
Abstract.
Glycogen synthase 2 (Gys-2) is the ratelimiting enzyme in the storage of glycogen in liver and adipose tissue, yet little is known about regulation of Gys-2 transcription. The peroxisome proliferator-activated receptors (PPARs) are transcription factors involved in the regulation of lipid and glucose metabolism and might be hypothesized to govern glycogen synthesis as well. Here, we show that Gys-2 is a direct target gene of PPARα, PPARβ/δ and PPARγ. Expression of Gys-2 is significantly reduced in adipose tissue of PPARα-/-, PPARβ/δ-/- and PPARγ+/- mice. Furthermore, synthetic PPARβ/δ, and γ agonists markedly up-regulate Gys-2 mRNA and protein expression in mouse 3T3-L1 adipocytes. In liver, PPARα deletion leads to decreased glycogen levels in the refed state, which is paralleled by decreased expression of Gys-2 in fasted and refed state. Two putative PPAR response elements (PPREs) were identified in the mouse Gys-2 gene: one in the upstream promoter (DR-1prom) and one in intron 1 (DR-1int). It is shown that DR-1int is the response element for PPARs, while DR-1prom is the response element for Hepatic Nuclear Factor 4 alpha (HNF4α). In adipose tissue, which does not express HNF4α, DR-1prom is occupied by PPARβ/δ and PPARγ, yet binding does not translate into transcriptional activation of Gys-2. Overall, we conclude that mouse Gys-2 is a novel PPAR target gene and that transactivation by PPARs and HNF4α is mediated by two distinct response elements.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Metabolic syndrome describes a combination of metabolic abnormalities that include central obesity, dyslipidemia, hypertension, insulin resistance, and a pro-inflammatory and pro-thrombotic state. An important group of pharmacological targets for the treatment of metabolic syndrome are the peroxisome proliferator activated receptors (PPARs). PPARs are ligand-activated transcription factors belonging to the superfamily of nuclear receptors, which include numerous cellular receptors for nutrients and steroids. So far, three PPAR isotypes (α, β/δ, γ) have been identified in a wide range of species, each displaying a different tissue distribution and ligand specificity [1]. PPARs share a similar structure and a common molecular mechanism of action by forming an obligate heterodimer with the 9-cis retinoic acid receptor RXR. PPAR-RXR heterodimers selectively bind genomic sequences consisting of a direct repeat of the hexameric nucleotide sequence AGGTCA separated by 1 nucleotide (Direct Repeat-1). These so-called peroxisome proliferator response elements (PPRE) are located in the promoter of PPAR target genes or in intronic regions [2–5].
The PPARα isotype (NR1C1) is highly expressed in liver, and governs the adaptive response to fasting [6–8]. PPARα is an extremely important regulator of hepatic nutrient metabolism including fatty acid oxidation (peroxisomal and mitochondrial), fatty acid uptake, amino acid metabolism, glycerol metabolism, and lipoprotein assembly and transport [9–11]. In addition, PPARα potently suppresses the hepatic inflammatory response [12, 13], an effect that is also observed in extra-hepatic tissues such as the vascular wall [14]. Much less is known about the role of PPARα in other tissues, although evidence is accumulating that PPARα induces cardiac and skeletal muscle fatty acid oxidation [15, 16]. Importantly, PPARα mediates the effects of hypolipidemic fibrate drugs, which decrease plasma triglycerides and increase plasma HDL concentrations. In contrast to PPARα, PPARγ (NR1C3) is highly expressed in white adipose tissue (WAT), where it promotes lipid storage. PPARγ is a key transcription factor in the adipogenesis program and is essential for adipocyte survival [17, 18]. It also serves as the molecular target for the thiazolidine-dione (TZD) class of insulin-sensitizing drugs that are widely used in the treatment of type 2 diabetes. PPARγ promotes whole body glucose utilization; however, it has been difficult to identify the molecular mechanisms behind this effect. Much of the attention has been focused on possible cross-talk between adipose tissue and skeletal muscle, as muscle is responsible for the major share of whole body glucose utilization. However, adipose tissue is a large organ, especially in the obese, and accordingly it can also be envisioned that the insulin-sensitizing effect of TZDs on glucose uptake is partially exerted at the adipose tissue level.
While PPARα and PPARγ have been extensively studied over many years, much less is known about the function of the PPARβ/δ isotype (NR1C2). Studies with genetically modified PPARδ/δ mice have illustrated the importance of this nuclear receptor in WAT and skeletal muscle, two organs that have a key role in glucose homeostasis [19–22]. It was shown that activation of PPARβ/δ in adipose tissue protects against adiposity and hyperlipidemia by inducing fatty acid catabolism [21]. Moreover, pharmacological activation as well as specific constitutive over-expression of PPARβ/δ leads to a shift in muscle fiber composition towards type I muscle fibers, resulting in increased muscle oxidative capacity [20, 22]. PPARβ/δ has also been shown to stimulate hepatic VLDL production, influence wound healing, and affect colon carcinogenesis [23–25]. However, whether PPARδ/δ has a functional role in glucose homeostasis, in analogy with other PPAR isotypes, remains to be firmly established.
Here we show that glycogen synthase 2 (Gys-2), the rate-limiting enzyme for glycogen synthesis in liver and adipose tissue, is a target gene of PPARα, PPARβ/δ and PPARγ. Transcriptional regulation is achieved via a PPAR response element present in the first intron. We show that an additional direct repeat response element identified in the Gys-2 promoter mediates transactivation by hepatic nuclear factor 4α (HNF4α).
Materials and methods
Chemicals. Wy14643 was obtained from Eagle Picher Technologies laboratories (Lenexa, KS, USA). Rosiglitazone was from Alexis (Breda, The Netherlands). SYBR Green was from Eurogentec (Seraing, Belgium). Dulbecco’s modified Eagles medium (DMEM), fetal calf serum (FCS), calf serum and penicillin/streptomycin/fungizone were from Cambrex Bioscience (Seraing). Otherwise, chemicals were from Sigma (Zwijndrecht, The Netherlands).
Animal experiments. PPARβ/δ mutant null mice (PPARβ/δ-/-) and PPARγ heterozygous mice (PPARγ+/−) were on a mixed background (Svl29/C57BL/6) and have been described previously [26, 27]. Wild-type littermates served as control animals. PPARα-/- mice and corresponding wild-type mice on an Svl29 background were purchased at Jackson Laboratories (Bar Harbor, ME, USA). Liver-specific hepatocyte nuclear factor 4α (HNF4α)-null mice were generated as described previously. Livers were collected from 45-day-old HNF4αflox/flox X albumin-Cre+ (KO) and HNF4αflox/flox X albumin-Cre− (FLOX) mice [28]. Mice were maintained at 20°C with a 12-h light-dark cycle. All mice were between 3 and 6 months of age. For the fasting experiment, 3-month-old male mice were fasted for different periods of time starting at the onset of the light cycle. For the refeeding experiment, mice were fasted for 24 h after which they were put back on chow for 7 h before sacrifice. After sacrificing the animals, tissues were immediately frozen in liquid nitrogen. The animal experiments were approved by the animal experimentation committee of Wageningen University or the Etat de Vaud (Switzerland).
