
Abstract
Cellular senescence is a process that can prevent tumour development in a cell autonomous manner by imposing a stable cell cycle arrest after oncogene activation. Paradoxically, senescence can also promote tumour growth cell non-autonomously by creating a permissive tumour microenvironment that fuels tumour initiation, progression to malignancy and metastasis. In a pituitary tumour known as adamantinomatous craniopharyngioma (ACP), cells that carry oncogenic β-catenin mutations and overactivate the WNT signalling pathway form cell clusters that become senescent and activate a senescence-associated secretory phenotype (SASP). Research in mouse models of ACP has provided insights into the function of the senescent cell clusters and revealed a critical role for SASP-mediated activities in paracrine tumour initiation. In this review, we first discuss this research on ACP and subsequently explore the theme of paracrine tumourigenesis in other tumour models available in the literature. Evidence is accumulating supporting the notion that paracrine signalling brought about by senescent cells may underlie tumourigenesis across different tumours and cancer models.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Almost over 60 years ago, it was first reported that continuous in vitro culturing of human cells results in a gradual but ultimately complete decay of their proliferative capacity [1, 2]. The term cellular senescence was then applied to describe this particular phenomenon as it was hypothesized to be the result of a deterioration in the cell’s homeostatic functions with time, a process resembling organismal aging [3]. However, recently acquired understanding of the complexity and heterogeneity of this phenomenon has revealed that senescent cells can be anything but a simple manifestation of decay and dysfunction, as their name might otherwise suggest.
The early concept of cellular senescence has now been expanded to describe a growing list of phenotypes initiated by damaging stimuli such as telomere attrition, ionizing radiation, chemotherapeutic compounds, reactive oxygen species (ROS), mitochondrial dysfunction and oncogenic signalling [4]. Importantly, all of these phenotypes share common hallmark features such as the activation of DNA-damage pathways, cell cycle arrest mediated by the p16INK4/Rb and p21CIP1/p53 pathways, the activation of anti-apoptotic mechanisms and the widespread secretion of growth factors, cytokines, chemokines and extracellular matrix components (collectively known as the senescence-associated secretory phenotype or SASP). The different types of senescent phenotypes and their underlying mechanisms have been thoroughly reviewed elsewhere [4, 5].
Senescent cells and the SASP can induce a vast array of context-dependent effects, playing significant roles in the regulation of normal tissue physiology but also in disease. Senescent cells can be found in several tissues during embryonic development and participate in the proper patterning of some organs and tissues [6,7,8,9]. After development, senescent cells are also involved in tissue regeneration and wound repair in several organs, although their exact role appears to be more complex and context dependent. While they have been reported to play beneficial roles in acute wound repair [10,11,12,13,14,15,16], the opposite has been observed during chronic wounding scenarios [17,18,19,20]. This detrimental aspect of long-term senescent cell accumulation has also been widely described in the development of several pathologies, including those related to organismal ageing (e.g. atherosclerosis, rheumatoid arthritis, metabolic dysfunction, diabetes and neurodegenerative diseases, among many others). It is possible that this dichotomy is related to a tight regulation of dynamic balances between contrasting SASP activities, such as the paracrine promotion of cellular plasticity and reprogramming on one side, and the induction of by-stander senescence and inflammation on the other [21, 22]. Importantly, there is evidence demonstrating that the SASP can lead to widespread effects beyond the microenvironment, such as driving systemic inflammation and haemostasis, as well as mediating several side effects of chemotherapy including decreased physical activity and strength, bone marrow suppression and cancer recurrence [23,24,25,26]. Both detrimental and beneficial activities of senescent cells and the SASP have previously been reviewed in detail [27,28,29].
