
Abstract
Mapping the epigenome is key to describe the relationship between chromatin landscapes and the control of DNA-based cellular processes such as transcription. Cleavage under targets and release using nuclease (CUT&RUN) is an in situ chromatin profiling strategy in which controlled cleavage by antibody-targeted Micrococcal Nuclease solubilizes specific protein-DNA complexes for paired-end DNA sequencing. When applied to budding yeast, CUT&RUN profiling yields precise genome-wide maps of histone modifications, histone variants, transcription factors, and ATP-dependent chromatin remodelers, while avoiding cross-linking and solubilization issues associated with the most commonly used chromatin profiling technique Chromatin Immunoprecipitation (ChIP). Furthermore, targeted chromatin complexes cleanly released by CUT&RUN can be used as input for a subsequent native immunoprecipitation step (CUT&RUN.ChIP) to simultaneously map two epitopes in single molecules genome-wide. The intrinsically low background and high resolution of CUT&RUN and CUT&RUN.ChIP allows for identification of transient genomic features such as dynamic nucleosome-remodeling intermediates. Starting from cells, one can perform CUT&RUN or CUT&RUN.ChIP and obtain purified DNA for sequencing library preparation in 2 days.
You have full access to this open access chapter, Download protocol PDF
Similar content being viewed by others

Key words
1 Introduction
Chromatin organization, or how DNA is packaged inside the nucleus, determines cell-type-specific gene expression and cellular identity. Nucleosomes, composed of DNA wrapped around an octameric core of histone proteins are the fundamental repeating units of chromatin. Chromatin organization is highly dynamic, and functionally distinct chromatin states are outcomes of the interplay between multiple proteins and proteins complexes including, but not limited to, histone chaperones, histone modifying enzymes, ATP-dependent nucleosome remodelers, and transcription factors (TF) [1]. Mutations affecting the functions of these proteins are associated with a variety of developmental disorders and cancers. The first step in understanding the mechanisms of chromatin organization is to accurately map the genomic locations and chromatin contexts where chromatin-regulatory proteins bind.
The most widely used technique for genome-wide mapping of chromatin-associated proteins is chromatin immunoprecipitation followed by deep sequencing (ChIP-seq) [2]. ChIP-seq generally involves formaldehyde fixation (cross-linking) of bulk chromatin within cells followed by mechanical shearing or enzymatic cleavage into smaller fragments and immunoprecipitation of the DNA cross-linked with the protein-factor of interest. Problems arise due to differential sensitivity of genomic regions to chromatin fragmentation approaches, the need to fragment the entire genome to solubilize chromatin resulting in high background noise, as well as epitope masking and false positives potentially due to chemical cross-linking [3,4,5,6]. Although successive technological and methodological advancements have greatly improved the resolution of ChIP-seq, these methods still rely on formaldehyde cross-linking to preserve the in vivo patterns [7,8,9]. Native ChIP minimizes epitope masking and cross-linking artifacts; however, problems still remain with incomplete extraction efficiency of protein-DNA complexes [10].
Chromatin-tethering of a variety of DNA enzymes including methyl transferases [11], nucleases [12,13,14], and transposases [15, 16] are attractive alternatives for chromatin profiling. Enzyme tethering methods fundamentally differ from ChIP-seq as they are carried out in vivo or in situ where the tethered enzyme acts upon the DNA in its local vicinity, and the modified DNA is directly extracted from permeabilized cells, thus eliminating the need to solubilize and recover chromatin. Cleavage Under Targets and Release Using Nuclease (CUT&RUN) is an enzyme-tethering method in which a protein A - Micrococcal Nuclease fusion (pA-MNase) is targeted to genomic regions via factor-specific antibodies in permeabilized native cells or nuclei (in the case of yeast) such that chromatin around the factor-binding sites is selectively cleaved [12] (Fig. 1). Micrococcal Nuclease (MNase) is an endo-exo nuclease from Staphylococcus aureus that has been used for decades for mapping nucleosomes. In the presence of calcium ions, MNase nibbles away unbound DNA to generate minimal protections of protein-DNA complexes. Subjecting DNA fragments obtained after MNase treatment of nuclei to paired-end short-read sequencing enables determination of the genome-wide footprints corresponding to both small proteins (e.g., transcription factors) as well as nucleosomes at base-pair resolution [17]. In CUT&RUN, MNase cleavage solubilizes factor-associated chromatin, which diffuse out cleanly into the supernatant, leaving the bulk of the undigested chromatin insoluble. As only the targeted fragments enter into solution and the vast majority of DNA is left behind, CUT&RUN has exceptionally high signal-to-noise and data quality, requiring only ~1/tenth the sequencing depth as ChIP-seq [18, 19].

