
Abstract
The main pathological characteristic of glomerulonephritis is diffuse mesangial cell proliferation. MiR-34a is associated with the proliferation of various organs and cancer cells. However, the role of miR-34a in renal proliferation diseases is not clear. Therefore, this study aimed to elucidate the mechanism of miR-34a in the regulation of renal mesangial cell proliferation. The miR-34a expression level at different time points in an anti-Thy1 mesangial proliferative nephritis rat model was determined by qRT-PCR. The cell proliferation rate and cell cycle changes were measured in the in vitro cultured rat mesangial cells (RMCs). Our results suggested that miR-34a expression was negatively correlated with the degree of cell proliferation in the anti-Thy1 nephritis model. MiR-34a could extend the G0/G1 phase and block cell proliferation in RMCs. Dual-luciferase assay results showed that there were binding sites of miR-34a at 3′-UTR of platelet-derived growth factor receptor-β (PDGFR-β). MiR-34a can inhibit PDGFR-β protein expression at a post-transcriptional level, suppress Ras/MAPK signaling pathways, and down-regulate expression of cell cycle proteins at the G0/G1 phase, such as cyclin D1, CDK4/CDK6. In addition, miR-34a may also inhibit RMC proliferation by directly targeting cyclin E and CDK2. MiR-34a inhibits exogenous stimuli-induced proliferation of mesangial cells. Expression levels of phospho-PDGFR-β and phospho-MEK1 (an important downstream molecule in PDGFR-β-induced signaling pathway) were significantly increased in the anti-Thy-1 nephritis rat model. These results suggest that miR-34a may regulate RMC proliferation by directly inhibiting expressions of PDGFR-β, MEK1, and cell cycle proteins, cyclin E and CDK2.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Mesangial proliferative glomerulonephritis (MsPGN) can be divided into IgA nephropathy and non-IgA nephropathy, which show diffuse hyperplasia of glomerular mesangial cells (GMCs) and different degrees of extracellular matrix (ECM) accumulation [1, 2]. Inhibition of mesangial cell proliferation is an important treatment strategy in proliferating glomerular diseases [3].
Anti-Thy1 nephritis is a rat model of human mesangial proliferative glomerulonephritis, involving a combination of Thy1 antibody and Thy1 antigen in mesangial cells. Macromolecular immune complexes are formed in situ and deposited in the mesangial area, leading to mesangial cell proliferation and inflammation [4, 5]. These inflammatory mediators in turn stimulate activation, shrinkage, and proliferation of mesangial cells; release a variety of inflammatory mediators and ECM components [6, 7]; and aggravate inflammation, all of which result in glomerular damages, including damage to the molecular barrier and charge barrier of the filtration membrane, and proteinuria [8].
MicroRNAs (miRNAs) are 21- to 25-nucleotide (nt)-long, single-stranded RNA molecules that serve as post-transcriptional regulators of gene expression in eukaryotes and that do not code for any protein [9–11]. miRNAs can regulate the expression of thousands of proteins by degrading target mRNA or inhibiting translation as a result of complementary matching between miRNAs and specific sites in target genes [12]. miRNAs are widely involved in a series of biological functions and are closely associated with human diseases, including kidney diseases [13].
miR-34 was first found in the worm Caenorhabditis elegans in 2001 [14]. In recent years, it has been shown that miR-34a is involved in tumor proliferation of neuroblastoma [15, 16], colon cancer [17], uveal melanoma [18], brain tumors [19], and cervical cancer [20] through regulation of different target genes. However, the role of miR-34a in mesangial proliferative glomerulonephritis is unclear. We thus aimed to investigate the role of miR-34a in renal proliferation diseases.
Materials and methods
Anti-Thy1 nephritis animal model
Male Wistar rats (Beijing Vital River Laboratory Animal Technology Co., Ltd., Beijing, China) weighing between 200 and 220 g were randomly allocated to the control and anti-Thy1.1 groups. Anti-Thy1.1 nephritis was induced by a single intravenous injection of a monoclonal anti-Thy1 antibody (2.5 mg/kg) produced by OX-7 cells. Controls were injected with an identical volume of normal saline. Anti-Thy1-treated animals were killed on days 3, 5, 7, 10, and 14 post-injection (n = 3 per time point), and the control animals were killed on day 0 (n = 3). All rats were provided a diet of standard laboratory chow and free access to water. The renal tissues were examined by routine periodic acid-Schiff staining.
Immunohistochemical analysis and evaluation
The renal tissues were fixed in formalin and embedded with paraffin. The histologic paraffin sections were cut to 3- to 4-μm thickness and mounted on poly-l-lysine-coated slides. Endogenous peroxidase was blocked with 3 % hydrogen peroxide. The sections were heated in a microwave oven for 10 min in sodium citrate buffer (pH 6.0), incubated with 1.5 % normal goat serum for 20 min, and incubated overnight with 1:100 diluted primary antibody (Ki-67). For a negative control, the sections were incubated with PBS. After removal of unbound primary antibody, the sections were incubated with a biotinylated secondary antibody for 60 min at room temperature. The sections were rinsed and incubated with avidin-biotinylated horseradish peroxidase (Vectastain Elite ABC Kit; Vector Laboratories, USA) for 60 min. Incubation with 3,3-diaminobenzidine tetrahydrochloride was performed for 10 min as a substrate chromogen solution to produce a brown color. Finally, the sections were counterstained with hematoxylin. Twenty glomeruli per section at each time point were evaluated under high-power light microscopy (×400) in a blinded fashion. The Ki67 labeling index was calculated as the number of Ki-67-positive cells to total glomerular cells.
Cell culture and transfection
Rat mesangial cells (RMCs) were purchased from the American Type Culture Collection (ATCC). Cells were cultured in RMPI 1640 medium (Gibco, USA) containing 10 % fetal bovine serum (FBS; Hyclone, Canada), 100 U/ml penicillin, and 100 μg/ml streptomycin (Gibco, USA) at 37 °C in a humidified incubator with an atmosphere containing 5 % CO2. Trypsin (0.25 %) was used for cell passages. RMCs (~50 % confluence) were transfected with miR-34a mimics (Dharmacon, Thermo Scientific, USA; cat. no. C-300551-07), miRNA control (Dharmacon cat. no. CN-001000-01), siPDGFR-β or siCon (Table 1) using jetPRIME (Polyplus-transfection, USA) for 48 h, followed by determination of protein and RNA levels.