In the case of cancer and neoplastic diseases, senescence can be induced cell autonomously by oncogene activation (i.e. oncogene-induced senescence, OIS) or through therapeutics such as DNA-damaging chemical compounds and ionizing radiation (i.e. therapy-induced senescence, TIS), which lead to the activation of DNA-damage pathways and the activation of a stable cell cycle arrest [30]. Additionally, the SASP can induce senescence cell non-autonomously in neighbouring cells (i.e. paracrine-induced senescence or bystander effect) or mediate cancer cell clearance by the immune system [31]. For this, cellular senescence has been widely regarded as an innately protective mechanism that restricts cancer cell proliferation and tumour growth [32, 33]. However, the paradigm of senescence as a tumour-suppressing mechanism has been challenged by studies showing that senescent cells and the SASP can represent a double-edged sword with serious negative effects in cancer and other diseases. In particular, there is mounting evidence showing that paracrine SASP signals can stimulate several pro-tumourigenic cellular and molecular processes such as cancer cell proliferation, progression to malignancy, immune system evasion, resistance to therapy-induced apoptosis, angiogenesis, formation and maintenance of metastatic niches, as well as increased cell invasiveness, migration and epithelial-to-mesenchymal transitions (EMT) [34,35,36,37,38,39,40,41], and even induce tumour formation cell non-autonomously [42, 43]. We refer the reader elsewhere for comprehensive reviews on the pro-tumourigenic activities of senescence and the SASP [30, 44,45,46].
The cell non-autonomous origin of some tumours stands in stark contrast to traditional models of carcinogenesis [47,48,49]. A review of the available evidence supporting this scarcely discussed mechanism could provide further insights into the role of senescence in cancer. In this manuscript, we discuss studies on senescence and the SASP which have improved our understanding of the origins and biology of a paediatric pituitary tumour known as adamantinomatous craniopharyngioma (ACP). We describe two genetically engineered mouse models of ACP and present evidence supporting a cell non-autonomous model of tumour formation driven by senescence. We further explore the literature to discuss existing examples of the widely unexplored phenomenon of paracrine tumour initiation and highlight studies that have also shown a major role for senescence and the SASP in this process.
Adamantinomatous craniopharyngioma and mouse ACP models
Human adamantinomatous craniopharyngioma (ACP): clinical aspects and pathology
Craniopharyngiomas (CPs) are benign epithelial tumours (WHO grade 1) of the sellar region, which is an anatomical structure located between the hypothalamus and the cranial base. CPs represent between 1.2 and 4.6% of all intracranial tumours, with an incidence of 0.5–2.5 new cases per 1 million population per year [50, 51]. There are two subtypes of CPs, the papillary and the adamantinomatous (PCP and ACP, respectively), which differ in their clinical, histological and molecular features [52]. Because of the proven relevance of senescence in ACP, in this review, we will focus only on this tumour type.
ACPs represent the most common non-neuroepithelial intracranial tumours in children and young adults [50, 53]. They are difficult to manage and can behave aggressively in the clinic. Additionally, treatments are non-specific (i.e. maximal safe surgical resection avoiding damage of the hypothalamus and visual pathways, followed by radiotherapy), non-curative and associated with high morbidity [53,54,55,56,57]. This morbidity is due to the tumour’s tendency to invade surrounding structures such as the pituitary, hypothalamus and optic chiasm. Consequences of both tumour growth and its treatment include pan-hypopituitarism with multiple neuroendocrine deficiencies, blindness and hypothalamic damage, which usually leads to obesity, subsequent type-2 diabetes and cardiovascular disease [58,59,60]. Furthermore, reduced psychosocial and neurocognitive function are common in survivors, mostly in patients of younger age [58, 61]. All of these comorbidities lead to poor quality of life and increased long-term mortality in survivors [62].
The histomorphological features of ACPs are well defined (Fig. 1). The tumour epithelium is surrounded by glial reactive tissue that is comprised of non-tumoural cells such as astrocytes, immune cells and fibroblasts [63]. The tumours themselves are usually comprised of solid and cystic components [64]. The solid part includes epithelial tumour cells, organised in well-defined structures such as the palisading epithelium, the stellate reticulum and whorl-like structures. The epithelial component of these tumours shows no sign of neuroendocrine differentiation (i.e. lack of expression of pituitary hormones, cell-lineage markers or neuroendocrine markers like synaptophysin). Additional solid components include wet keratin (i.e. eosinophilic areas of keratinised cells without nuclei) and calcification foci. In addition to these solid structures, ACP tumours usually contain one or multiple cysts filled with a dark fluid enriched in inflammatory mediators and lipids [65,66,67].