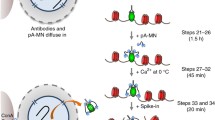
Schematic of the CUT&RUN(.ChIP) approach. Permeabilized yeast nuclei attached to ConA beads are successively treated with the targeting antibody and pA-MNase fusion, which diffuse through the nuclear pores. Ca++ induces cleavage of target DNA by immobilized MNase. The reaction is done at 0 °C which limits diffusion of released fragments, thereby minimizing background cleavage. Because there is no cross-linking, cleaved chromatin particles are efficiently released into solution at 37 °C. In the ChIP step, the first antibody is competed off from the released particles, antibody for a second epitope is added, and immune-complexes are enriched by binding to protein A/G beads. DNA extracted from the CUT&RUN supernatant and from ChIP are used to prepare libraries for paired-end deep-sequencing
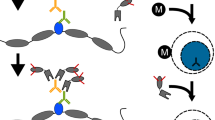
As an in situ method, CUT&RUN is inherently capable of probing the local chromatin environment such as the binding sites of TFs and the positions of flanking nucleosomes from the same experiment [12] (Fig. 2). This is possible by computationally filtering the sequence data based on fragment lengths, as nucleosomes typically protect ~150 bp of DNA from MNase cleavage and TFs usually protect short DNA fragments (<80 bp). Most chromatin profiling methods are capable of mapping only one epitope at a time and are therefore not able to directly test protein co-occupancies on chromatin that forms the basis of gene-regulatory mechanisms, while most conclusions are based on correlation. To overcome this limitation, we took advantage of the native chromatin complexes that are solubilized in CUT&RUN and used as input for immunoprecipitation with a second antibody under native conditions (CUT&RUN.ChIP) (Fig. 1) [20]. This required us to compete off the CUT&RUN antibody from the released particles using the immunizing peptide so as to avoid nonspecific enrichment in the ChIP step. When applied to budding yeast, CUT&RUN.ChIP mapped histone co-occupancies genome-wide with exceptional high efficiency and resolution (Fig. 3), and revealed that the ATP-dependent chromatin remodeler RSC and the general regulatory TFs Abf1 and Reb1 are associated with partially unwrapped dynamic nucleosomal intermediates at gene promoters [20].

Simultaneous mapping of TFs and flanking nucleosomes. Top: pA-MNase tethered to TFs bound at gene promoter nucleosome-depleted regions digests DNA around the bound TF as well as reach beyond the immediately flanking nucleosomes to solubilize both the TF- and flanking nucleosome-bound chromatin. Bottom: Separation of mapped DNA fragments by size from CUT&RUN targeting the general regulatory TF Abf1 shows crisp TF peaks (<120 bp size class) over Abf1 DNA-binding motifs (shown in red) [10] and the positions of the flanking nucleosomes (>150 bp size class) from the same experiment

CUT&RUN.ChIP maps histone co-occupancies. Blue tracks show spike-in normalized CUT&RUN.ChIP profiles (>150 bp or nucleosomal reads) of histone modifications in nucleosomes containing the histone variant H2A.Z. Supernatant from Htz1-FLAG CUT&RUN experiment was used as input for the subsequent native ChIP. IgG is a nonspecific negative control for ChIP. ChIP-seq of H2A.Z from cross-linked MNase-digested chromatin (green, top) is shown for comparison [22]. Results show that H2A.Z nucleosomes are present only at the beginning of genes and totally absent from gene bodies unlike profiles observed in conventional MNase-ChIP-seq. H2A.Z nucleosomes are enriched in histone H3 with trimethylation of lysine 4 (H3K4me3) and histone H4 with acetylated N-terminal tail (H4ac) but lack histone H3 trimethylated at lysine 36 (H3K36me3), commonly associated with gene-body nucleosomes. CUT&RUN.ChIP tracks were scaled to the same intensity (y-axes). The positions of the annotated yeast open reading frames are shown at the bottom with the gene names
In summary, CUT&RUN has several advantages for chromatin profiling in budding yeast: (1) The method is performed in situ in non–cross-linked nuclei and does not require chromatin fragmentation or solubilization; (2) The intrinsically low background allows low sequencing depth and identification of low-signal genomic features invisible to ChIP-seq; (3) The simple procedure can be completed in as little as two days and is suitable for robotic automation [21]; (4) Combining CUT&RUN with native ChIP allows mapping of protein co-occupancies on chromatin; (5) A simple spike-in strategy can be used for accurate quantitation of protein-DNA interactions. Although there is no published report of CUT&RUN profiling of the fission yeast Schizosaccharomyces pombe epigenome, the protocol described here starting from native nuclei should work as efficiently in principle.
2 Materials
Prepare all solutions using ultrapure water (prepared by purifying deionized water to attain a sensitivity of 18 MΩ-cm at 25 °C) and analytical grade reagents.
2.1 Yeast Culture and Nuclei Preparation
-
1.
Saccharomyces cerevisiae cultured on Yeast Peptone Dextrose (YPD) agar.
-
2.
Yeast Peptone Dextrose (YPD) media: 10 g/L yeast extract, 10 g/L bactopeptone, 20 g/L glucose. Autoclaved.
-
3.
1 M PIPES, pH 6.5: Mix 15.11 g PIPES in 30 mL water, add 10 N Sodium Hydroxide dropwise (takes about 5 mL) to adjust pH to 6.5, adjust final volume to 50 mL with water, filter-sterilize (0.2 μm), store at 4 °C.
-
4.
Resuspension buffer (make fresh before use): 1.2 M Sorbitol, 100 mM Potassium Phosphate buffer, pH 7.5, 0.5 mM Calcium Chloride, 0.5 mM 2-Mercaptoethanol.
-
5.
Zymolyase: Dissolve 1 mg/mL Zymolyase in 1.0 M Sorbitol and 10 mM PIPES, pH 6.5, store aliquots at −20 °C.
-
6.