Quantitative reverse-transcriptase polymerase chain reaction (qRT-PCR)
TaqMan miRNA assays were performed to quantify mature miR-34 levels. Briefly, cDNA was synthesized using Taqman RT reagents (Applied Biosystems, USA), followed by TaqMan-based quantitative PCR using specific mirVana qRT-PCR primer sets. The primary transcript levels of PDGFR-β, cyclin D1, cyclin E, CDK2, CDK4, CDK6, and p27kip1 (Table 2) were determined with the SYBR Green Real-time qPCR Master Mix (Toyobo, Japan). The qRT-PCR analyses were conducted using an Applied Biosystems ABI Prism 7300 Sequence Detection System, and qRT-PCR reactions were performed using the following parameters: 95 °C for 10 min, followed by 40 cycles at 95 °C for 15 s and 60 °C for 1 min. The relative RNA levels were calculated using the ΔΔC t method and normalized to U6 snRNA control for rat miR-34a or to GAPDH for RMC mRNAs. Sequence-specific primers for the indicated genes were designed using the Primer Premier software, version 5.0 (PREMIER Biosoft International, USA).
Cell proliferation assay
As a measure of cell proliferation, the number of viable cells was detected using the Cell Counting Kit-8 (Beyotime, China). RMCs were seeded in 96-well plates at a density of 2,000/well, synchronized by incubation in FBS(−) medium for 12 h, and then transfected with miR-34a mimics or a miRNA control. At 24, 48, and 72 h post-transfection, the number of viable cells was measured by recording the optical density at 450 nm and generating growth curves. Experiments were performed in triplicate, and sextuplet wells were used.
Cell cycle analysis
RMCs were seeded in six-well plates at a density of 1.0 × 105/well, synchronized by incubation in FBS(−) medium for 12 h, and then transfected with miR-34a mimics or a miRNA control for 48 h. The cells were washed twice with PBS, suspended in 75 % ethanol, and fixed by incubation in 75 % ethanol at 4 °C overnight. Fixed cells were collected by centrifugation, washed with PBS, treated with RNase (50 μg/ml; Sigma, USA), and stained with propidium iodide (50 μg/ml; Sigma, USA). A FACS flow cytometer (BD Co., USA) was used to determine cellular DNA contents. The percentage of cells in G0/G1, S, and G2/M phases were determined using the Cell FIT Cell Cycle Analysis software (version 2.01.2; BD).
Western-blot analysis
Proteins were extracted from RMCs using RIPA lysis buffer (50 mM Tris–HCl, pH 7.5, 150 mM NaCl, 0.5 % deoxycholate, 1 % Nonidet P-40, 0.1 % SDS, 1 mM PMSF, and protease cocktail at 1 μg/ml). Protein concentrations were determined using a BCA kit (Pierce, USA). Protein samples (60 μg per lane) were separated by 10 or 12 % SDS-PAGE electrophoresis and transferred to NC membranes. After staining with Ponceau S, the membranes were incubated overnight in 5 % non-fat milk at 4 °C, followed by incubation with primary antibodies against PDGFR-β, p-PDGFR-β, and p27kip1 (Cell Signaling Technology, USA); cyclin D1, cyclin E, CDK2, CDK4, CDK6, ERK1/2, and p-ERK1/2 (Abcam, USA); MEK1 and p-MEK1 (Abcam, USA); Raf1, p-Raf1 and K-ras(Abnova, USA); or β-actin (Sigma, USA). Immunoreactive bands were visualized using ECL reagent (Santa Cruz Biotechnology), according to the manufacturer’s instructions, and exposure to X-ray film. Protein band intensities were quantified using Quantity One software (Bio-Rad, USA).
Plasmids
PDGFR-β, cyclin E, and CDK2 cDNAs were amplified from RMCs by PCR. The 1521-bp fragment of the PDGFR-β 3′-UTR, the 426-bp fragment of the cyclin E 3′-UTR, or the 999-bp fragment of the CDK2 3′-UTR (Table 3), all containing the miR-34a targeting sequence, was cloned into the psiCHECK dual-luciferase reporter plasmid (Promega, USA; cat. no. C8021) to produce psiCHECK-WT-PDGFR-β, psiCHECK-WT-cyclin E, or psiCHECK-WT-CDK2, respectively, using PCR. Sequence-specific primers for the indicated genes were designed using the Primer Premier software, version 5.0. The sequences of all constructs were verified.
Dual-luciferase reporter assay
For reporter assays, RMCs were cultured to approximately 80 % confluence in a 24-well plate and then co-transfected with a dual-luciferase reporter plasmid (psiCHECK-PDGFR-β, or psiCHECK-cyclin E, or psiCHECK-CDK2) and miR-34a mimics for 48 h. The activities of firefly and Renilla luciferases were determined using the Dual-Luciferase Reporter Assay System (Promega cat. no. E1910), and firefly luciferase activity was normalized to that of Renilla luciferase.
Statistical analyses
Measurement data are expressed as mean ± SEM, unless otherwise stated. Statistical analyses were performed using the SPSS 15.0 software package (SPSS, Inc., USA). Comparison among groups was conducted with ANOVA. Statistical differences were evaluated with analysis of variance. A p value <0.05 denoted a statistically significant difference.
Results
Pathological changes in rat model of anti-Thy1 mesangial proliferative glomerulonephritis
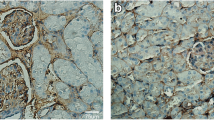
We injected Thy1 antibody into Wistar rats to create an anti-Thy1 mesangial proliferative glomerulonephritis rat model. Following injection of anti-Thy1 antibodies, partial complement-dependent mesangiolysis appeared on day 3; mesangial cell proliferation and ECM accumulation occurred on day 5 and peaked on day 7. On day 10, recovery from glomerular injury began to decrease, and ECM accumulation attenuated on day 14 (Fig. 1a). We also detected the expression changes of cell proliferation marker Ki-67 by immunohistochemistry (Fig. 1b, c). We found higher levels of Ki-67 at every time point during anti-Thy1 nephritis compared with the control, which suggests that the cell cycle remained active from days 3 to 10. Ki-67 increased on days 3 and 5, peaked on day 7, and decreased from days 10 to 14. This suggests that cell cycle activity increased from days 3 to 7 and subsequently gradually decreased from days 10 to 14 (p < 0.05)(Fig. 1b, c).