Oligonucleotide micro-array. Total RNA was prepared from epididymal WAT of wild-type and PPARβ/δ-/- mice (five animals of each genotype) using TRIzol reagent (Invitrogen, Breda, The Netherlands) and subsequently pooled per group. Pooled RNA was further purified using Qiagen RNeasy columns, and the quality was verified using Bioanalyzer 2100 (Agilent, Amsterdam). For one cycle cRNA synthesis (Affymetrix, Santa Clara, USA) 10 µg of RNA was used. Hybridization, washing and scanning of Affymetrix GeneChip mouse genome 430 2.0 arrays was according to standard Affymetrix protocols. Fluorimetric data were processed by Affymetrix GeneChip operating software and the gene chips were globally scaled to all the probe sets with an identical target intensity value. Further analysis was performed by Data Mining Tool (Affymetrix).
3T3-L1 adipogenesis assay. 3T3-L1 fibroblasts were grown in DMEM plus 10 % calf serum and plated for final differentiation in DMEM plus 10 % FCS. At 2 days after reaching confluence, the medium was changed and the following compounds were added: isobutyl methylxanthine (0.5 mM), dexamethasone (1 µM), and insulin (5 µg/mL). After 3 days, the medium was changed to DMEM plus 10% FCS and insulin (5 µg/mL). After 6 days the medium was changed to DMEM plus 10% FCS, which was changed every 3 days.
Primary mouse or rat hepatocyte isolation. Primary mouse and rat hepatocytes were isolated as described previously [29]. Briefly, after cannulation of the portal vein, the liver was perfused with calcium-free HBSS, which was pre-gassed with 95 % O2 /5 % CO2. Next, the liver was perfused with a collagenase solution until swelling and degradation of the internal liver structure was observed. The hepatocytes were released, filtered and washed several times using Krebs buffer. The viability was assessed by tryptan blue staining and was at least 80 %. Cells were cultured in William’s Medium E supplemented with 10% FCS, penicillin/streptomycin/fungizone, insulin and dexamethasone. Cells were plated in collagen (Serva Feinbiochemica, Heidelberg, Germany) -coated wells with a density of 0.5×106 cells/ml. After 4 h of incubation, the medium was removed and replaced with fresh medium. The next day, hepatocytes were used for experiments.
RNA isolation, reverse-transcription, and real-time quantitative PCR. Total RNA was extracted from tissues with TRIzol reagent (Invitrogen); 1 µg total RNA was then reverse-transcribed with iScript (Bio-Rad, Veenendaal, The Netherlands). cDNA was PCR-amplified with Platinum Taq DNA polymerase (Invitrogen) on a Bio-Rad iCycler or MylQ PCR machine. Primers were designed to generate a PCR amplification product of 100–200 bp and were taken from Primerbank (http://pga.mgh.harvard.edu/primerbank/). Specificity of the amplification was verified by melt curve analysis and evaluation of efficiency of PCR amplification. Expression was related to the control gene 36B4, which did not change under any of the experimental conditions studied.
The following primer pairs were used: mGys-2 (forward): CCAGCTTGACAAGTTCGACA, mGys-2 (reverse): AT-CAGGCTTCCTCTTCAGCA, m36B4 (forward): AGCGCGTCCTGGCATTGTGTGG, m36B4 (reverse): GGGCAGCAGTGGTGGCAGCAGC, mPPARα (forward): TATTCGGCTGAAGCTGGTGTAC, mPPARα (reverse): CTGGCATTTGTTCCGGTTCT, mPPARβ (forward): TTGAGCCCAAGTTCGAGTTTG, mPPARβ (reverse): CGGTCTCCACACAGAATGATG, mPPARγ (forward): CA-CAATGCCATCAGGTTTGG, mPPARγ (reverse): GCTGGTCGATATCACTGGAGATC.
Transactivation assay. The proximal part of the mouse Gys-2 promoter was PCR amplified from mouse genomic DNA (strain C57BL/6) using the forward primer: 5′ CTTGCTGCCTTTCAG-GAGAGGGCAG 3′ and reverse primer: 5′ TTCTCTTTAGC-CATTA AG ATAG 3′. The resulting 553-bp fragment was used for a second PCR amplification step introducing Hindlll and Kpnl sites, which were used for subcloning into the pGL-3 Basic vector (Invitrogen).
A 156-bp nucleotide fragment surrounding the putative PPRE within the mGys-2 promoter was PCR amplified from mouse genomic DNA (strain C57BL/6) and subcloned into Kpnl/Bg/II sites of the pGL3 SV40 promoter vector (pGL3-tk-LUC, Promega, Leiden, The Netherlands) using the forward primer: 5′AAATCG-CAGCTGAAACCT 3′, and reverse primer: 5′ CTCCTGCTTGTGCTTCTGC 3′. A 314-nucleotide fragment surrounding the putative PPRE within intron 1 of the mouse Gys-2 gene was PCR amplified from mouse genomic DNA (strain C57BL/6) and subcloned into the Kpnl and Bglll sites of the pGL-Tk-Luc reporter gene. Reporter vectors were transfected into human hepatoma HepG2 cells, together with an expression vector (pSG5) for mPPARα, mPPARβ, or mPPARyl, in the presence or absence of Wy14643 (50 µM), L-165041 (5 µM), or rosiglitazone (5 µM), respectively. Transfections were carried out using the calcium-phosphate precipitation method. A β-galactosidase reporter vector was co-transfected to normalize for differences in transfection efficiency. Luciferase activity was measured 24 h post transfection using the Promega luciferase assay kit (Promega) on a Fluoroskan Ascent Fl apparatus (Thermo labsystems, Breda, The Netherlands). β-Galactosidase activity was measured in the cell lysate by a standard assay using 2-nitrophenyl-βD-galactopyrano-side as a substrate. To disable the mouse Gys-2 PPRE within the mGys-2 promoter, two separate (A and B) partially overlapping PCR fragments were generated using the wild-type mGys-2 promoter as a template. Primers sets used to generate part A of the mutated mGys-2 promoter fragment were: 5′-TTTGGTCTAAAGGCCTTTGGCCAAAGG-3′ and 5′-CTTGCTGCCTTTCAGGAGAGGGC
AG-3′. Primers sets used to generate part B of the mutated mGys-2 promoter fragment were: 5′-CCTTTGGCCAAAGGCCTTTA-GACCAAA-3′ and 5′-TTCTCTTTAGCCATTAAGATAGG-GATTG-3′. PCR was carried-out using the two upper DNA fragments and the following primers: 5′- cccaagcttCTTGCTGCCTTTCAGGAG-3′ and 5′- ggggtaccTTCTCTTTAGCCATTAAGATAG-3′. The PCR fragment was subsequently cloned into the pGL3 basic reporter vector (HindIII/KpnI cloning site) and verified by automated sequencing. The hHNF4α expression plasmid was constructed by amplifying human hepatoma HepG2 cDNA using the following primers, forward primer 5′-GAATGCGACTCTCCAAAACC-3′ and reverse primer 5′-ATCCTTCCCATTCCTGCTCT-3′, followed by subcloning of the resulting PCR product into an pGEM-Teasy vector (Promega). The insert was excised by NotI digestion and further subcloned into pcDNA3.1/V5-HisA (Invitrogen). The nucleotide sequence was verified by automated sequencing.