SPC buffer: 1 M Sorbitol, 20 mM PIPES, pH 6.5, 0.1 mM Calcium Chloride. SPC + protease inhibitor: Add one Roche Protease inhibitor cocktail tablet per 50 mL buffer, and PMSF to 1 mM before use.
-
7.
Ficoll buffer: 9% (w/v) Ficoll 400, 20 mM PIPES, pH 6.5, 0.5 mM calcium chloride. Add Ficoll gradually to half the final volume of water, stir at room temperature, bring up to final volume with water when Ficoll dissolves, filter-sterilize (0.2 μm).
2.2 CUT&RUN and Sequencing
-
1.
Concanavalin A–coated (ConA) magnetic beads.
-
2.
Binding buffer: 20 mM HEPES-KOH, pH 7.9, 10 mM Potassium Chloride, 1 mM Calcium Chloride, 1 mM Manganese Chloride.
-
3.
5% w/v Digitonin
-
4.
Wash buffer: 20 mM Sodium HEPES, pH 7.5, 150 mM Sodium Chloride, 0.5 mM Spermidine, add one Roche Protease inhibitor cocktail tablet per 50 mL buffer. Wash buffer can be stored for up to 7 days at 4 °C, but spermidine might degrade over time. For Dig-Wash, add Digitonin to 0.05% final concentration (10 μL of 5% digitonin in water to 10 mL buffer).
-
5.
Antibody buffer: Dig-Wash buffer supplemented with 2 mM ethylenediaminetetraacetic acid (EDTA).
-
6.
Antibody to an epitope of interest, for example, rabbit anti-histone Htz1 (H2A.Z) antibody or mouse anti-FLAG M2 antibody (for FLAG-tagged proteins).
-
7.
Secondary antibody, for example, guinea pig anti-rabbit IgG or rabbit anti-mouse IgG.
-
8.
Isotype control IgG (negative control antibody).
-
9.
Protein A-Micrococcal nuclease (pA-MNase) or Protein A-Protein G-Micrococcal nuclease (pAG-MNase) fusion proteins purified from overexpression in Escherichia coli (see [14] and [18] for protein purification protocols), or obtained commercially (e.g., CUTANA pAG-MNase, Epicypher; or CUT&RUN pAG-MNase, Cell Signaling Technology). pA-MNase and pAG-MNase expression plasmids can be obtained from Addgene.
-
10.
Heterologous spike-in DNA, for example DNA purified from MNase-treated chromatin from Drosophila S2 cells (see refs. [12, 19]).
-
11.
2× STOP buffer: Dig-Wash buffer supplemented with 20 mM EDTA, 4 mM EGTA (ethylene glycol-bis(β-aminoethyl ether)-N,N,N′,N′-tetraacetic acid), 50 μg/ml RNase A (DNase free), 50 pg/mL heterologous spike-in DNA.
-
12.
Immunizing peptide for competing off CUT&RUN antibody (only for CUT&RUN.ChIP), for example 3×-FLAG peptide.
-
13.
Protein A- or Protein-G coated magnetic beads, for example Dynabeads Protein A.
-
14.
10% sodium dodecyl sulfate (SDS).
-
15.
20 mg/mL Proteinase K.
-
16.
Phenol–chloroform–isoamyl alcohol 25:24:1.
-
17.
Chloroform–isoamyl alcohol 24:1.
-
18.
2 mg/mL Glycogen.
-
19.
Ethanol.
-
20.
10 M Tris–HCl, pH 8.0.
-
21.
Fluorometric DNA quantification assay, for example, Qubit NGS equipment and Qubit dsDNA HS kit.
-
22.
DNA capillary electrophoresis equipment and reagents, for example, TapeStation 4200 system and D1000 high-sensitivity Screen Tapes and reagents.
-
23.
SPRI paramagnetic beads (e.g., Agencourt AMPure XP, Beckman Coulter).
-
24.
NGS library preparation reagents, for example, KAPA HyperPrep kit.
-
25.
Illumina TruSeq-compatible adapters and indexing primers.
2.3 General Equipment
-
1.
Centrifuges, swinging bucket rotors that can accommodate 50 mL tubes at speeds up to 3000 × g, and tabletop refrigerated centrifuges that can accommodate 1.5 mL tubes at speeds up to 16,000 × g.
-
2.
Microcentrifuge tubes (non–low-bind) and PCR tubes/96-well plates.
-
3.
Magnetic separator that can hold 1.5 mL tubes and PCR tubes.
-
4.
Nutating and end-over-end mixers.
-
5.
Heating blocks or water baths at 37 °C.
-
6.
Phase-lock tubes (e.g., Qiagen MaXtract High Density).
3 Method
3.1 Yeast Nuclei Preparation
-
1.
Inoculate single S. cerevisiae colony into 10 mL YPD, grow overnight at 30 °C, use 5 mL–10 mL to inoculate 500 mL YPD next morning.
-
2.
Grow 500 mL S. cerevisiae cultures to OD600 = 0.6–0.8 in YPD at 30 °C (expect about 5 × 108 cells).
-
3.
Transfer culture to centrifugation bottles.
-
4.
Centrifuge for 10 min at 2700 × g and 4 °C.
-
5.
Discard the supernatant, wash cell pellets with 50 mL cold ultrapure H2O, and transfer to 50 mL conical tubes.
-
6.