adapted from Martinez-Barbera JP, Andoniadou CL (2020) Biological Behaviour of Craniopharyngiomas. Neuroendocrinology 1–8, with permission of S. Karger AG, Basel
Histopathology of human adamantinomatous craniopharyngioma (ACP). TE tumour epithelium, GRT glial reactive tissue, PE palisading epithelium, SR stellate reticulum, WL whorl-like epithelial cell groups. Immunostaining for β-catenin showing nucleo-cytoplasmic accumulation in cells of the WL, whilst the rest of the tumour cells show normal membranous staining. Scale bar 200 μm. The figure is
Molecularly, ACPs are driven by the overactivation of the WNT/β-catenin signalling pathway [68, 69]. This pathway is heavily involved in normal development and physiology as well as in cancer [70]. Figure 2 depicts a schematic and description of the pathway. Research from the last 2 decades has demonstrated that mutations in exon 3 of CTNNB1, the gene encoding for β-catenin, are the most common molecular alterations associated with ACP tumourigenesis [68, 69]. These mutations are predicted to prevent protein degradation and cause nucleo-cytoplasmic accumulation of β-catenin and activation of the pathway [71]. In agreement, immunohistochemistry against β-catenin has shown the presence of sporadic epithelial tumours cells with cytoplasmic and nuclear staining, either dispersed throughout the tumour or grouped in whorl-like epithelial structures (also known as clusters) (Figs. 1, 3) [72]. Despite β-catenin accumulation being restricted to a minority of cells, CTNNB1 mutations have been identified in all of the epithelial tumour cells in a large cohort of ACPs by combining laser capture microdissection with deep sequencing [73]. Three-dimensional imaging of human ACP tumours has revealed that these β-catenin-accumulating cell clusters are located within finger-like protrusions of tumour epithelium that invade the brain and surrounding structures, suggesting a potential role in tumour invasion [74]. Importantly, murine studies have demonstrated that mutations in CTNNB1 are tumour drivers and provided important insights into the role of the nucleocytoplasmic β-catenin cell clusters in ACP tumourigenesis (see below) [43, 75]. The cellular origin of human ACP is still a matter of debate, with the most prominent hypothesis being that it arises from embryonic oral ectoderm and in particular from remnants of Rathke’s pouch epithelium, a proposition derived from the observation of a common expression of certain cytokeratins between ACPs and oral epithelium [71, 76, 77]. In support of this, a recent RNA sequencing study found that human ACPs share a common transcriptional profile with tissues present during normal tooth development [67].

Schematic of main components of the canonical WNT signalling pathway. In the absence of WNT ligands, β-catenin, (encoded by the CTNNB1 gene) is normally recruited in a destruction complex containing several proteins including APC (adenomatous polyposis coli), AXIN, CKIα (casein kinase 1 alpha) and GSK3β (glycogen synthase kinase 3β). This results in β-catenin phosphorylation of specific amino acids encoded by CTNNB1 exon 3 and protein degradation by the ubiquitin–proteasome pathway. Consequently, levels of β-catenin protein concentration are low in the cytoplasm and nucleus, hence keeping the target genes in a repressed state. At the same time, two surface E3 ubiquitin ligases, RNF43 and ZNFR3, regulate levels of the WNT ligand–receptor Frizzled through its ubiquitination which leads to its endosomal internalization and degradation. Binding of WNT ligands to their receptor, Frizzled, leads to the formation of a complex alongside coreceptors LRP and Dishevelled (DVL), which captures and disassembles the β-catenin destruction complex and thus prevents β-catenin phosphorylation and degradation. This causes protein stabilization, nucleo-cytoplasmic accumulation of β-catenin and the activation of target genes. Examples of WNT target genes encode for LGR receptors (LGR4-6), which upon binding of R-spondins (Rspo), recruit the RNF43/ZNFR3 complex and, therefore, allows the accumulation of Frizzled in the membrane. This leads to positive feedback and amplification of the WNT signalling pathway. Mutations in exon 3 of CTNNB1, containing the regulatory amino acids of β-catenin responsible for its degradation, prevent β-catenin phosphorylation by the destruction complex. This leads to its nucleo-cytoplasmic accumulation and constitutive overactivation of the WNT/β-catenin pathway even in the absence of WNT ligands and R-spondins. Created with BioRender.com