Gel shift. hRXRα and mPPARγ proteins were generated from pSG5 expression vectors, using the TNT coupled in vitro transcription/translation system (Promega). The following oligos were annealed to generate the double-stranded DNA probe; for DR-lint: 5′-CAGGACTTTGGTGACCTCTGGCCTATAT-3′ and 5′-ACACATATAGGCCAGAGGTCACCAAAGTC-3′. For nonspecific competition, the following primers were used: etsF 5′-TGGAATGTACCGGAAATAACACCA-3′, etsR 5′- TGGTGTTATTTCCGGTACATTCCA-3′. Oligonucleotides were annealed and labeled by Klenow filling (New England Biolabs, Leusden, The Netherlands) using Redivue [α-32P]dCTP (3000 Ci/mmol; Amersham, Roosendaal, The Netherlands). In vitro-translated proteins (0.5–0.8 µl/reaction) were pre-incubated for 15 min on ice in 1× binding buffer (80 mM KCl, 1 mM DTT, 10 mM Tris-HCl pH 7.4, 10 % glycerol, plus protease inhibitors) in presence of 2 µg poly[dI.dC], 5 µg sonicated salmon sperm DNA and competitor oligonucleotides in a final volume of 20 µl. Then 1 ng (1 ng/µl) of radiolabeled oligo was added and incubation was continued for another 10 min at room temperature. Complexes were separated on a 4 % Polyacrylamide gel (acrylamide/bisacrylamide 37.5:1) equilibrated in 0.5× TBE at 25 mA.
Chromatin immunoprecipitation. Chromatin immunoprecipitation (ChIP) on 3T3-L1 cells and mouse liver was carried out as described previously [11]. Sequences of primers used for PCR were 5′-TCTGGCAGGCATAAGGACCCGAGTT-3′ and 5′- GGAAGCCAGGACAGAGTGCAAATACAAT-3′ for DR-lint (Intron). For DR-lprom, the following primers were used: 5′-AAAACTGCTTGTGTCTGAGGGAAAC-3′ and 5′-AGAG-GACAGACTGAGCATGACAAGAG-3′. Control primers used were 5′-GCTGCGAGATCCATCACCCACTAAAC-3′ and 5′-AGCCATCTCACCAGCCCCAACTT-3′. Antibodies against PPARs were from Santa Cruz Biotechnology (Santa Cruz, CA, USA). ChIP on rat hepatocytes was done using a commercially available kit (Active Motif, Rixensart, Belgium). The HNF4α antibody was from Tebu-Bio (Heerhugowaard, The Netherlands). The primers used to amplify the sequence surrounding the DR-lprom were 5-GAATGCCGCTGTGCCTGAGGGAAAC-3′ and 5′-AGAGGACAGAAGAAGAGTGACAAGAG-3′. For DR-lint: 5′-TCTGTCAGGCATAAGGACCTGGGTT-3′ and 5′-ATTGTATTTGAACTCTGTCCTGGTCTCT-3′.
Histology. Liver tissue from wild-type and PPARα-/- mice was embedded in Tissue-Tek O.C.T compound from Sakura Finetek (Zoeterwoude, The Netherlands) and frozen. Cryosections of 5 µm from frozen liver were made and analyzed for glycogen accumulation using the periodic acid-Schiff (PAS) reaction. Hematoxylin and eosin staining of liver cryosections was done using standard protocols.
Western blot. A mouse anti-glycogen synthase monoclonal antibody was used (clone GS-7H5 MAB3106) (Chemicon International, Hampshire, UK). Western blotting was carried out as previously described [4]. The primary antibody was used at a dilution of 1:1000 and the secondary antibody (anti-mouse IgG, Dako, Glostrup, Denmark) was used at a dilution of 1:8000.
Results
Expression of Gys-2 in WAT is regulated by PPARs.
Our initial aim was to identify novel putative target genes of PPARβ/δ in WAT. Accordingly, we compared gene expression in WAT of wild-type versus PPARβ/δ-/- mice using Affymetrix micro-array analysis. The expression of several genes involved in glucose and lipid metabolism was down-regulated in PPARβ/δ-/-mice, including PPARγ, PGC-lα, and GLUT4, which was confirmed for several genes by real-time quantitative PCR (qPCR) (Table 1). Expression of Gys-2 was most significantly down-regulated in PPARβ/δ-/-mice, and therefore Gys-2 was selected for more detailed investigation. Q-PCR confirmed the marked down-regulation of Gys-2 mRNA in WAT of PPARβ/δ-/- mice (Fig. la). Furthermore, expression of Gys-2 also appeared to be down-regulated in WAT of PPARγ+/− and PPARα-/- mice, although the former result did not achieve statistical significance. In PPARβ/δ-/- mice, the decrease in Gys-2 mRNA was paralleled by a significant down-regulation of PPARα and PPARγ expression, while in PPARα-/- mice expression of PPARβ/δ was significantly down-regulated (Fig. lb). These data show that PPARs are crucial for maintaining Gys-2 expression in fat, although it is difficult to ascertain which PPAR isotype is the main regulator of Gys-2 expression in WAT.

Expression of Glycogen synthase 2 (Gys-2) in white adipose tissue (WAT) is regulated by peroxisome proliferator-activated receptors (PPARs). Expression of Gys-2 (a) and PPARs (b) in WAT of PPARβ/δ-/- mice, PPARγ+/− mice, and PPARα-/-mice, as determined by qPCR. The effects of PPAR deletion were evaluated by Student’s Mest (* p<0.05; ** p<0.01). Error bars represent SEM (n=5). (c) Differentiated 3T3-L1 adipocytes were treated with the synthetic PPARγ agonists rosiglitazone or ciglitazone, or the PPARβ/δ agonist L165041 for 24 h. Expression of Gys-2 was determined by qPCR. Expression of cells treated with vehicle (DMSO) was set at 1. Data shown are representative results from three independent experiments, (d) Gys protein expression was analyzed in lysates from 3T3-L1 adipocytes treated with either PPARβ/δ (L165041, 2.5 µM) or PPARγ (rosiglitazone, 1 µM) agonist.
To investigate whether expression of Gys-2 in adipocytes is under direct control of PPARs, the effect of PPAR ligands on Gys-2 mRNA was studied in differentiated mouse 3T3-L1 adipocytes. It was observed that the PPARβ/δ agonist L165041, and the PPARγ agonists ciglitazone and rosiglitazone significantly induced Gys-2 mRNA levels (Fig. 1c). This effect was confirmed at the protein level (Fig. 1d). Thus, Gys-2 may represent a direct target gene of PPARγ and PPARβ/δ in adipocytes.
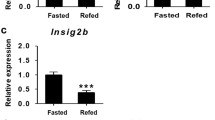
PPARα governs Gys-2 hepatic expression. A link between PPARs and glycogen has been previously made. It was observed that liver glycogen levels were reduced in refed PPARα-/- mice compared to refed wild-type mice [30, 31], which we confirmed using histochemical staining (Fig. 2). Furthermore, hepatic expression of Gys-2 was decreased in PPARα-/- mice [32]. Expression of Gys-2 is highest in liver, followed by WAT (our unpublished data). We confirm that hepatic Gys-2 mRNA is markedly reduced in PPARα-/- mice; however, only in the 24-h fasted and refed state (Fig. 3a). To further examine the role of PPARα in Gys-2 expression, primary hepatocytes from wild-type and PPARα-/- mice were treated with the synthetic PPARα agonist Wy14643, allowing for a direct evaluation of the effect of PPARα activation on Gys-2 expression. Basal expression of Gys-2 was about fourfold reduced in PPARα-/- hepatocytes, indicating a requirement for PPARα (Fig. 3b). Furthermore, Wy14643 stimulated Gys-2 expression in wild-type but not in PPARα-/- hepatocytes (Fig. 3b). Induction of Gys-2 expression by synthetic PPARα agonists was also observed in rat primary hepatocytes (Fig. 3c). Together, these data suggest a direct role of PPARα in governing hepatic Gys-2 expression.