Centrifuge for 10 min at 2700 × g and 4 °C.
-
7.
Resuspend cells in 36 mL Resuspension buffer.
-
8.
Incubate for 15 min in 37 °C water bath.
-
9.
Add 500 μL 1 mg/mL Zymolase (freshly prepared in Resuspension buffer) and incubate for 10–20 min in 37 °C water bath (see Note 1).
-
10.
Transfer to ice immediately once spheroplasting is complete and quickly proceed to next step.
-
11.
Centrifuge for 5 min at 1000 × g and 4 °C.
-
12.
Wash pellets (resuspend very gently with a 1 mL pipette) with 25 mL cold SPC buffer with protease inhibitors.
-
13.
Centrifuge for 5 min at 1000 × g and 4 °C.
-
14.
Repeat steps 10 and 11 once, resuspend pellets in 0.5 mL SPC buffer without protease inhibitors (Pause point: If you are preparing nuclei from more than one yeast culture, it is okay to hold the spheroplasts in SPC buffer on ice for other samples to catch up).
-
15.
Mix with cold 25 mL Ficoll buffer.
-
16.
Transfer to 50 mL glass beaker and stir very gently (slowest setting) on a magnetic stirrer with a small (8–10 mm) magnetic bead at 4 °C for 15 min.
-
17.
Transfer to 50 mL conical tubes and centrifuge for 10 min at 7500 × g rcf and 4 °C.
-
18.
Wash pellet with 10 mL cold SPC buffer with protease inhibitors.
-
19.
Centrifuge for 10 min at 4200 × g and 4 °C.
-
20.
Repeat steps 18 and 19 once, resuspend each nuclei pellet in total 5 mL to 10 mL cold SPC buffer with protease inhibitors. Take out a small aliquot (~50 μL) and count nuclei concentration. Adjust volume of SPC to get 5 × 107 nuclei per mL. Slow-freeze nuclei in an isopropyl alcohol chamber at −80 °C. Nuclei can be stored at −80 °C (see Note 2).
3.2 CUT&RUN
This protocol is recommended for 5 × 107 yeast nuclei. We start with this high number so that the supernatant from CUT&RUN can be used for ChIP with a second antibody for up to 10 targets. Alternatively, for example for a time-course experiment, the starting material can be used for up to 10 time points. Scale down/up as necessary. Use freshly prepared or frozen nuclei. If using frozen nuclei, quick-thaw in a 37 °C water bath for 1–2 min until the frozen content starts to melt, and quickly transfer to ice to thaw completely. Avoid multiple freeze–thaw cycles, which are detrimental to nuclear integrity. Because it is crucial that DNA breakage is minimized throughout the protocol, we recommend that cavitation during resuspension and vigorous vortexing be avoided.
-
1.
Equilibrate 100 μL Concanavalin A–coated (ConA) magnetic beads with 1 mL binding buffer in 1.5 or 1.7 mL microcentrifuge tubes (see Note 3). We recommend using 20 μL beads for 1 × 107 yeast nuclei.
-
2.
Place on a magnet stand to clear and pull off the liquid.
-
3.
Repeat steps 1 and 2 twice, and bring up beads in 100 μL binding buffer.
-
4.
Add beads to thawed yeast nuclei (1 mL, 5 × 107 nuclei), rotate gently at room temperature for 10 min. Remove liquid from the cap and side with a quick spin on a microcentrifuge (no more than 100 × g for 10 s). Place on a magnet stand to clear (<1 min) and pull off the liquid (see Note 4).
-
5.
Wash beads (with the bound nuclei) with 1 mL Antibody buffer. Resuspend beads by inverting tubes a few times, or use a 1 mL pipette to break clumps very gently if necessary. Place on a magnet stand to clear, pull off the liquid, and resuspend beads in 1 mL Antibody buffer (see Note 5). From this step keep samples cold (on ice) as much as possible.
-
6.
Add primary antibody against your target of interest (e.g., mouse anti-FLAG for a FLAG-tagged protein; recommended antibody dilution = 1:100 unless determined empirically; see Note 6), gently rotate for 1 h at room temperature or overnight at 4 °C (recommended, to obtain higher yields). To evaluate success of the procedure without requiring library preparation, include in parallel a positive control antibody (e.g., anti-H2A.Z or H3K4me3) and a negative control antibody (e.g., rabbit isotype-control IgG) (see Note 7). Go to step 8 if not using a secondary antibody (see Note 8).
-
7.
Collect the contents of the tubes to the bottom with a quick spin on a microcentrifuge (no more than 100 × g for 10 s). This will minimize carryover of reagents that could result in overall background cleavages during the digestion step.
-
8.
Place tubes on a magnet stand to clear, pull off the liquid and wash beads with 1 mL cold Dig-Wash buffer. Place tubes on a magnet stand to clear, pull off the liquid, add 1 mL cold Dig-Wash buffer with secondary antibody (e.g., rabbit anti-mouse IgG) at 1:100 dilution. Rotate for 1 h at 4 °C.
-
9.
Quick spin on a microcentrifuge (no more than 100 × g for 10 s), place tubes on a magnet stand to clear, pull off the liquid and wash beads with 1 mL cold Dig-Wash buffer. Repeat once more.
-
10.