reproduced with permission from Carreno G, Gonzalez-Meljem JM, Haston S, Martinez-Barbera JP (2016) Stem cells and their role in pituitary tumorigenesis. Mol Cell Endocrinol 445:27–34
Human adamantinomatous craniopharyngioma (ACP) and ACP murine models contain nucleo-cytoplasmic β-catenin-accumulating cell clusters. a Immunofluorescent staining in human ACP showing the nucleo-cytoplasmic accumulation of β-catenin in cell groups known as “clusters” (arrows), a defining characteristic of these tumours. b Expression of oncogenic β-catenin in Rathke’s Pouch progenitors leads to the formation of clusters in a Hesx1 Cre/+; Ctnnb1 lox(ex3)/+ pre-tumoural pituitary. c Clusters also form upon inducible expression of oncogenic β-catenin in adult pituitary stem cells in Sox2 CreERT2/+; Ctnnb1 lox(ex3)/+ mice. A 16-week post-tamoxifen induction pituitary is shown. Scale bars 100 μm. AL anterior lobe, IL intermediate lobe. The figure is
Mouse models of ACP: insights into tumour initiation and pathogenesis
Two genetically engineered mouse models of ACP have been developed by expressing oncogenic β-catenin in either HESX1 + embryonic precursors of the developing pituitary (embryonic model; Hesx1 Cre/+; Ctnnb1 lox(ex3)/+ mouse line) or in SOX2 + adult pituitary stem cells (inducible model; Sox2 CreERT2/+; Ctnnb1 lox(ex3)/+ mouse line) (Fig. 4a, b, respectively) [43, 75]. These mouse models utilise the Cre/loxP technology to induce the expression of a murine oncogenic form of β-catenin that is functionally equivalent to those identified in human ACPs. Specifically, Cre recombinase expression in either Hesx1 or Sox2-expressing cells leads to the deletion of exon 3 from the Ctnnb1lox(Ex3) allele, which then codes for a mutant version of β-catenin missing several important regulatory amino acids located in the N-terminal end of the protein. Degradation of this mutant β-catenin is impaired, as the protein cannot be phosphorylated by its destruction complex but can still activate transcription of target genes (Fig. 2). In agreement, both of these ACP murine models show over-activation of the WNT/β-catenin pathway (e.g. expression of target genes such as Axin2 and Lef1) in the developing (embryonic) or adult (inducible) pituitary leading to the formation of tumours resembling human ACP [72, 78].

adapted from Gonzalez-Meljem JM, Haston S, Carreno G, et al. (2017) Stem cell senescence drives age-attenuated induction of pituitary tumours in mouse models of paediatric craniopharyngioma. Nat Commun 8:1819, which is an open-access article licensed under a Creative Commons Attribution 4.0 International License
Lineage tracing in mouse ACP models shows the cell non-autonomous origin of tumours. Mouse ACP models were crossed with R26 YFP/+ lineage reporter mice which allows labelling of cells and their descendants upon expression of Cre recombinase. a Double immunofluorescent staining in Sox2 CreERT2/+; Ctnnb1 lox(ex3)/+; R26 YFP/+ pituitaries showing cell clusters (arrowheads) and a large tumoural lesion (asterisk) that accumulate β-catenin. The tumour cells (asterisk) do not express the lineage reporter YFP, indicating they are not descendants of SOX2 + stem cells. Note that the clusters (arrowheads) co-express nucleocytoplasmic β-catenin and YFP, demonstrating that they derive from SOX2 + stem cells. b In Hesx1 Cre/+; Ctnnb1 lox(ex3)/+; R26 YFP/+ mice, most cells of the anterior lobe of the pituitary descend from HESX1 + Rathke’s pouch precursor cells, as shown by YFP expression in a 5-week-old pituitary. After a period of latency, pituitary tissue is displaced by developing tumour tissue that does not express YFP. Scale bar 5 mm. c The absence of