Pathological changes and expression of Ki-67 in anti-Thy1 nephritis model rats. a Periodic acid-Schiff stain (original magnification ×400). b Expression of Ki-67 in anti-Thy1 nephritis model rats. Expression of Ki-67 was detected with immunohistochemistry analysis. c Ki-67 labeling index was calculated by Ki-67-positive cells to total glomerular cells. N: normal group rats; day 3, 5, 7, 14, and 28: anti-Thy1 nephritis rats at day 3, 5, 7, 14, 28, separately. *p < 0.05 vs. N (normal group rats). # p < 0.05 vs. Day7 group, n = 6
MiR-34a expression in the kidney tissues of anti-Thy1 nephritis rat model
MiR-34a expression was detected in the kidney tissues of the anti-Thy1 nephritis rat model at various time points (days 3, 5, 7, 10, and 14) by real-time qPCR. Compared with the normal group, miR-34a expression gradually decreased as pathological changes progressed from day 3 (p < 0.05). Expression levels and pathologic outcomes became normal on day 14 (p < 0.05) (Fig. 2).

MiR-34a expression levels were detected with qRT-PCR in anti-Thy1 model rats. *p < 0.05 vs. N (normal group), n = 6; # p < 0.05 vs. day 7 group, n = 6
Effect of miR-34a on mesangial cell proliferation rate and cell cycle
RMCs were transfected with miR-34a mimics (mimics group) or a miRNA-negative control (Con group), and the proliferation rate of miR-34a mimic-transfected cells was determined using the Cell Counting Kit-8 (Fig. 3a). Proliferation of RMCs was reduced at 24 h (p > 0.05), significantly reduced at 48 h (p < 0.05), and more obvious at 72 h (p < 0.01) in miR-34a mimics-transfected group, compared with the normal group and the miRNA-negative control group, indicating that miR-34a mimics influence RMC proliferation. Cellular DNA content was further assessed by flow cytometry. The results showed that miR-34a mimics markedly extended the G0/G1 phase and reduced the G2/M phase (Fig. 3b; Table 4), compared with the normal group and the miRNA-negative control group (p < 0.05).

Changes of proliferation activity and cell cycle in the RMCs transfected with miR-34a mimics. a Proliferation activity of RMCs was detected by Cell Counting Kit-8 after transfection with miR-34a mimics or miRNA-negative control. *p < 0.05 vs. normal (normal group) and Con (miRNA-negative control group) at 48 h, n = 3; # p < 0.05 vs. normal group and Con group at 72 h, n = 3. Mimics: miR-34a mimics. b Analysis results of cell cycle by flow cytometry method (FCM) in the RMCs transfected with microRNA-34a mimics or miRNA-negative control
Influence of miR-34a on possible target gene, PDGFR-β
By using online biology software miRGen (http://www.diana.pcbi.upenn.edu/miRGen.html), we hypothesized that PDGFR-β may be a target gene of miR-34a (Table 5). To confirm whether miR-34a interacts with the 3′-UTR of PDGFR-β and regulates target gene expression at a post-transcriptional level, we cloned the 3′-UTR fragment of PDGFR-β into the dual-luciferase reporter vector psiCHECK to construct a psiCHECK-PDGFR-β plasmid, which was co-transfected into RMCs with miR-34a mimics or miRNA-negative control. MiR-34a mimics significantly down-regulated firefly luciferase activity in psiCHECK-PDGFR-β-transfected cells (p < 0.05) (Fig. 4a). To further confirm the direct action of miR-34a, levels of PDGFR-β mRNA and protein were determined in the miR-34a mimics-transfected cells. There was no significant difference between miR-34a mimic-transfected cells and control cells in terms of PDGFR-β mRNA expression level (p > 0.05) (Fig. 4b). Western blot showed that protein expressions of PDGFR-β and phospho-PDGFR-β were significantly decreased in the miR-34a mimic-transfected cells compared with the control cells (p < 0.05) (Fig. 4c). These results indicate that PDGFR-β may be a target gene of miR-34a. Moreover, miR-34a likely inhibits PDGFR-β primarily through post-transcriptional regulation. At the same time, we detected protein expressions of PDGFR-β and phospho-PDGFR-β (activated PDGFR-β) in the anti-Thy-1 nephritis rat model. Compared with day 0, on day 7 (the most obvious pathological change), phospho-PDGFR-β was significantly increased (p < 0.05) (Fig. 4d), indicating that PDGFR-β plays a key regulative role in proliferation development in the anti-Thy-1 nephritis.