Pull off the liquid on a magnet stand and resuspend beads in 1 mL cold Dig-Wash buffer with a final concentration of 500–1000 ng/mL pA-MNase or pAG-MNase (see Note 9, or follow manufacturer’s recommendation for enzyme obtained from commercial sources), and rotate for 1 h at 4 °C.
-
11.
Quick spin on a microcentrifuge (no more than 100 × g for 10 s), place tubes on a magnet stand to clear, pull off the liquid and wash beads with 1 mL cold Dig-Wash buffer. Repeat twice more.
-
12.
Pull off liquid on a magnet stand and resuspend beads in 1 mL cold Dig-Wash buffer. Hold tubes on a metallic tube-holder set an ice-water bath (0 °C) for 10 min.
-
13.
Add 20 μL 100 mM Calcium Chloride (2 mM final concentration), mix quickly by placing tubes on a vortex mixer set at a very low speed. Immediately replace tubes to 0 °C. Incubate for 5–30 min for digestion (see Notes 10 and 11).
-
14.
Stop digestion by adding 1 mL 2× STOP buffer containing 50 pg/mL Drosophila melanogaster spike-in DNA (see Note 12), mix by inverting tubes a few times. If there are multiple time points, remove 100 μL of reaction and add to 100 μL 2×STOP.
-
15.
Incubate 20 min in 37 °C water bath to release CUT&RUN fragments from the insoluble nuclear chromatin.
-
16.
Centrifuge 5 min at 4 °C and 16,000 × g, place on magnet stand and transfer liquid to a fresh tube.
-
17.
If you are performing CUT&RUN.ChIP, jump to the next Subheading 3.3, or else proceed to DNA extraction (following steps).
-
18.
To each sample or time point (200 µL) add 100 µL Wash buffer at room temperature, 3 μL 10% SDS (to 0.1%), and 2.5 μL 20 mg/mL Proteinase K. Mix by vortexing and incubate for 1 h at 50 °C.
-
19.
Add 300 μL phenol–chloroform–isoamyl alcohol (25:24:1) and mix by full-speed vortexing ~2 s.
-
20.
Transfer to a phase-lock tube (e.g., Qiagen MaXtract), and centrifuge 5 min at room temperature and 16,000 × g.
-
21.
Add 300 μL chloroform–isoamyl alcohol (24:1) on top of the aqueous phase, invert ~10× to mix, and centrifuge for 5 min at room temperature and 16,000 × g.
-
22.
Remove the aqueous phase by pipetting into a fresh tube containing 2 μL 10 mg/mL glycogen.
-
23.
Add 750 μL chilled 100% ethanol, mix by inverting tubes, chill on ice for 10 min or longer at −20 °C, and centrifuge for 40 min at 16,000 × g and 4 °C (see Note 13).
-
24.
Pour off the liquid and drain on a paper towel.
-
25.
Rinse the pellet in 1 mL chilled 100% ethanol and centrifuge 5 min 16,000 × g and 4 °C.
-
26.
Carefully pour off the liquid, drain on a paper towel, and let air dry by leaving the caps open.
-
27.
When the pellet is dry, dissolve in 30 μL 10 mM Tris–HCl pH 8, and proceed to sequencing-library preparation (see Subheading 3.4).
3.3 Native ChIP with CUT&RUN Supernatant as Input (CUT&RUN.ChIP)
-
1.
For CUT&RUN.ChIP, supplement 2× STOP buffer with 20 μg/mL of the immunizing/blocking peptide for the primary CUT&RUN antibody used in step 6 (e.g., FLAG peptide) (see Note 14).
-
2.
Add 1 mL 2× STOP to 1 mL CUT&RUN reaction after 0 °C incubation for the desired duration (similar to step 14 of Subheading 3.2), and mix by inverting tubes a few times.
-
3.
Incubate 20 min in 37 °C water bath to release CUT&RUN fragments from the insoluble nuclear chromatin.
-
4.
Centrifuge 5 min at 4 °C and 16,000 × g, place on magnet stand and transfer liquid to a fresh tube.
-
5.
Remove 200 μL for each ChIP experiment, add 2 μL antibody against the epitope of interest, rotate 1 h at room temperature or overnight at 4 °C. Add an isotype control IgG to one sample as a negative control.
-
6.
Save 200 μL CUT&RUN supernatant as the “ChIP input” sample at 4 °C.
-
7.
Take 200 μL protein A– or protein G–coated magnetic beads (see Note 15), place on a magnet stand to clear and pull off the liquid.
-
8.
Wash beads with 1 mL Wash buffer (without digitonin), place on a magnet stand to clear and pull off the liquid.
-
9.
Repeat step 8 twice more, resuspend beads in 200 μL Wash buffer.
-
10.
Add 20 μL beads to each ChIP experiment, except the “input” fraction.
-
11.
Rotate for 30 min at 4 °C.
-
12.
Quick spin on a microcentrifuge at 1000 × g for 10 s. Place on a magnet stand to clear and pull off the liquid.
-
13.
Wash beads once with 1 mL Wash buffer, quick spin on a microcentrifuge, place on a magnet stand to clear and pull off the liquid.
-
14.
Resuspend beads in 300 μL Wash buffer. Add 100 μL Wash buffer to the “input” fraction.
-
15.
Proceed to DNA extraction (steps 18–27 of Subheading 3.2) with each ChIP and input sample.
3.4 Library Preparation and Paired-End Illumina Sequencing
-
1.