Staining for hepatic glycogen is higher in refed wild-type mice compared to refed PPARα-/- mice, (a) Representative hematoxylin and eosin staining of liver from two wild-type and two PPARα-/- mice, (b) Representative PAS staining of liver from two wild-type and two PPARα-/- mice. Mice were fasted for 24 h followed by refeeding for 7 h before sacrifice.

PPARα governs hepatic expression of Gys-2. (a) Relative expression of Gys-2 in fed, fasted and refed wild-type and PPARα-/- mice, as determined by qPCR. Mice were fasted for 0,6, 12 or 24 h, with refeeding for 7 h following 24 h fasting. Significant differences between wild-type and PPARα-/- mice were observed in the 24-h fasted and the refed state (b) Relative expression of Gys-2 in freshly isolated wild-type and PPARα-/- hepatocytes treated for 24 h with vehicle (DMSO) or Wy14643 (10 µM), as determined by qPCR. Significant effects were observed by two-way ANOVA for genotype (p<0.001), and for the interaction between the genotype and Wy14643 (p<0.05). (c) Relative expression of Gys-2 in freshly isolated rat hepatocytes which were treated for 24 h with either vehicle (DMSO), Wy14643 (50 uM) or fenofibrate (50 µM). The effects of Wy14643 and fenofibrate were statistically significant (Student’s Mest: p<0.01). Error bars represent SEM.
Identification of a putative PPRE in the proximal promoter of the mouse Gys-2 gene. To determine what genomic region could be responsible for the PPAR-induced up-regulation of Gys-2 mRNA, the mouse Gys-2 gene was scanned for potential PPREs (NUBIScan algorithm and Hidden Markov Model framework) [33, 34]. A Direct Repeat-1 motif (DR-lprom) was localized to the proximal Gys-2 gene promoter, about 169 bp upstream from the transcription start site. With the exception of two nucleotides, DR-lprom is identical to the consensus sequence, suggesting that this sequence could serve as a functional PPRE (Fig. 4a). DR-lprom was conserved between mouse and rat.

The DR-1 present in the Gys-2 promoter does not mediate PPAR-dependent transactivation. (a) Alignment of the consensus PPRE sequence with the sequence of rat and mouse Gys-2 DR-lprom and Gys-2 DR-lint. (b) HepG2 cells were transfected with a reporter vector containing a 553-nucleotide fragment of the proximal mouse Gys-2 promoter gene and PPAR expression vectors, (c) HepG2 cells were transfected with a SV40 reporter vector containing an isolated 156-nucleotide fragment surrounding DR-lprom of the proximal mouse Gys-2 promoter gene and PPAR expression vectors. Luciferase and β-galactosidase activities were determined 24 h after exposure of the cells to different PPAR agonists: 50 µM Wy14643, 5 µM L-165041 and 10 µM of rosiglitazone. Error bars represent SEM. Chromatin immunoprecipitation of DR-lprom using antibodies against mPPARγ (d) or mPPARδ/δ (e). The gene sequence spanning DR-lprom and a random control sequence were analyzed by PCR in the immunoprecipitated chromatin of 3T3-L1 preadipocytes and mature adipocytes. Preimmune serum was used as a control. PI, preimmune serum, Cntl, random control sequence. (f) Schematic overview of localization of primers used for amplification of immunoprecipitated DNA.
To examine whether the promoter region containing the putative PPRE is responsible for PPAR-dependent up-regulation of Gys-2 expression, a 553-nucleotide fragment of the mouse Gys-2 promoter gene was cloned in front of a luciferase reporter gene and transactivation studies were carried out in HepG2 cells. Surprisingly, co-transfection of PPARα or PPARγl expression vectors in combination with PPAR agonists slightly decreased luciferase activity, while PPARβ/δ activation had little effect (Fig. 4b). Transactivation assays performed with a small genomic fragment surrounding DR-lprom cloned in front of SV40-luciferase led to a similar overall PPAR-mediated repression for PPARα and PPARγl, while PPARγ/δ had little effect (Fig. 4c). Co-transfection of RXR or of different co-activators such as CBP and PGClα did not change this pattern (data not shown). Thus, the PPRE identified in the Gys-2 promoter probably does not mediate the effect of PPARs on Gys-2 expression. Nevertheless, ChIP experiments carried out in 3T3-L1 cells indicated that (1) PPARγ was bound to DR-lprom in mature adipocytes, but not in pre-adipocytes (Fig. 4d), and (2) PPARβ/δ was bound to DR-lprom in pre-adipocytes and, more strongly, in mature adipocytes (Fig. 4e). Thus, despite DR-lprom behaving poorly as a PPRE in classical transactivation assay, it binds both PPARγ and PPARδ/δ in adipocytes. This suggests that in vivo binding of PPARγ and PPARδ/δ to DR-lprom does not translate into transcriptional activation of the Gys-2 gene, and accordingly that activation of Gys-2 expression by PPARs may be mediated by another genomic region. It should be mentioned that ChIP did not reveal any binding of PPARα to DR-lprom in hepatocytes (data not shown).
Interestingly, using the same strategy as described above, a putative PPRE that is homologous to the consensus DR-1 sequence was identified in intron 1 of the mouse Gys-2 gene (Fig. 4a). To assess whether DR-lint was able to mediate PPAR-dependent transactivation, a 314-nucleotide genomic fragment surrounding DR-lint was cloned in front of the SV40 promoter followed by a luciferase reporter gene. In HepG2 cells treatment with the synthetic PPARα agonist Wy14643 induced reporter activity and this activation was further enhanced upon co-transfection of mPPARα (Fig. 5a). Similar inductions of reporter activity were observed for PPARβ/δ and PPARγ and their respective agonists (Fig. 5a). Thus, DR-lint is able to mediate PPAR-dependent transactivation, irrespective of the PPAR isotype, suggesting that it may at least be partially responsible for PPAR-dependent regulation of Gys-2 expression.

Gys-2 up-regulation by PPARs is mediated by a PPRE present in intron 1 of the Gys-2 gene (DR-lint). (a) HepG2 cells were transfected with a 314-nucleotide fragment of intron 1 of the mouse Gys-2 gene and PPAR expression vectors. Luciferase and β-galactosidase activities were determined 24 h after exposure of the cells to different PPAR agonists: 50 µM Wy14643, 5 µM L-165041 and 10 µM rosiglitazone. Error bars represent SEM. (b) Binding of the PPAR/RXR heterodimers to DR-lint as determined by gel shift assays. A double-stranded response element containing Gys-2 DR-lint was incubated with in vitro transcribed/translated mPPARα protein (left panel), mPPARβ/δ protein (middle panel) and mPPARγl protein (right panel) together with in vitro transcribed/translated hRXRα protein. Fold excess of specific (malic enzyme PPRE) or nonspecific (ETS oligonucleotide) cold probe is indicated.