Effect of miR-34a on expression of PDGFR-β. a Effect of miR-34a on expression of PDGFR-β by using Dual-Luciferase Reporter plasmids. The 3′-UTR of PDGFR-β was inserted into 3′ end of luciferase coding sequence in psiCHECK vector and recombinant plasmid was transfected into RMCs with miR-34a mimics or miRNA-negative control. Firefly luciferase activity was normalized to Renilla luciferase activity. *p < 0.01 vs. PDGFR-β-Mock (psiCHECK vector) and PDGFR-β-Con (psiCHECK-PDGFR-β+miRNA-negative control). PDGFR-β-Mimics: psiCHECK-PDGFR-β+miR-34a mimics. b Expression level of PDGFR-β mRNA was detected with real-time qPCR after transfection with miR-34a mimics or miRNA-negative control. Normal: normal group; Con: miRNA-negative control group; Mimics: miR-34a mimics. c Protein expression levels of PDGFR-β and phospho-PDGFR-β were detected with Western blot after transfection with miR-34a mimics or miRNA-negative control. Data are means of three separate experiments ± SD. *, # p < 0.05 vs. normal group and Con group, n = 3. Normal: normal group; Con: miRNA-negative control group; Mimics: miR-34a mimics. d Protein expression levels of PDGFR-β and phospho-PDGFR-β were detected with Western blot on day 0, day 7, and day 14 in anti-Thy1 model rats. Data are means of three separate experiments ± SD. *p < 0.05 vs. day 0 and day 14, n = 3
Effects of miR-34a on expressions of cell cycle-related mRNAs and proteins
Compared with the normal group and Con group, cyclin E mRNA level was significantly decreased (p < 0.05), and that of P27kip1 was significantly increased (p < 0.05); however, there were no changes in mRNA levels of cyclin D1, CDK2, CDK4, or CDK6 in the miR-34a-transfected cells (p > 0.05) (Fig. 5a). Levels of cell cycle proteins (cyclins D1 and E), CDK proteins (CDK2, CDK4, and CDK6), and CKI (p27kip1) were also determined. Levels of cyclin D1, cyclin E, CDK2, CDK4, and CDK6 were significantly reduced (p < 0.05), while that of p27kip1 was significantly increased (p < 0.05) in the miR-34a-transfected cells, compared with the normal group and Con group (Fig. 5b). Two cell-cycle complexes, cyclin D1/CDK4/CDK6 and cyclin E/CDK2, mainly play a role during the mid G0/G1 stage. Previous studies showed that cyclin D1 [21], CDK4 [9], and CDK6 [21, 22] are target genes of miR-34a. Therefore, we hypothesized that cyclin E and CDK2 may be target genes of miR-34a. We cloned the 3′-UTRs of cyclin E and CDK2 into psiCHECK-2 to construct two plasmids, psiCHECK-cyclin E and psiCHECK-CDK2. Both miR-34a mimics and the two plasmids were co-transfected into cells. MiR-34a significantly down-regulated firefly luciferase activity in psiCHECK-2-cyclin E-transfected cells (Fig. 5c) and psiCHECK-2-CDK2-transfected RMC cells (Fig. 5d), suggesting cyclin E and CDK2 to be target genes of miR-34a.

Expression of G0/G1 phase cell cycle-related proteins in RMCs after transfection with miR-34a mimics. a MiRNA expression levels of cyclin D1, cyclin E, CDK2, CDK4, CDk6, p27kip1 were detected with qRT-PCR. *p < 0.05 vs. normal (normal group) and Con (miRNA-negative control group). b Proteins expression levels of cyclin D1, cyclin E, CDK2, CDK4, CDk6, p27kip1 were detected with Western blot. Data are means of three separate experiments ± SD. a,b,c,d,e,f p < 0.05 vs. normal group and Con group, n = 3. Mimics represented miR-34a mimics. c Effect of miR-34a on expression of cyclin E. Data are means of three separate experiments ± SD. *p < 0.01 vs. Mock (psiCHECK-vector) and Con (psiCHECK-cyclin E + negative control). Mimics: psiCHECK-cyclin E + miR-34a mimics. d Effect of miR-34a on expression of CDK2. Data are means of three separate experiments ± SD. # p < 0.01 vs. Mock (psiCHECK-vector) and Con (psiCHECK-CDK2 + negative control). Mimics: psiCHECK-CDK2 + miR-34a mimics
Influence of miR-34a on the molecules in the Ras/MAPK signaling pathway
RMCs were transfected with miR-34a mimics or a miR-negative control, and expression levels of two key targets in the Ras/MAPK signaling pathways were determined. The total MEK1 protein level was significantly decreased (p < 0.05), but those of Raf1 and ERK1/2 were unchanged (p > 0.05) (Fig. 6a), indicating that MEK1 is the target gene of miR-34a, which has also been demonstrated by others [23]. Moreover, protein levels of K-ras, p-Raf1, p-MEK1, and p-ERK1/2 were significantly down-regulated (p < 0.05) (Fig. 6a). We also detected MEK1, which is the most important molecule in the RAS/MAPK signal pathway in the anti-Thy1 mesangial proliferative glomerulonephritis rat model. Compared with 0 days, phospho-MEK1 at 7 days was significantly increased (p < 0.05) (Fig. 6b). The above results suggested that Ras/MAPK signaling pathway plays a key role in the development of rat mesangial proliferative glomerulonephritis.

Expression levels of Ras/MAPK signaling pathway-related proteins were detected with Western blot in RMCs after transfection with miR-34a mimics. a Protein expression levels of K-ras, Raf-1, p-Raf-1, MEK1, p-MEK1, ERK, p-ERK were detected with Western blot. Data are means of three separate experiments ± SD. a,b,c,d p < 0.05 vs. normal group and Con group (miRNA-negative control group), n = 3. Mimics: miR-34a mimics. Renilla luciferase activity was normalized to firefly luciferase activity. b Protein expression levels of MEK1 and p-MEK1 were detected with Western blot. Data are means of three separate experiments ± SD. *p < 0.01 vs. day 0 and day 14, n = 3
Effects of siPDGFR-β on RMC cell cycle, cell cycle proteins, and the Ras/MAPK signaling pathway
To further determine whether miR-34a can directly target PDGFR-β, we evaluated the influences of siPDGFR-β on the cell cycle, cell cycle-related proteins, and Ras/MAPK signaling pathway molecules using an RNAi technique. After siPDGFR-β transfection, proliferation of RMC cells was suppressed (Fig. 7a; Table 6); and the G0/G1 phase was extended and the G2/M phase was reduced (Fig. 7b). Protein levels of cyclin D1, cyclin E, CDK2, CDK4, and CDK6 were significantly decreased after siPDGFR-β transfection (p < 0.05) (Fig. 7c). Levels of the Ras/MAPK signaling pathway proteins, K-ras, Raf1, MEK1, and ERK1/2, were determined. There were no changes in protein levels of total Raf1, MEK1, and ERK1/2 (p > 0.05) (Fig. 7d). However, the protein levels of K-ras, p-Raf1, p-MEK, and p-ERK1/2 were significantly decreased (p < 0.05) (Fig. 7d). These results indicate that siPDGFR-β directly suppressed PDGFR-β and then influenced the cell cycle through the Ras/MAPK signaling pathway. The actions of miR-34a and siPDGFR-β on both PDGFR-β and the cell cycle were similar.