Cre-mediated recombination in the tumours is further demonstrated using the mT/mG dual reporter mouse line, in which unrecombined cells express membrane TdTomato protein (red) while pituitary-lineage cells express GFP (green). Note that in the Hesx1 Cre/+; R26 mTmG/+ control pituitary, the anterior lobe (al) tissue is green (recombined) and the posterior lobe (pl) is red (unrecombined since the pl is not derived from the Hesx1 lineage). In an ACP Hesx1 Cre/+; Ctnnb1 lox(ex3)/+; R26 mTmG/+ mouse tumour, most of the tumour cells express TdTomato. Scale bar 1 mm. d Double immunofluorescent staining against the proliferation marker Ki67 and YFP revealing that although most of the cells in the pituitary anterior lobe (al) of a 5-week-old ACP embryonic model are YFP + ve, the tumours in a 20-week-old mouse develop from YFP-ve cells that show a high proliferative activity (asterisk). In very advanced tumours (35 weeks) most of the YFP + ve cells are missing and only sporadic cells are detected in the periphery. Panel a is adapted with permission from Andoniadou CL, Matsushima D, Mousavy-gharavy SN, et al. (2013) The Sox2 + population of the adult murine pituitary includes progenitor/stem cells with tumour-inducing potential. Cell Stem Cell 13:433–445. b–d are
In the embryonic model, the expression of oncogenic β-catenin is driven into early precursors of Rathke’s pouch (RP) expressing the homeobox transcription factor Hesx1 [75]. RP is the primordium of the anterior pituitary and these precursors give rise to all of the pituitary hormone-producing cells (e.g. somatotrophs expressing growth hormone and corticotrophs expressing adrenocorticotrophic hormone, among others) [79]. Sporadic cells and cell clusters accumulating nucleo-cytoplasmic β-catenin are observed in the developing pituitary soon after the initial expression of oncogenic β-catenin in RP precursors and prior to any sign of cell transformation or tumour development (Fig. 3). Mice are born without pituitary tumours but the clusters are detectable for the first several weeks of postnatal life [80]. Analysis of tumour growth dynamics in the embryonic model has revealed a latency period of approximately 18 weeks from birth until the appearance of proliferative tumours [81]. It is possible that human ACPs follow similar a behaviour as there is a bimodal peak distribution for the age at diagnosis; first at 5–15 years (paediatric ACP) and at 45–60 (adult ACP), while a number of neonatal and embryonic cases have also been reported [58, 82,83,84,85]. As in human ACP, cells accumulating β-catenin are a minority despite the DNA mutation (i.e. Ctnnb1 exon 3 deletion) being present throughout the developing pituitary cells as shown by laser capture microdissection and PCR [75]. Mouse tumours contain solid and cystic components, do not express markers of neuroendocrine differentiation and show histological and imaging features resembling human ACP [81].
An interesting finding from the analysis of the embryonic model is that if oncogenic β-catenin is expressed in committed or differentiated pituitary embryonic cell types, rather than in Hesx1-expressing undifferentiated precursors, clusters do not form and tumours never develop [75]. Because the RP precursors are multipotent, this finding suggests that tumour formation requires a stem-like cell to express oncogenic β-catenin. This possibility has been further explored in the inducible model. Expression of oncogenic β-catenin in SOX2 + ve cells of the postnatal pituitary, which contain bona fide organ-specific stem cells results in the initial formation of β-catenin-accumulating cell clusters and subsequent development of ACP-like tumours after a latency period of 3–6 months (Fig. 4a) [43]. In conclusion, data from these mouse models support the idea that the CTNNB1 mutations identified in human ACP are drivers of tumourigenesis and have provided evidence that ACP likely originates from embryonic RP precursors. Additionally, the dynamics of tumour development suggests that the formation of β-catenin-accumulating cell clusters precede cell transformation and tumour initiation.