In agreement with the transactivation data, PPARα, PPARβ/δ and PPARγ proteins were able to specifically bind DR-lint in gel shift experiments. A retarded heterodimeric complex was observed only in the presence of both PPAR and obligate binding partner RXRα (Fig. 5b). The complex disappeared in the presence of an excess of cold specific oligonucleotide, but not nonspecific oligonucleotide.
Examination of in vivo PPAR binding to DR-lint by ChIP yielded very similar results as for DR-lprom: PPARγ was bound to DR-lint in mature 3T3-L1 adipocytes, but not in pre-adipocytes (Fig. 6a), whereas PPARβ/δ was bound to DR-lint in both pre- and mature adipocytes (Fig. 6b). In liver, ChIP analysis demonstrated binding of PPARα to DR-lint in wild-type but not PPARα-/- mice, and binding was enhanced by fasting and Wy14643 (Fig. 6c). Together, these data indicate that mouse Gys-2 is a direct PPAR target gene and that regulation by PPARs is at least partially mediated by a PPRE present in intron 1.

PPARγ, PPARβ/δ, and PPARα bind to the Gys-2 DR-lint in vivo. Chromatin immunoprecipitation of Gys-2 DR-lint using antibodies against mPPARγ (a), mPPARβ/δ (b) or mPPARα (c). The gene sequence spanning DR-lint and a random control sequence were analyzed by PCR in the immunoprecipitated chromatin of 3T3-L1 preadipocytes and mature adipocytes (a, b) or mouse liver (c). Preimmune serum was used as a control. PI, preimmune serum, Cntl, random control sequence, (d) Schematic overview of localization of primers used for amplification of immunoprecipitated DNA.
Gys-2 is a novel direct target of the liver enriched factor HNF4α. As explained above, PPARα caused a reduction in Gys-2 promoter activity via DR-lprom (Fig. 4b, c). A similar decrease of promoter activity in response to PPARα despite the presence of a putative PPRE has been reported for other genes. Indeed, it was found that PPARα decreases expression of the apoCIII and transferrin genes via competition with the HNF4α. Since HNF4α is known to recognize DR-1 sequences as well, we examined whether HNF4α might control the expression of Gys-2 in liver, possibly via DR-lprom. Gys-2 mRNA levels were markedly decreased in liver-specific HNF4αnull mice, thus supporting a role for HNF4α in regulating Gys-2 expression (Fig. 7a). In transactivation assays using the Gys-2 promoter, HNF4α markedly activated reporter activity, suggesting the presence of a HNF4α response element within the 0.55-kb promoter fragment (Fig. 7b). Mutating DR-lprom resulted in an approximately 50 % reduction in HNF4α-dependent activation of the Gys-2 promoter (Fig. 7c), which suggests that (a) HNF4α responsiveness is partially mediated by DR-lprom, or (b) the mutations with DR-lprom only partially disabled HNF4α responsiveness. Regardless of these explanations, these data suggest that HNF4α directly regulates the hepatic expression of Gys-2 at least partially via DR-lprom. Finally, ChIP clearly showed HNF4α binding to DR-lprom but not DR-lint in rat primary hepatocytes (Fig. 7d).

Gys-2 DR-lprom is a binding site for the nuclear receptor HNF4α. (a) Gys-2 mRNA levels in liver of liver-specific HNF4α-null (HNF4α-/-) and wild-type (FLOX) mice were analyzed by qPCR (n=3 per group). (b) HepG2 cells were transfected with a reporter vector containing 553-bp of the mouse Gys-2 proximal promoter and increasing amounts of hHNF4α expression vector, (c) HepG2 cells were transfected with a reporter vector containing 553 bp of the wild-type and mutated mGys-2 proximal promoter and hHNF4α expression vector, (d) Chromatin immu-noprecipitation of Gys-2 DR-lprom using antibodies against HNF4α. The gene sequence spanning DR-lprom and a control sequence were analyzed by PCR in the immunoprecipitated chromatin of rat primary hepatocytes. (e) HepG2 cells were transfected with a reporter vector containing 553 bp of the mouse Gys-2 proximal promoter, an expression vector for hHNF4α, and increasing amounts of mPPARα expression vector. Normalized luciferase activity of the mGys-2 reporter vector in the absence of mPPARα, hHNF4α and Wy 14643 was set at 1. Error bars represent SEM.
Cross-talk between PPARα and HNF4α in the transcriptional control of Gys-2. Whereas HNF4α activates the Gys-2 promoter via DR-lprom, PPARα does the opposite, suggesting that PPARα may interfere with Gys-2 promoter activation by HNF4α. To examine whether this is the case, the effect of PPARα on HNF4α-mediated transactivation of the 0.55-kb Gys-2 promoter was studied. PPARα activation significantly reduced HNF4α-dependent transactivation, indicating competition between HNF4α and PPARα in the regulation of the Gys-2 promoter (Fig. 7e). As already mentioned above, we failed to find any evidence for binding of PPARα to DR-lprom in hepatocytes. Thus, the inhibitory effect of PPARα on transcriptional activation of Gys-2 by HNF4α likely does not occur via competition with HNF4α for actual binding to DR-lprom.
Discussion
In the present study, we have identified the mouse Gys-2 gene as a direct PPAR and HNF4α target gene. We have shown that the effects of PPARs and HNF4α on Gys-2 expression occur via two distinct response elements. Indeed, while transcriptional activation of the Gys-2 gene by PPARs was found to be mediated by a PPRE present in intron 1 of the mGys-2 gene (DR-lint), the stimulatory effect of HNF4α was mediated by a response element in the immediate upstream promoter (DR-lprom).
Our data are suggestive of the following scenario. In liver, which expresses high amounts of HNF4α, DR-lprom is occupied by HNF4α but not PPARα, while DR-lint is bound by PPARα but not HNF4α. Hence, HNF4α and PPARα activate Gys-2 expression via different response elements. Nevertheless, important negative cross-talk between the two nuclear receptors was observed. In the absence of any mutual binding to the response elements, it can be hypothesized that competition may take place at the level of binding to common co-activator proteins in a mechanism that is often referred to as squelching. In adipose tissue, which does not express HNF4α, DR-lprom is occupied by PPARβ/δ and PPARγ, but this does not result in transcriptional activation. Rather, transactivation occurs via binding of PPARβ/δ and PPARγ to DR-lint.
Our data indicate that HNF4α is an extremely powerful activator of mouse Gys2 transcription, explaining the marked reduction in hepatic Gys-2 expression in liver-specific HNF4α-null mice [35]. As mentioned above, regulation of mGys-2 expression by HNF4α at least partially occurs via DR-lprom. A recent study that combined ChIP with promoter micro-arrays showed that the Gys-2 promoter is bound by HNF4α in human liver [36], thus establishing Gys-2 as a direct target of HNF4α in human as well. It is not very clear why disabling the DR-lprom reduced HNF4α-dependent trans-activation by only 50 %. It is possible that the 0.55-kb Gys-2 promoter fragment contains an additional HNF4α response element, although in silico analysis failed to reveal such an element. Alternatively, it is possible that mutating the wild-type DR-lprom (AGGCCAAAGGCCA) into a mutated DR-lprom (AGGCCTTTGGCCA) only partially disabled the response element.