Effects of siPDGFR-β on proliferation, cell cycle, cell cycle proteins, and Ras/MAPK signaling pathway molecules in RMCs. a Proliferation activity of RMCs after transfection with siPDGFR-β or negative control was detected by the Cell Counting Kit-8. *p < 0.05 vs. normal group and siCON group at 48 h, n = 3; # p < 0.01 vs. normal group and siCON group at 72 h, n = 3. N: normal group; siCon: negative control group; siPR-β: siPDGFR-β. b FCM analysis of cell cycles of RMCs transfected with siPDGR-β or negative control. *p < 0.05 vs. normal group and siCon. group, n = 3. Normal: normal group; siCon: negative control group; siPR-β: siPDGFR-β. c Protein expression levels of PDGFR-β, p-PDGFR-β, cyclinD1, cyclinE, CDK2, CDK4, CDk6, p27kip1, K-ras, Raf-1, p-Raf-1, MEK1, p-MEK1, ERK1/2, p-ERK1/2 were detected with Western blot. Data are means of three separate experiments ± SD. a,b,c,d,e,f,g,h,i,j,k.l.m p < 0.05 vs. normal group and siCon group. Normal: normal group; siCon: negative control group; siPR-β: siPDGFR-β
MiR-34a suppressed the proliferation of RMCs that were stimulated by 20 % FBS
We have demonstrated that miR-34a can inhibit the proliferation of normal mesangial cell by directly inhibiting target gene PDGFR-β. Here, we stimulated mesangial cell proliferation with exogenous factors (20 % FBS), and then observed if miR-34a could antagonize this phenomenon. The results showed that, after 20 % FBS treatment, the proliferation rate of cultured mesangial cells was significantly higher than 10 % FBS-treated mesangial cells (p < 0.05) (Fig. 8a). After miR-34a mimics transfection, the proliferation rate of mesangial cells cultured with 20 % FBS was significantly lower than the control group (p < 0.05) and no difference with 10 % FBS culture group (p > 0.05) (Fig. 8a). Compared with the 10 % FBS group and 20 % FBS control group, the G0/G1 phase of RMCs cultured with 20 % FBS after miR-34a mimics transfection was extended, and the G2/M phase was reduced (Fig. 8b; Table 7). Compared with 20 % FBS control group, protein levels of cyclin D1, cyclin E, CDK2, CDK4, and CDK6 were significantly decreased in 20 % FBS-cultured RMC after miR-34a mimics transfection (p < 0.05) (Fig. 8c). Levels of the Ras/MAPK signaling pathway proteins, K-RAS, Raf1, MEK1, and ERK1/2, were determined. Compared with the 20 % FBS control group, K-RAS and total MEK1 protein levels of miR-34a mimics were decreased (p < 0.05), and the phosphorylated protein levels of Raf1, MEK1, ERK1/2 were significantly decreased (p < 0.05) (Fig. 8c). Compared with 20 % FBS-cultured group, which was transfected with miR-34a mimics, p27kip1 in the RMCs cultured with 20 % FBS was decreased (p < 0.05) (Fig. 8c). These results indicate that miR-34a directly suppressed the 20 % FBS-stimulated RMC proliferation.

Effect of miR-34a on cell cycle, cell cycle proteins, and the Ras/MAPK signaling pathway molecules in the RMCs, which were stimulated to proliferate by 20 % FBS. a miR-34a mimics antagonize 20 % FBS-stimulate mesangial cell proliferation. RMCs cultured by 10 % FBS or 20 % FBS, and transfected with miR-34a mimics or negative control. The proliferation rate was detected by Cell Counting Kit-8. a,b p < 0.05 vs. 20 % FBS group and 20 % FBS + Con group at 48 h, n = 3; c,d p < 0.01 vs. 20 % FBS group and 20 % FBS + Con group at 72 h, n = 3. 10 % FBS: 10 % FBS group; 20 % FBS: 20 % FBS group; 20 % FBS + Con: 20 % FBS + negative control; 20 % FBS + miR-34a: 20 % FBS + miR-34a mimics. b FCM analysis of cell cycles of RMCs transfected with miR-34a mimics or negative control, n = 3. 10 % FBS: 10 % FBS group; 20 % FBS: 20 % FBS group; 20 % FBS + Con: 20 % FBS + negative control; 20 % FBS + miR-34a:20 % FBS + miR-34a mimics. c Protein expression levels of PDGFR-β, p-PDGFR-β, cycliyD1, cyclinE, CDK2, CDK4, CDk6, p27kip1, K-ras/Raf-1, p-Raf-1, MEK1, p-MEK1, ERK1/2, p-ERK1/2 were detected with Western blot. Data are means of three separate experiments ± SD. a,b,c,d,e,f,g,h,i,j,k,l p < 0.05 vs. 20 % FBS + mimics group. 10 % FBS: 10 % FBS group; 20 % FBS: 20 % FBS group; 20 % FBS + Con: 20 % FBS + negative control; 20 % FBS + miR-34a: 20 % FBS + miR-34a mimics
Discussion
MsPGN is divided into IgA and non-IgA nephropathy, and the main pathological changes associated with this condition are diffuse proliferation of GMCs and different degrees of ECM accumulation [1, 2, 24]. IgA nephropathy is the most common type of glomerulonephritis globally [25–27], and occurs mainly among Asians, especially the Japanese and Chinese [28–36]. A study of 13,519 Chinese renal biopsies found that primary MsPGN accounted for 70.88 % of all cases of glomerular diseases. IgA nephropathy comprised 45.26 % of the primary glomerular diseases, while non-IgA mesangial proliferative lesions comprised 25.62 % [37]. The risk factors associated with inflammatory reactions have not been determined. Mesangial cells are not only target cells of immune injury, but also proliferate under the influence of various stimulatory molecules, release inflammatory factors, and ECM components, and further promote kidney damage, ultimately leading to glomerular sclerosis and interstitial fibrosis.
miR-34 was first discovered in C. elegans in 2001 [14] and is associated with a variety of organ and tumor hyperplasias [15–20]. Therefore, this study aimed to investigate the role of miR-34a in proliferative glomerulonephritis. We first established an anti-Thy1 MsPGN rat model. In the anti-Thy1 glomerulonephritis rat model, we detected miR-34a expression in kidney tissues at various time points and found that miR-34a level gradually decreased as proliferation increased, then returned to normal levels when mesangial proliferation normalized. This indicates that miR-34a likely plays a suppressive role in RMC proliferation.