Optional: Use 1–2 μL of the DNA obtained from CUT&RUN or CUT&RUN.ChIP (as in step 27 of Subheading 3.2) to quantify double-stranded (ds) DNA concentration, for example, using fluorescence detection with a Qubit instrument (see Note 16).
-
2.
Optional: Evaluate the presence and size distribution of cleaved fragments by capillary electrophoresis with fluorescence detection, for example by using a TapeStation 4200 instrument with D1000 high-sensitivity reagents, following the manufacturer’s instructions (see Note 16). CUT&RUN for a highly abundant chromatin-associated protein such as the histone variant H2A.Z (with an anti-FLAG antibody targeting Htz1-3XFLAG) typically shows a nucleosomal ladder; and DNA fragments corresponding to mononucleosomes get specifically enriched upon native ChIP for H2A.Z using an antibody targeting a different epitope on the protein (Fig. 4).
-
3.
Prepare barcoded libraries for Illumina sequencing with Tru-Seq compatible adapters and indexing primers using a single-tube protocol, following the manufacturer’s instructions (also see published protocols [12, 19] and Note 16). Good results have been obtained using the Hyper-prep kit from KAPA Biosystems (https://sequencing.roche.com/en-us/products-solutions/by-category/library-preparation/dna-library-preparation/kapa-hyperprep.html). Remove un-ligated adapters after the adapter ligation step by SPRI paramagnetic bead-based sequential cleanups (e.g., with AMPure XP beads) using 1.2× and 1.4× vol/vol ratios of sample to bead suspension. To minimize the contribution of large DNA fragments and to minimize PCR duplicates, PCR cycles should be between 8 and 14 cycles, preferably with a 10 s at 60 °C combined annealing/extension step (see Table 1 and Note 17). Remove excess PCR primers by two rounds of post-PCR-amplification cleanup using 1.4× vol/vol ratio of sample to bead suspension.
-
4.
Quantify library yield using a dsDNA-specific assay as in step 1.
-
5.
Determine the size distribution of libraries by capillary electrophoresis as in step 2, and mix libraries to achieve equal representation as desired aiming for a final concentration as recommended for the sequencing platform.
-
6.
Perform paired-end 25 × 25 base pair Illumina sequencing on the barcoded libraries for 25 cycles using an Illumina HiSeq 2500 or another massively parallel DNA sequencer following the manufacturer’s instructions (also see published protocol [12, 19] and Note 18).

TapeStation analysis of CUT&RUN(.ChIP) experiments. Example gel images (left) and densitometric scanning (right) show the fragment length profiles of DNA from Htz1-FLAG CUT&RUN (blue) and H2A.Z ChIP targeting a separate epitope after Htz1-FLAG CUT&RUN (orange), before (panel A) and after (panel B) sequencing-library preparation. CUT&RUN produces a nucleosomal ladder, while mononucleosomal fragments are specifically enriched upon the subsequent ChIP. Note the increase in fragment sizes post library preparation due to adapter ligation (~120 bp)
3.5 Data Processing and Analysis
-
1.
Align paired-end Saccharomyces cerevisiae reads to the sacCer3/V64 genome using Bowtie2 version 2.4.1 with parameters “--end-to-end --very-sensitive --no-mixed --no-discordant -q --phred33 -I 10 -X 700”. Align spike-in fragments to repeat-masked FlyBase r6.06 (Drosophila) genome, with parameters “--end-to-end --very-sensitive --no-overlap --no-dovetail --no-mixed --no-discordant -q --phred33 -I 10 -X 700” to avoid cross-mapping of the experimental genome to that of the spike-in DNA (see Note 19). Extract properly paired reads from the alignments.
-
2.
Make scaled fractional count or spike-in calibrated BedGraph for each sample using genomecov (with the “-bg” option) in BEDtools v2.30.0. For scaled fractional count, compute the fraction of all aligned bases at each base pair and scale it by the size of the sacCer3 genome (121,507,105 bp), so if the counts were uniformly distributed the value would be one at each base pair. For spike-in calibration compute the fraction of all aligned bases at each base pair, scale it by the total number of reads mapped to the spike-in genome, and divide by a large number (e.g., 10,000) (see Note 19). Unix-compatible scripts for processing CUT&RUN data and spike-in calibration are available from https://github.com/Henikoff/Cut-and-Run
-
3.
To analyze TF-binding and the position of flanking nucleosomes from the same experiment, separate sacCer3 reads into ≤120 bp and ≥ 150 bp size classes for generating BedGraph (see Note 20).
-
4.
Data shown in Figs. 2 and 3 are BedGraph files displayed as genomic tracks using Integrated Genome Browse (IGB) version 2.3.32.
4 Notes
-
1.
The extent of spheroplasting can be monitored by removing 20 μL aliquots from the spheroplasting reaction into 1 mL of 1% SDS and measuring OD600 (vs. 1% SDS blank). Spheroplasting is typically complete when OD600 is 10–20% of the starting value (usually in 10–15 min at 37 °C). Spheroplasting should be carefully monitored, do not over- or under-digest. Work with one yeast pellet at a time.
-
2.
We do not recommend flash freezing, as this can cause background DNA breakage that may impact final data quality.
-
3.
Although low-retention pipette tips are preferred for accurate solution transfer, use only conventional (not low-binding) microcentrifuge tubes to avoid loss of beads between wash steps and buffer exchanges.
