed by the Research and Development Initiative for Scientific Innovation of New Generation Batteries (RISING) project of the New Energy and Industrial Technology Development Organization (NEDO). Supercomputing time at the Academic Center for Computing and Media Studies (ACCMS) at Kyoto University is gratefully acknowledged. Further information regarding the materials and methods is included in the supplementary materials.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Open Access This chapter is licensed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license and indicate if changes were made.
The images or other third party material in this chapter are included in the chapter’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the chapter’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.
Copyright information
© 2018 The Author(s)
About this chapter

Cite this chapter
Kanno, R., Kobayashi, G., Suzuki, K., Hirayama, M., Mori, D., Tamura, K. (2018). Synthesis and Structures of Novel Solid-State Electrolytes. In: Tanaka, I. (eds) Nanoinformatics. Springer, Singapore. https://doi.org/10.1007/978-981-10-7617-6_13
Download citation
DOI: https://doi.org/10.1007/978-981-10-7617-6_13
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-10-7616-9
Online ISBN: 978-981-10-7617-6
eBook Packages: Chemistry and Materials ScienceChemistry and Material Science (R0)