While functional PPREs are commonly located within regulatory sequences, i.e., proximal promoters, PPREs have also been identified in intronic sequences. Examples are PPREs within intron 3 of human/mouse Angptl4 and rat peroxisomal thiolase B genes, and within intron 1 of the rat acyl-CoA binding protein gene and human carnitine palmitoyltransferase 1A [2–5]. Our data demonstrate that regulation of Gys-2 expression by PPARs is also mediated by an intronic PPRE.
In the past few years, our understanding of the function of PPARβ/δ in vivo has improved greatly thanks to studies using various transgenic mouse models [19, 37]. At the level of metabolism, PPARβ/δ over-expression promotes skeletal muscle fatty acid oxidation and type I fiber content in mice, resulting in improved endurance exercise performance [22]. Conversely, deletion of PPARβ/δ in cardiomyocytes is associated with impaired fatty acid oxidation and expression of fatty acid oxidative genes, whereas glucose uptake is increased [38]. In WAT, PPARβ/δ stimulates fatty acid oxidation and uncoupling, thereby diminishing adiposity [21]. It is thus clear that PPARβ/δ plays a pivotal role in governing fatty acid oxidation in a variety of tissues. In contrast, data linking PPARβ/δ to regulation of glucose homeostasis remain scarce [20, 39, 40]. Our data reveal that PPARβ/δ is a critical regulator of the adipose expression of the Gys-2 gene. Furthermore, micro-array and qPCR analysis indicated that expression of numerous other genes involved in lipid and glucose metabolism was markedly down-regulated in PPARβ/δ-null mice, including GLUT4, p85, and CD36. Since PPARγ and PGC-lα were significantly down-regulated as well, it is possible that many of the observed changes are not linked to the absence of PPARβ/δ per se but rather reflect indirect effects mediated via decreased PPARγ and PGC-lα mRNA. Although such an effect may contribute to some extent to the down-regulation of Gys-2 in PPARδ/δ-null mice, the in vitro studies leave no doubt that Gys-2 is a direct target gene of PPARβ/δ, as well as of PPARγ.
Glycogen is stored in many tissues, yet it is particularly abundant in liver, muscle, and adipose tissue. In liver, glycogen serves to maintain blood glucose levels between meals, while skeletal muscle glycogen is used to fuel muscle contractions. In contrast, adipose tissue glycogen serves as a source of glycerol 3-phosphate, which is required for (re)-esterification of fatty acids into triglycerides [41]. Several alternative pathways exist to produce glycerol 3-phosphate, including synthesis from glucose, and conversion of gluconeogenic precursors (glyceroneogenesis). Since expression and activity of glycerol kinase are very low in adipose tissue [42], direct phosphorylation of glycerol is not considered as a major pathway to generate glycerol 3-phosphate. However, recent studies suggest that this may change after treatment with synthetic PPARγ agonists, which markedly up-regulate glycerol kinase expression in human and mouse adipocytes [11, 42]. In fact, it has been hypothesized that stimulation of glycerol kinase expression by TZDs, resulting in increased fatty acid re-esterification, may at least partially account for the suppressive effect of TZDs on plasma free fatty acid levels. Stimulation of fatty acid esterification is part of a general lipogenic and adipogenic effect of PPARγ in the adipocyte. Since adipose glycogen stores yield glycerol 3-phosphate as a precursor for fatty acid (re-)esterification, up-regulation of Gys-2 expression by PPARγ can be placed in the context of the lipogenic role of PPARγ in the adipocyte, which is aimed at promoting energy storage.
Besides contributing to lipogenesis, synthesis of glycogen permits continued uptake of glucose into cells. Accordingly, it can be speculated that up-regulation of adipose Gys-2 by PPARγ might partially account for the stimulation of glucose uptake into adipocytes by PPARγ agonists.
It is currently still ambiguous whether PPARβ/δ serves a general anabolic or catabolic function in the adipocyte. On the one hand, it has been reported that PPARβ/δ promotes fatty acid oxidation in adipocytes [20, 21]. On the other hand, PPARβ/δ also seems to have a facilitative, yet important role in lipo- and adipogenesis [43]. As discussed above for PPARγ, up-regulation of Gys-2 expression by PPARβ/δ may indicate a role for PPARβ/δ in fatty acid (re-)esterification, thus contributing to a lipogenic role for PPARβ/δ.
The highest levels of glycogen are found in liver and fluctuate with nutritional status. The hepatic synthesis of glycogen from glucose is catalyzed by Gys-2 [44]. Remarkably, expression of Gys-2 in liver increases during fasting, at the same time when glycogen stores are actively broken down [32]. The reason behind this seemingly counterintuitive regulation is not very clear, but it may serve to prime the glucose synthesizing system for when dietary glucose becomes available again. In the absence of PPARα, we observed that the expression of Gys-2 drops markedly during prolonged fasting and refeeding. The reduced Gys-2 expression is likely responsible for the diminished rate of glycogen formation upon refeeding, as observed by us and previously by others. Indeed, the effect of PPARα deletion on liver glycogen is minor except under conditions of refeeding [30–32]. It has been reported that after a short-term fast the gluconeogenic flux in PPARα-null mice is directed more towards glycogen, leading to a decrease in hepatic glucose output. However, it is unclear what happens to the gluconeogenic flux toward glycogen in the fasted-refed state, although our and other data clearly indicate that total glycogen synthesis is decreased in PPARα-/- mice. Mutation of the GYS2 gene in humans leads to lower hepatic glycogen levels and fasting hypoglycemia [45], biochemical features that are also observed in PPARα-/- animals [31]. However, opposite to that observed in patients with a dysfunctional GYS2 gene, PPARα-/- mice show low plasma ketones, which is explained by the stimulatory effect of PPARα on fatty acid oxidation and ketogenesis.
Overall, our data suggest that the decreased hepatic glycogen levels in PPARα-/- and liver-specific HNF4α-null mice [28, 35] may be due to decreased activation of Gys-2 expression via DR-lint and DR-lprom, respectively. Although PPARα and HNF4α stimulate Gys-2 expression via different response elements, important interplay exist between signaling of the two nuclear receptors.
In conclusion, we show that Gys-2 is a direct target gene of PPARs. Transcriptional regulation is achieved via a PPRE present in the first intron. An additional direct repeat response element identified in the Gys-2 promoter mediates transactivation by HNF4α.
References
Desvergne, B. and Wahl, W. (1999) Peroxisome proliferatoractivated receptors: nuclear control of metabolism. Endocr. Rev. 20, 649–688.
Hansmannel, F., Clemencet, M. C., Le Jossic-Corcos, C., Osumi, T., Latruffe, N. and Nicolas-Frances, V. (2003) Functional characterization of a peroxisome proliferator responseelement located in the intron 3 of rat peroxisomal thiolase B gene. Biochem. Biophys. Res. Commun. 311, 149–155.
Helledie, T., Grontved, L., Jensen, S. S., Kiilerich, P., Rietveld, L., Albrektsen, T., Boysen, M. S., Nohr, J., Larsen, L. K., Fleckner, J., Stunnenberg, H.G., Kristiansen, K. and Mandrup, S. (2002) The gene encoding the Acyl-CoA-binding protein is activated by peroxisome proliferator-activated receptor gamma through an intronic response element functionally conserved between humans and rodents. J. Biol. Chem. 277, 26821–26830.