We found that the cell proliferation rate was lower in the miR-34a-transfected RMC group than in the control group, indicating that miR-34a inhibits the proliferation of RMCs. We then used flow cytometry to evaluate the influences of miR-34a on the cell cycle. In the miR-34a-transfected cells, G1/G0 was lengthened and the G2+M and S phases were shortened. Thus miR-34a can extend the G1 phase and inhibit cell proliferation. The above results are consistent with those reported for miR-34a in other tissues and tumors [16, 38, 39].
The most critical elements that positively regulate the G1 phase are cell cycle proteins (cyclins D1 and E). Cell cycle proteins and cell cycle protein kinases (CDK2, CDK4, and CDK6) are assembled into two types of complex: cyclin D1/CDK4/CDK6 and cyclin E/CDK2 [40, 41]. The cell cycle protein kinase inhibitor (CKI) p27kip1 inhibits cyclinD1/CDK4/CDK6 and cyclinE/CDK2 complexes, negatively regulating the cell cycle [42]. In the normal glomerulus, p27kip1 is highly expressed to maintain a steady state in the kidney and avoid over-proliferation [43]. The p27kip1 level dropped sharply and returned to baseline in the MsPGN model [43]. There was a similar change in PDGF-induced cell proliferation [44]. According to flow cytometry, miR-34a affects mainly the G0/G1 phase of the cell cycle; the main G0/G1 phase proteins cyclin D1, cyclin E, CDK2, CDK4, CDK6, and p27kip1 were evaluated in this study [1, 45, 46].
MiRanda, TargetScanS, and PicTar were integrated into MiRGen online biology prediction software (http://www.diana.pcbi.upenn.edu/miRGen.html), which is the bioinformatics software used most commonly to predict miRNA target genes. MiRGen prediction results suggested that PDGFR-β may be a direct target gene of miR-34a.
PDGFR is a receptor protein of PDGF. PDGF is classified into four subtypes, of which PDGF-B and PDGF-D are involved in the regulation of cell proliferation through PDGFR-β [46]. PDGF and PDGFR-β binding activates the Ras/MAPK signaling pathway as well as many downstream target genes, such as cyclins D1 and E [1], which control cell proliferation. The selective PDGFR tyrosine blocker STI157 can block mesangial cell proliferation [47].
We found that the PDGFR-β protein level was markedly lower in the miR-34a-transfected RMCs, while the mRNA level did not change. The results of the dual-luciferase assay indicated that PDGFR-β is a direct target gene of miR-34a. We assumed that cyclin E and CDK2 were target genes of miR-34a. The results of the dual-luciferase assay confirmed this hypothesis. MiR-34a not only regulates PDGFR-β expression but also acts directly in conjunction with cyclin E/CDK2 to regulate the cell cycle and influence proliferation. Other studies have confirmed that cyclin D1 [21], CDK4 [9], and CDK6 [21, 22] are target genes of miR-34a. In our study, there were no changes in the mRNA levels of cyclin D1, CDK2, or CDK6 while the proteins expression was significantly reduced after miR-34a transfection; however, the mRNA level of cyclin E was decreased in the miR-34a-transfected RMCs. Moreover, both p27kip1 protein and mRNA levels were increased in the miR-34a-transfected group, which is consistent with the effect on mesangial cell proliferation [44, 48].
To further define whether miR-34a regulates downstream target genes through the Ras/MAPK signaling pathway, we used miR-34a mimics to transfect RMCs. The total Raf1 and ERK1/2 protein level did not change, but those of K-RAS, p-Raf1, MEK1, p-MEK1, and p-ERK1/2 decreased. A previous study showed that the total Raf1, ERK1/2, or MEK1 protein level was unchanged when RAS/MAPK signaling pathways were activated. We hypothesize that the change in the total MEK1 protein level occurred because MEK1 is a target gene of miR-34a [23]. PDGFR-β activates the RAS/MAPK signal pathway molecules, K-RAS, p-Raf1, p-MEK1, and p-ERK1/2, thus affecting the G1 phase [49–52]. Therefore, miR-34a may directly suppress PDGFR-β and so regulate the expression of cell-cycle proteins (CDK and CKI) through the RAS/MAPK signaling pathway [1, 45].
To demonstrate that miR-34a modulates the cell cycle by directly inhibiting PDGFR-β, we used siPDGFR-β to inhibit the expression of PDGFR-β. The results showed that the changes in both cell proliferation and the cell cycle were identical to those in the miR-34a-transfected group after siPDGFR-β transfection. The cell cycle proteins also showed the same trend as in the miR-34a group. SiPDGFR-β and miR-34a exert similar influences on PDGFR-β and the cell cycle. Therefore, miR-34a can affect the G0/G1 phase through inhibition of PDGFR-β.
We have confirmed that miR-34a can directly inhibit the target gene PDGFR-β and in turn affect the normal RMC proliferation function by suppressing the G0/G1 phase. Here we use high concentrations of serum (20 % serum) to stimulate mesangial cell proliferation, and then use miR-34a to antagonize this phenomenon. The results are clear that high concentrations of serum could accelerate cell proliferation, however, the use of miR-34a together with a high concentration of serum in the cell, the cell proliferation rate return to normal levels for 10 % serum culture group. Thus, miR-34a can antagonize proliferation of the high concentration serum group.
In conclusion, we found that miR-34a regulates mesangial cell proliferation by directly inhibiting expressions of PDGFR-β and factor (MEK1) in the RAS/MAPK signal pathway, and by influencing the G0/G1 phase. MiR-34a may also affect cell proliferation by directly suppressing the expression of cyclin E and CDK2 cell cycle proteins in the G0/G1 phase (Fig. 9).