-
4.
Take the supernatant after bead binding and count nuclei using a cell counter to determine the percent of nuclei bound to the beads. Alternatively, take a small volume of the nuclei suspension before bead binding and the supernatant after bead binding, and compare nuclei counts under a microscope using a cell counting device such as a hemocytometer.
-
5.
EDTA in antibody buffer removes excess divalent cations used to activate the ConA, because carryover of Ca++ from the beads can prematurely initiate strand cleavage after addition of pA-MNase. Chelation of divalent cations when cells are permeabilized also serves to quickly halt metabolic processes and prevent endogenous DNase activity. Washing out the EDTA before pA-MNase addition avoids inactivating the enzyme. Spermidine in the wash buffer is intended to compensate for removal of Mg++, which might otherwise affect chromatin properties.
-
6.
The primary antibody should be used at a dilution recommended for immunofluorescence-type experiments. If a working dilution is not known, we recommend 1:100 as a starting point. Although unbound antibodies are washed away in the subsequent washing steps, using excess antibodies can potentially lead to higher background and/or nonspecific signals.
-
7.
Do not include a no-antibody control, as the lack of tethering may allow any unbound pA-MNase to act as a “time-bomb” and digest accessible DNA, resulting in a background of DNA-accessible sites.
-
8.
The binding efficiency of Protein A to the primary antibody depends on host species and IgG isotype. For example, Protein A binds well to rabbit and guinea pig IgG but poorly to mouse and goat IgG, and so for these latter antibodies, a secondary antibody, such as rabbit anti-mouse IgG is recommended. The hybrid Protein A-Protein G-MNase fusion (pA/G-MNase) binds avidly to a broad range of antibody species and subtypes (e.g., mouse antibodies) [18]. Nevertheless, using a secondary antibody is likely to provide higher yield, and is therefore recommended.
-
9.
Enzymatic activity of purified recombinant pA-MNase or pAG-MNase can be determined empirically by enzyme titration for in vitro digestion of plasmid DNA, or purified budding yeast chromatin to obtain mostly mononucleosomal fragments, and by CUT&RUN using an antibody against an abundant epitope. We found that when our in-house purified pAG-MNase was used in excess, there was no increase in release of background fragments, indicating that the washes are effective in removing excess unbound fusion protein [18].
-
10.
MNase binds DNA but only cleaves when Ca++ is present, so that digestion is a zero-order reaction that seems to be less temperature-dependent than the subsequent diffusion of released pA-MNase-bound particles, which can digest accessible regions of the genome. Efficient cleavage and release of particles can be obtained at 0 °C while minimizing background cleavages attributable to diffusion. We found that digestion at ambient or higher temperatures result in unacceptable background cleavage levels.
-
11.
Digestion time needs to be optimized for the factor of interest. For stable chromatin features such as histone posttranslational modifications on nucleosomes, longer digestion (up to 30 min at 0 °C) may increase yield without significantly increasing background cleavages. However, for proteins that weakly interact with DNA, such as certain transcription factors, a shorter digestion time might be more suitable as longer digestion with the tethered MNase may lead to their eviction from chromatin due to extensive digestion of the underlying DNA.
-
12.
Heterologous spike-in DNA should be present in the 2× STOP to calibrate DNA amounts, for example to compare treatments or digestion time points. This is especially important for CUT&RUN as there is too little background cleavage for normalization of samples.
-
13.
Long incubation at −20 °C and long centrifugation for DNA precipitation may result in better recovery of small DNA fragments.
-
14.
It is extremely important to compete off bound antibodies from the released targets to prevent their recapture by Protein A– or Protein G–coated magnetic beads during the ChIP step. To evaluate successful competition of the antibodies used in CUT&RUN and specificity of the ChIP antibody, compare spike-in DNA-calibrated ChIP and IgG negative control samples versus CUT&RUN supernatant (used as ChIP input). Also see Fig. 3.
-
15.
The choice of Protein A- or Protein-G coated magnetic beads depends on the host species and isotypes of ChIP antibodies.
-
16.
Some long undigested DNA will leak through in CUT&RUN and dominate Qubit fluorescence, particularly in CUT&RUN of typical transcription factors. For these, the amount of targeted DNA recovered is too low to be detected, even by TapeStation analysis. For such samples, we often use the entire amount of recovered DNA for making sequencing libraries. We do not recommend size selection of DNA before library preparation.
-
17.
Rapid PCR cycles favor exponential amplification of the desired CUT&RUN fragments over linear amplification of large DNA fragments that are too long for polymerase to complete. Higher than 14 PCR cycles may result in unacceptable proportions of PCR duplicates in the sequenced libraries. Higher ratio of beads during the cleanup steps ensure that small sub-nucleosomal size DNA fragments (such as those resulting from CUT&RUN of transcription factors) are retained in the library.
-
18.
Paired-end sequencing is recommended for CUT&RUN, as single-end sequencing sacrifices resolution and discrimination between transcription factors and neighboring nucleosomes. Because of the very low background with CUT&RUN, typically one–three million paired-end reads suffices for transcription factors or nucleosome modifications in budding yeast.
-
19.