Mandard, S., Zandbergen, F., Tan, N. S., Escher, P., Patsouris, D., Koenig, W., Kleemann, R., Bakker, A., Veenman, F., Wahli, W., Muller, M. and Kersten, S. (2004) The direct peroxisome proliferator-activated receptor target fasting-induced adipose factor (FIAF/PGAR/ANGPTL4) is present in blood plasma as a truncated protein that is increased by fenofibrate treatment. J. Biol. Chem. 279, 34411–34420.
Napal, L., Marrero, P. F. and Haro, D. (2005) An intronic peroxisome proliferator-activated receptor-binding sequence mediates fatty acid induction of the human carnitine palmitoyltransferase 1A. J. Mol. Biol. 354, 751–759.
Escher, P., Braissant, O., Basu-Modak, S., Michalik, L., Wahli, W. and Desvergne, B. (2001) Rat PPARs: quantitative analysis in adult rat tissues and regulation in fasting and refeeding. Endocrinology 142, 4195–4202.
Kersten, S., Seydoux, J., Peters, J. M., Gonzalez, F. J., Desvergne, B. and Wahli, W. (1999) Peroxisome proliferatoractivated receptor alpha mediates the adaptive response to fasting. J. Clin. Invest. 103, 1489–1498.
Leone, T.C., Weinheimer, C. J. and Kelly, D. P. (1999) Acritical role for the peroxisome proliferator-activated receptor alpha (PPARalpha) in the cellular fasting response: the PPARalphanull mouse as a model of fatty acid oxidation disorders. Proc. Natl. Acad. Sci. USA 96, 7473–7478.
Kersten, S., Mandard, S., Escher, P., Gonzalez, F. J., Tafuri, S., Desvergne, B. and Wahli, W. (2001) The peroxisome proliferator- activated receptor alpha regulates amino acid metabolism. FASEB J. 15, 1971–1978.
Mandard, S., Muller, M. and Kersten, S. (2004) Peroxisome proliferator-activated receptor alpha target genes. Cell. Mol. Life Sci. 61, 393–416.
Patsouris, D., Mandard, S., Voshol, P. J., Escher, P., Tan, N. S., Havekes, L. M., Koenig, W., Marz, W., Tafuri, S., Wahli, W., Muller, M. and Kersten, S. (2004) PPARalpha governs glycerol metabolism. J. Clin. Invest. 114, 94–103.
Gervois, P., Kleemann, R., Pilon, A., Percevault, F., Koenig, W., Staels, B. and Kooistra, T. (2004) Global suppression of IL- 6-induced acute phase response gene expression after chronic in vivo treatment with the peroxisome proliferator-activated receptor-alpha activator fenofibrate. J. Biol. Chem. 279, 16154–16160.
Nakajima, T., Kamijo, Y., Tanaka, N., Sugiyama, E., Tanaka, E., Kiyosawa, K., Fukushima, Y., Peters, J. M., Gonzalez, F. J. and Aoyama, T. (2004) Peroxisome proliferator-activated receptor alpha protects against alcohol-induced liver damage. Hepatology 40, 972–980.
Li, A.C., Binder, C. J., Gutierrez, A., Brown, K. K., Plotkin, C. R., Pattison, J.W., Valledor, A. F., Davis, R.A., Willson, T. M., Witztum, J. L., Palinski, W. and Glass, C.K. (2004) Differential inhibition of macrophage foam-cell formation and atherosclerosis in mice by PPARalpha, beta/delta, and gamma. J. Clin. Invest. 114, 1564–1576
Finck, B. N., Bernal-Mizrachi, C., Han, D. H., Coleman, T., Sambandam, N., LaRiviere, L. L., Holloszy, J. O., Semenkovich, C. F. and Kelly, D. P. (2005) A potential link between muscle peroxisome proliferator- activated receptor-alpha signaling and obesity-related diabetes. Cell. Metab. 1, 133–144.
Finck, B. N., Lehman, J. J., Leone, T. C., Welch, M. J., Bennett, M. J., Kovacs, A., Han, X., Gross, R. W., Kozak, R., Lopaschuk, G.D. and Kelly, D. P. (2002) The cardiac phenotype induced by PPARalpha overexpression mimics that caused by diabetes mellitus. J. Clin. Invest. 109, 121–130.
Imai, T., Takakuwa, R., Marchand, S., Dentz, E., Bornert, J. M., Messaddeq, N., Wendling, O., Mark, M., Desvergne, B., Wahli, W., Chambon, P. and Metzger, D. (2004) Peroxisome proliferator-activated receptor gamma is required in mature white and brown adipocytes for their survival in the mouse. Proc. Natl. Acad. Sci. USA 101, 4543–4547.
Rosen, E. D., Sarraf, P., Troy, A. E., Bradwin, G., Moore, K., Milstone, D. S., Spiegelman, B. M. and Mortensen, R. M. (1999) PPAR gamma is required for the differentiation of adipose tissue in vivo and in vitro. Mol. Cell 4, 611–617.
Luquet, S., Lopez-Soriano, J., Holst, D., Fredenrich, A., Melki, J., Rassoulzadegan, M. and Grimaldi, P. A. (2003) Peroxisome proliferator-activated receptor delta controls muscle development and oxidative capability. FASEB J. 17, 2299–2301.
Tanaka, T., Yamamoto, J., Iwasaki, S., Asaba, H., Hamura, H., Ikeda, Y., Watanabe, M., Magoori, K., Ioka, R. X., Tachibana, K., Watanabe, Y., Uchiyama, Y., Sumi, K., Iguchi, H., Ito, S., Doi, T., Hamakubo, T., Naito, M., Auwerx, J., Yanagisawa, M., Kodama, T. and Sakai, J. (2003) Activation of peroxisome proliferator-activated receptor delta induces fatty acid betaoxidation in skeletal muscle and attenuates metabolic syndrome. Proc. Natl. Acad. Sci. USA 100, 15924–15929.
Wang, Y. X., Lee, C. H., Tiep, S., Yu, R. T., Ham, J., Kang, H. and Evans, R. M. (2003) Peroxisome-proliferator-activated receptor delta activates fat metabolism to prevent obesity. Cell 113, 159–170.
Wang, Y. X., Zhang, C. L., Yu, R. T., Cho, H. K., Nelson, M.C., Bayuga-Ocampo, C. R., Ham, J., Kang, H. and Evans, R. M. (2004) Regulation of muscle fiber type and running endurance by PPARdelta. PLoS Biol. 2, E294.
Akiyama, T. E., Lambert, G., Nicol, C. J., Matsusue, K., Peters, J. M., Brewer, H. B. Jr. and Gonzalez, F. J. (2004) Peroxisome proliferator-activated receptor beta/delta regulates very low density lipoprotein production and catabolism in mice on a Western diet. J. Biol. Chem. 279, 20874–20881.
Di, Poi N., Tan, N. S., Michalik, L., Wahli, W. and Desvergne, B. (2002) Antiapoptotic role of PPARbeta in keratinocytes via transcriptional control of the Akt1 signaling pathway. Mol. Cell. 10, 721–733.
Gupta, R. A., Wang, D., Katkuri, S., Wang, H., Dey, S. K. and DuBois, R. N. (2004) Activation of nuclear hormone receptor peroxisome proliferator-activated receptor-delta accelerates intestinal adenoma growth. Nat. Med. 10, 245–247.
Nadra, K., Anghel, S. I., Joye, E., Tan, N. S., Basu-Modak, S., Trono, D., Wahli, W. and Desvergne, B. (2006) Differentiation of trophoblast giant cells and their metabolic functions are dependent on peroxisome proliferator-activated receptor beta/delta. Mol. Cell. Biol. 26, 3266–3281.