Proposed mechanisms by which miR-34a regulates mesangial cell proliferation
References
Cove-Smith A, Hendry BM (2008) The regulation of mesangial cell proliferation. Nephron Exp Nephrol 108:e74–e79
Schlondorff D, Banas B (2009) The mesangial cell revisited: no cell is an island. J Am Soc Nephrol 20:1179–1187
Eddy AA (2005) Progression in chronic kidney disease. Adv Chronic Kidney Dis 12:353–365
Kriz W, Hahnel B, Hosser H, Ostendorf T, Gaertner S, Kranzlin B, Gretz N, Shimizu F, Floege J (2003) Pathways to recovery and loss of nephrons in anti-Thy-1 nephritis. J Am Soc Nephrol 14:1904–1926
Ono T, Liu N, Makino T, Nogaki F, Muso E, Honda G, Kita T (2005) Suppressive mechanisms of Sairei-to on mesangial matrix expansion in rat mesangioproliferative glomerulonephritis. Nephron Exp Nephrol 100:e132–e142
Ikezumi Y, Kanno K, Karasawa T, Han GD, Ito Y, Koike H, Toyabe S, Uchiyama M, Shimizu F, Kawachi H (2004) The role of lymphocytes in the experimental progressive glomerulonephritis. Kidney Int 66:1036–1048
Wang Y, He Q, Qin H, Xu J, Tong J, Gao L, Xu J (2006) The complement C5b-9 complexes induced injury of glomerular mesangial cells in rats with Thy-1 nephritis by increasing nitric oxide synthesis. Life Sci 79:182–192
Wan Y, Gu L, Suzuki K, Karasawa T, Fujioka Y, Han GD, Koike H, Kawachi H, Shimizu F (2005) Multi-glycoside of Tripterygium wilfordii Hook f. ameliorates proteinuria and acute mesangial injury induced by anti-Thy1.1 monoclonal antibody. Nephron Exp Nephrol 99:e121–e129
He L, He X, Lim LP, de Stanchina E, Xuan Z, Liang Y, Xue W, Zender L, Magnus J, Ridzon D, Jackson AL, Linsley PS, Chen C, Lowe SW, Cleary MA, Hannon GJ (2007) A microRNA component of the p53 tumour suppressor network. Nature 447:1130–1134
Selbach M, Schwanhausser B, Thierfelder N, Fang Z, Khanin R, Rajewsky N (2008) Widespread changes in protein synthesis induced by microRNAs. Nature 455:58–63
Krol J, Loedige I, Filipowicz W (2010) The widespread regulation of microRNA biogenesis, function and decay. Nat Rev Genet 11:597–610
Friedman RC, Farh KK, Burge CB, Bartel DP (2009) Most mammalian mRNAs are conserved targets of microRNAs. Genome Res 19:92–105
Li JY, Yong TY, Michael MZ, Gleadle JM (2010) Review: the role of microRNAs in kidney disease. Nephrology (Carlton) 15:599–608
Lau NC, Lim LP, Weinstein EG, Bartel DP (2001) An abundant class of tiny RNAs with probable regulatory roles in Caenorhabditis elegans. Science 294:858–862
Welch C, Chen Y, Stallings RL (2007) MicroRNA-34a functions as a potential tumor suppressor by inducing apoptosis in neuroblastoma cells. Oncogene 26:5017–5022
Cole KA, Attiyeh EF, Mosse YP, Laquaglia MJ, Diskin SJ, Brodeur GM, Maris JM (2008) A functional screen identifies miR-34a as a candidate neuroblastoma tumor suppressor gene. Mol Cancer Res 6:735–742
Tazawa H, Tsuchiya N, Izumiya M, Nakagama H (2007) Tumor-suppressive miR-34a induces senescence-like growth arrest through modulation of the E2F pathway in human colon cancer cells. Proc Natl Acad Sci USA 104:15472–15477
Yan D, Zhou X, Chen X, Hu DN, Dong XD, Wang J, Lu F, Tu L, Qu J (2009) MicroRNA-34a inhibits uveal melanoma cell proliferation and migration through downregulation of c-Met. Invest Ophthalmol Vis Sci 50:1559–1565
Guessous F, Zhang Y, Kofman A, Catania A, Li Y, Schiff D, Purow B, Abounader R (2010) microRNA-34a is tumor suppressive in brain tumors and glioma stem cells. Cell Cycle 9:1031–1036
Pang RT, Leung CO, Ye TM, Liu W, Chiu PC, Lam KK, Lee KF, Yeung WS (2010) MicroRNA-34a suppresses invasion through downregulation of Notch1 and Jagged1 in cervical carcinoma and choriocarcinoma cells. Carcinogenesis 31:1037–1044
Ichimura A, Ruike Y, Terasawa K, Shimizu K, Tsujimoto G (2010) MicroRNA-34a inhibits cell proliferation by repressing mitogen-activated protein kinase kinase 1 during megakaryocytic differentiation of K562 cells. Mol Pharmacol 77:1016–1024
Sun F, Fu H, Liu Q, Tie Y, Zhu J, Xing R, Sun Z, Zheng X (2008) Downregulation of CCND1 and CDK6 by miR-34a induces cell cycle arrest. FEBS Lett 582:1564–1568
Li Y, Guessous F, Zhang Y, Dipierro C, Kefas B, Johnson E, Marcinkiewicz L, Jiang J, Yang Y, Schmittgen TD, Lopes B, Schiff D, Purow B, Abounader R (2009) MicroRNA-34a inhibits glioblastoma growth by targeting multiple oncogenes. Cancer Res 69:7569–7576
Chen X, Qiu Q, Tang L, Liu S, Cai G, Liu H, Xie Y (2004) Effects of co-administration of urokinase and benazepril on severe IgA nephropathy. Nephrol Dial Transplant 19:852–857
D’Amico G (1987) The commonest glomerulonephritis in the world: IgA nephropathy. Q J Med 64:709–727
Julian BA, Waldo FB, Rifai A, Mestecky J (1988) IgA nephropathy, the most common glomerulonephritis worldwide. A neglected disease in the United States. Am J Med 84:129–132
Levy M, Berger J (1988) Worldwide perspective of IgA nephropathy. Am J Kidney Dis 12:340–347
Jennette JC, Wall SD, Wilkman AS (1985) Low incidence of IgA nephropathy in blacks. Kidney Int 28:944–950