We found that pAG-MNase purified over a nickel-agarose column using the protocol as described in Meers et al., 2019 [18] contains enough carryover Escherichia coli DNA which can be used for spike-in calibration, and shows close correspondence with adding heterologous spike-in DNA at the end of a CUT&RUN experiment. Align paired-end E. coli reads to NCBI Ecoli (Escherichia coli str. K-12 substr. MG1655 U00096.3) with parameters as described in Subheading 3.5, step 1, and use the data for spike-in calibration of SerCer3 reads as described in Subheading 3.5, step 2.
-
20.
This analysis provides mapping of the local vicinity of a DNA-binding protein, but can vary depending on the steric access to the DNA by the tethered MNase.
References
Lai WKM, Pugh BF (2017) Understanding nucleosome dynamics and their links to gene expression and DNA replication. Nat Rev Mol Cell Biol 18:548–562
Barski A, Cuddapah S, Cui K et al (2007) High-resolution profiling of histone methylations in the human genome. Cell 129:823–837
Park D, Lee Y, Bhupindersingh G et al (2013) Widespread misinterpretable ChIP-seq bias in yeast. PLoS One 8:e83506
Teytelman L, Thurtle DM, Rine J et al (2013) Highly expressed loci are vulnerable to misleading ChIP localization of multiple unrelated proteins. Proc Natl Acad Sci U S A 110:18602–18607
Teves SS, An L, Hansen AS et al (2016) A dynamic mode of mitotic bookmarking by transcription factors. elife 5:e22280
Jain D, Baldi S, Zabel A et al (2015) Active promoters give rise to false positive ‘Phantom Peaks’ in ChIP-seq experiments. Nucleic Acids Res 43:6959–6968
Rossi MJ, Kuntala PK, Lai WKM et al (2021) A high-resolution protein architecture of the budding yeast genome. Nature 592:309–314
He Q, Johnston J, Zeitlinger J (2015) ChIP-nexus enables improved detection of in vivo transcription factor binding footprints. Nat Biotechnol 33:395–401
Skene PJ, Henikoff S (2015) A simple method for generating high-resolution maps of genome-wide protein binding. elife 4:e09225
Kasinathan S, Orsi GA, Zentner GE et al (2014) High-resolution mapping of transcription factor binding sites on native chromatin. Nat Methods 11:203–209
Van Steensel B, Delrow J, Henikoff S (2001) Chromatin profiling using targeted DNA adenine methyltransferase. Nat Genet 27:304–308
Skene PJ, Henikoff S (2017) An efficient targeted nuclease strategy for high-resolution mapping of DNA binding sites. eLife 6:e21856
Grunberg S, Zentner GE (2017) Genome-wide mapping of protein-DNA interactions with ChEC-seq in saccharomyces cerevisiae. J Vis Exp 124:55836
Schmid M, Durussel T, Laemmli UK (2004) ChIC and ChEC; genomic mapping of chromatin proteins. Mol Cell 16:147–157
Carter B, Ku WL, Kang JY et al (2019) Mapping histone modifications in low cell number and single cells using antibody-guided chromatin tagmentation (ACT-seq). Nat Commun 10:3747
Kaya-Okur HS, Wu SJ, Codomo CA et al (2019) CUT&tag for efficient epigenomic profiling of small samples and single cells. Nat Commun 10:1930
Henikoff JG, Belsky JA, Krassovsky K et al (2011) Epigenome characterization at single base-pair resolution. Proc Natl Acad Sci U S A 108:18318–18323
Meers MP, Bryson TD, Henikoff JG et al (2019) Improved CUT&RUN chromatin profiling tools. Elife 8:e46314
Skene PJ, Henikoff JG, Henikoff S (2018) Targeted in situ genome-wide profiling with high efficiency for low cell numbers. Nat Protoc 13:1006–1019
Brahma S, Henikoff S (2019) RSC-associated subnucleosomes define MNase-sensitive promoters in yeast. Mol Cell 73(238–249):e233
Janssens DH, Wu SJ, Sarthy JF et al (2018) Automated in situ chromatin profiling efficiently resolves cell types and gene regulatory programs. Epigenetics Chromatin 11:74
Weiner A, Hsieh TH, Appleboim A et al (2015) High-resolution chromatin dynamics during a yeast stress response. Mol Cell 58:371–386
Acknowledgments
We thank Christine Codomo for preparing Illumina sequencing libraries, the Fred Hutch Genomics Shared Resource for sequencing, and Jorja Henikoff for help with data analysis. Research reported in this publication was supported by the National Institute of General Medical Sciences of the National Institutes of Health under award number K99GM138920 (S.B.) and the Howard Hughes Medical Institute (S.H.).
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Open Access This chapter is licensed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license and indicate if changes were made.
The images or other third party material in this chapter are included in the chapter's Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the chapter's Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.
Copyright information
© 2022 The Author(s)
About this protocol

Cite this protocol
Brahma, S., Henikoff, S. (2022). CUT&RUN Profiling of the Budding Yeast Epigenome. In: Devaux, F. (eds) Yeast Functional Genomics. Methods in Molecular Biology, vol 2477. Humana, New York, NY. https://doi.org/10.1007/978-1-0716-2257-5_9
Download citation
DOI: https://doi.org/10.1007/978-1-0716-2257-5_9
Published:
Publisher Name: Humana, New York, NY
Print ISBN: 978-1-0716-2256-8
Online ISBN: 978-1-0716-2257-5
eBook Packages: Springer Protocols