Rieusset, J., Seydoux, J., Anghel, S. I., Escher, P., Michalik, L., Soon, Tan N., Metzger, D., Chambon, P., Wahli, W. and Desvergne, B. (2004) Altered growth in male peroxisome proliferator-activated receptor gamma (PPARgamma) heterozygous mice: involvement of PPARgamma in a negative feedback regulation of growth hormone action. Mol. Endocrinol. 18, 2363–2377.
Hayhurst, G. P., Lee, Y. H., Lambert, G., Ward, J. M. and Gonzalez, F. J. (2001) Hepatocyte nuclear factor 4alpha (nuclear receptor 2A1) is essential for maintenance of hepatic gene expression and lipid homeostasis. Mol. Cell. Biol. 21, 1393–1403.
Kuipers, F., Jong, M. C., Lin, Y., Eck, M., Havinga, R., Bloks, V., Verkade, H. J., Hofker, M. H., Moshage, H., Berkel, T. J., Vonk, R. J. and Havekes, L. M. (1997) Impaired secretion of very low density lipoprotein-triglycerides by apolipoprotein Edeficient mouse hepatocytes. J. Clin. Invest. 100, 2915–2922.
Sugden, M. C., Bulmer, K., Gibbons, G. F., Knight, B. L. and Holness, M. J. (2002) Peroxisome-proliferator-activated receptor- alpha (PPARalpha) deficiency leads to dysregulation of hepatic lipid and carbohydrate metabolism by fatty acids and insulin. Biochem. J. 364, 361–368.
Xu, J., Xiao, G., Trujillo, C., Chang, V., Blanco, L., Joseph, S. B., Bassilian, S., Saad, M. F., Tontonoz, P., Lee, W. N. and Kurland, I. J. (2002) Peroxisome proliferator-activated receptor alpha (PPARalpha) influences substrate utilization for hepatic glucose production. J. Biol. Chem. 277, 50237–50244.
Bandsma, R. H., Van Dijk, T. H., Harmsel At, A., Kok, T., Reijngoud, D. J., Staels, B. and Kuipers, F. (2004) Hepatic de novo synthesis of glucose 6-phosphate is not affected in peroxisome proliferator-activated receptor alpha-deficient mice but is preferentially directed toward hepatic glycogen stores after a short term fast. J. Biol. Chem. 279, 8930–8937.
Podvinec, M., Kaufmann, M. R., Handschin, C. and Meyer, U. A. (2002) NUBIScan, an in silico approach for prediction of nuclear receptor response elements. Mol. Endocrinol. 16, 1269–1279.
Sandelin, A. and Wasserman, W. W. (2005) Prediction of nuclear hormone receptor response elements. Mol. Endocrinol. 19, 595–606.
Parviz, F., Matullo, C., Garrison, W.D., Savatski, L., Adamson, J. W., Ning, G., Kaestner, K. H., Rossi, J. M., Zaret, K. S. and Duncan, S. A. (2003) Hepatocyte nuclear factor 4alpha controls the development of a hepatic epithelium and liver morphogenesis. Nat. Genet. 34, 292–296.
Odom, D. T., Zizlsperger, N., Gordon, D. B., Bell, G. W., Rinaldi, N. J., Murray, H. L., Volkert, T. L., Schreiber, J., Rolfe, P. A., Gifford, D. K., Fraenkel, E., Bell, G. I. and Young, R. A. (2004) Control of pancreas and liver gene expression by HNF transcription factors. Science 303, 1378–1381.
Tan, N. S., Michalik, L., Noy, N., Yasmin, R., Pacot, C., Heim, M., Fluhmann, B., Desvergne, B. and Wahli, W. (2001) Critical roles of PPAR beta/delta in keratinocyte response to inflammation. Genes Dev. 15, 3263–3277.
Cheng, L., Ding, G., Qin, Q., Huang, Y., Lewis, W., He, N., Evans, R.M., Schneider, M.D., Brako, F.A., Xiao, Y., Chen, Y. E. and Yang, Q. (2004) Cardiomyocyte-restricted peroxisome proliferator-activated receptor-delta deletion perturbs myocardial fatty acid oxidation and leads to cardiomyopathy. Nat. Med. 10, 1245–1250.
Dressel, U., Allen, T. L., Pippal, J. B., Rohde, P. R., Lau, P. and Muscat, G. E. (2003) The peroxisome proliferator-activated receptor beta/delta agonist, GW501516, regulates the expression of genes involved in lipid catabolism and energy uncoupling in skeletal muscle cells. Mol. Endocrinol. 17, 2477–2493.
Terada, S., Wicke, S., Holloszy, J. O. and Han, D. H. (2006) PPARdelta activator GW-501516 has no acute effect on glucose transport in skeletal muscle. Am. J. Physiol. Endocrinol. Metab. 290, E607–611.
Antwi, D., Youn, J. H., Shargill, N. S., Lesikar, D. D. and Kaslow, H. R. (1988) Regulation of glycogen synthase in muscle and adipose tissue during fasting and refeeding. Am. J. Physiol. 254, E720–725.
Guan, H. P., Li, Y., Jensen, M. V., Newgard, C. B., Steppan, C. M. and Laza, M. A. (2002) Afutile metabolic cycle activated in adipocytes by antidiabetic agents. Nat. Med. 8, 1122–1128.
Matsusue, K., Peters, J. M. and Gonzalez, F. J. (2004) PPARbeta/delta potentiates PPARgamma-stimulated adipocyte differentiation. FASEB J. 18, 1477–1479.
Orho, M., Bosshard, N. U., Buist, N. R., Gitzelmann, R., Aynsley-Green, A., Blumel, P., Gannon, M. C., Nuttall, F. Q. and Groop, L. C. (1998) Mutations in the liver glycogen synthase gene in children with hypoglycemia due to glycogen storage disease type 0. J. Clin. Invest. 102, 507–515.
Weinstein, D. A., Correia, C. E., Saunders, A. C. and Wolfsdorf, J. I. (2006) Hepatic glycogen synthase deficiency: an infrequently recognized cause of ketotic hypoglycemia. Mol. Genet. Metab. 87, 284–288.
Acknowledgement
We would like to thank Marco Alves for the synthesis of LI65041 compound, Mechteld Grootte-Bromhaar and Jolanda van der Meijde for performing the micro-array experiment. This study was supported by the Netherlands Organization for Scientific Research (NWO), with additional support from the Dutch Diabetes Foundation, the Royal Netherlands Academy of Art and Sciences (KNAW), Wageningen Center for Food Sciences, the Center for Human Nutrigenomics, the Swiss National Science Foundation and the Human Frontier Science Program.
Author information
Authors and Affiliations
Corresponding author
Additional information
Received 8 January 2007; received after revision 28 February 2007; accepted 20 March 2007
S. Mandard, R. Stienstra: These authors equally contributed to this work.
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License ( https://creativecommons.org/licenses/by-nc/2.0 ), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Mandard, S., Stienstra, R., Escher, P. et al. Glycogen synthase 2 is a novel target gene of peroxisome proliferator-activated receptors. Cell. Mol. Life Sci. 64, 1145 (2007). https://doi.org/10.1007/s00018-007-7006-1
Published:
DOI: https://doi.org/10.1007/s00018-007-7006-1