Rambausek M, Rauterberg EW, Waldherr R, Demaine A, Krupp G, Ritz E (1987) Evolution of IgA glomerulonephritis: relation to morphology, immunogenetics, and BP. Semin Nephrol 7:370–373
Seedat YK, Nathoo BC, Parag KB, Naiker IP, Ramsaroop R (1988) IgA nephropathy in blacks and Indians of Natal. Nephron 50:137–141
Okada K, Funai M, Kawakami K, Kagami S, Yano I, Kuroda Y (1990) IgA nephropathy in Japanese children and adults: a comparative study of clinicopathological features. Am J Nephrol 10:191–197
Simon P, Ramee MP, Autuly V, Laruelle E, Charasse C, Cam G, Ang KS (1994) Epidemiology of primary glomerular diseases in a French region. Variations according to period and age. Kidney Int 46:1192–1198
Stratta P, Segoloni GP, Canavese C, Sandri L, Mazzucco G, Roccatello D, Manganaro M, Vercellone A (1996) Incidence of biopsy-proven primary glomerulonephritis in an Italian province. Am J Kidney Dis 27:631–639
Koyama A, Igarashi M, Kobayashi M (1997) Natural history and risk factors for immunoglobulin A nephropathy in Japan. Research Group on Progressive Renal Diseases. Am J Kidney Dis 29:526–532
Woo KT, Chiang GS, Pall A, Tan PH, Lau YK, Chin YM (1999) The changing pattern of glomerulonephritis in Singapore over the past two decades. Clin Nephrol 52:96–102
Li PK, Ho KK, Szeto CC, Yu L, Lai FM (2002) Prognostic indicators of IgA nephropathy in the Chinese–clinical and pathological perspectives. Nephrol Dial Transplant 17:64–69
Li LS, Liu ZH (2004) Epidemiologic data of renal diseases from a single unit in China: analysis based on 13,519 renal biopsies. Kidney Int 66:920–923
Wei JS, Song YK, Durinck S, Chen QR, Cheuk AT, Tsang P, Zhang Q, Thiele CJ, Slack A, Shohet J, Khan J (2008) The MYCN oncogene is a direct target of miR-34a. Oncogene 27:5204–5213
Li N, Fu H, Tie Y, Hu Z, Kong W, Wu Y, Zheng X (2009) miR-34a inhibits migration and invasion by down-regulation of c-Met expression in human hepatocellular carcinoma cells. Cancer Lett 275:44–53
Hunter T (1993) Braking the cycle. Cell 75:839–841
Sherr CJ (1993) Mammalian G1 cyclins. Cell 73:1059–1065
Funk JO, Galloway DA (1998) Inhibiting CDK inhibitors: new lessons from DNA tumor viruses. Trends Biochem Sci 23:337–341
Shankland SJ, Hugo C, Coats SR, Nangaku M, Pichler RH, Gordon KL, Pippin J, Roberts JM, Couser WG, Johnson RJ (1996) Changes in cell-cycle protein expression during experimental mesangial proliferative glomerulonephritis. Kidney Int 50:1230–1239
Lang S, Hartner A, Sterzel RB, Schocklmann HO (2000) Requirement of cyclin D1 in mesangial cell mitogenesis. J Am Soc Nephrol 11:1398–1408
Schocklmann HO, Lang S, Sterzel RB (1999) Regulation of mesangial cell proliferation. Kidney Int 56:1199–1207
Floege J, Eitner F, Alpers CE (2008) A new look at platelet-derived growth factor in renal disease. J Am Soc Nephrol 19:12–23
Johnson RJ, Raines EW, Floege J, Yoshimura A, Pritzl P, Alpers C, Ross R (1992) Inhibition of mesangial cell proliferation and matrix expansion in glomerulonephritis in the rat by antibody to platelet-derived growth factor. J Exp Med 175:1413–1416
Liu S, Xie Y, Lv Y, Qin F, Fu B, Shi S, Yin Z, Hong Q, Zhang X, Wang J, Chang M, Chen X (2010) A novel target of mizoribine inhibiting mesangial cell proliferation: S phase kinase-associated protein 2. Am J Nephrol 32:447–455
Khwaja A, Sharpe CC, Noor M, Hendry BM (2006) The role of geranylgeranylated proteins in human mesangial cell proliferation. Kidney Int 70:1296–1304
Bokemeyer D, Panek D, Kitahara M, Trzaskos JM, Muller CE, Hockemeyer J, Kunter U, Boor P, Floege J, Kramer HJ, Ostendorf T (2007) The map kinase ERK regulates renal activity of cyclin-dependent kinase 2 in experimental glomerulonephritis. Nephrol Dial Transplant 22:3431–3441
Huang Y, Noble NA, Zhang J, Xu C, Border WA (2007) Renin-stimulated TGF-beta1 expression is regulated by a mitogen-activated protein kinase in mesangial cells. Kidney Int 72:45–52
Kato M, Arce L, Natarajan R (2009) MicroRNAs and their role in progressive kidney diseases. Clin J Am Soc Nephrol 4:1255–1266
Acknowledgments
This research was supported by a grant (2013BAI09B05) from the National Clinical Research Center for Kidney Disease, a grant (2012AA02A512) from the National High Technology Research and Development Program, a grant (2011BA110B03) from the National Key Technology R&D Program, a grant (81330019) from the Major Program of National Natural Science Foundation of China, a grant (81270794) from the National Natural Science Foundation of China, grants (D131100004713003) from the Science and Technology Project of Beijing, a grant (31200589) from the National Natural Science Foundation of China. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Conflict of interest
All the authors declared no competing interests.
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article
Cite this article
Chen, D., Li, Y., Mei, Y. et al. miR-34a regulates mesangial cell proliferation via the PDGFR-β/Ras-MAPK signaling pathway. Cell. Mol. Life Sci. 71, 4027–4042 (2014). https://doi.org/10.1007/s00018-014-1599-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00018-014-1599-y