Paracrine tumourigenesis in the ACP mouse models
An interesting question that has been investigated using the ACP murine models is to interrogate the fate of the cells expressing and accumulating oncogenic β-catenin. This can be addressed in mice by permanently labelling these cells and their progeny with the expression of a fluorescent reporter protein (e.g. yellow fluorescent protein, YFP), an approach known as genetic lineage tracing [86]. Genetic tracing has been initially carried out in the inducible model to reveal that the ACP-like tumours do not derive from SOX2 + pituitary stem cells expressing oncogenic β-catenin [43]. Instead, targeted SOX2 + stem cells give rise to β-catenin-accumulating cell clusters, while the tumours originate from a different cell lineage as they do not express the lineage reporter, nor do they contain a Ctnnb1 exon 3 deletion (Fig. 4a). These findings are rather unexpected, especially when considering that in other systems (e.g. intestinal tumours), stem cells have been shown to become the tumour cell-of-origin when targeted with similar oncogenic mutations that result in the overactivation of the WNT/β-catenin signalling pathway [87, 88]. In agreement, it has been shown that SOX2 + pituitary stem cells can form tumours cell autonomously when YAP/TAZ signalling is enhanced [89]. The cell non-autonomous origin of the ACP-like tumours has been further corroborated in the embryonic model by lineage tracing (Fig. 4b–d) and DNA sequencing, which have shown that the tumours derive from a different cell lineage and contain novel somatic mutations not present in the germline [42]. Therefore, the expression oncogenic β-catenin in either SOX2 + adult stem cells or HESX1 + embryonic progenitors leads first to the formation of clusters, which are similar to those found in human ACP, and then promote tumour formation in a cell non-autonomous manner. Importantly, in both ACP models and in human ACP, these clusters are non-proliferative and express a multitude of growth factors (e.g. WNTs, FGFs, BMPs, EGF, among others) and inflammatory mediators (e.g. IL1, IL6, CXCL1, CXCL20, among others) [42, 43, 90]. Of note, recent research indicates that SOX2 + stem cells in the normal pituitary have an important paracrine signalling function by mediating the expansion of neighbouring committed progenitor cells, suggesting that oncogenic β-catenin and the senescence programme (discussed below) may consolidate the secretory phenotype of SOX2 + stem cells [91]. These data support a model of paracrine tumourigenesis, whereby non-dividing cluster cells are bestowed with the capacity to initiate tumours in a cell non-autonomous manner.
The role of senescence and its paracrine signals in ACP
Cellular senescence mediates paracrine tumourigenesis in mouse ACP models
Molecular profiling and genetic approaches have shown that these β-catenin accumulating clusters are enriched in senescent cells in both mouse and human ACP. Specifically, human and mouse cluster cells exhibit several hallmark features of senescence: (1) they are viable and non-proliferative (Ki67 and EdU negative); (2) express cell cycle inhibitors (e.g. p21CIP1); (3) exhibit DNA damage and activation of a DNA damage response; (4) have an enlarged lysosomal compartment (e.g. they show elevated expression of GLB1, the enzyme responsible for the widely employed senescence-associated β-galactosidase staining); (5) activate the NF-κB pathway and a SASP [42]. Corroborating the expression data above, unbiased molecular analyses have revealed that the cluster cells are analogous cellular structures in human ACP and both mouse ACP models, which share a common signature of senescence and SASP activation [42].
We have also reported that the presence of an activated SASP results in substantial microenvironmental changes in the pre-tumoural pituitary of the embryonic model. These changes include an excess production of ECM proteins (Fig. 5a), as well as the recruitment of YFP-ve (i.e. not targeted with oncogenic β-catenin) proliferative cells that coexpress the endothelial marker endomucin (EMCN) and the stem cell marker SOX9, which closely interact with the senescent clusters (Fig. 5b, c). Although these changes are less apparent in the inducible model, the presence of senescent β-catenin-accumulating cell clusters in this model also leads to increased proliferation in nearby non-cluster cells (i.e. increased Ki67 mitotic index) [42]. Notably, the close interaction of clusters with SOX9-expressing cells occurs in both mouse models as well as in human ACP [43, 75].

adapted from Gonzalez-Meljem JM, Haston S, Carreno G, et al. (2017) Stem cell senescence drives age-attenuated induction of pituitary tumours in mouse models of paediatric craniopharyngioma. Nat Commun 8:1819, which is an open-access article licensed under a Creative Commons Attribution 4.0 International License
The β-catenin accumulating senescent cell clusters modify the tumour microenvironment (TME) in the pre-tumoural pituitary of the ACP embryonic model. a Immunostaining against fibronectin, laminin and endomucin (EMCN) showing TME alterations prior to tumour initiation in the ACP mouse model. Scale bar 100 μm. b Double immunostaining for the lineage tracing reporter YFP showing an expanded population of EMCN-expressing cells that is not derived from the Hesx1 cell lineage targeted with oncogenic β-catenin. Note that clusters of YFP + ve cells are often surrounded by EMCN + ve cells (arrows). Scale bar 100 μm. c Triple immunostaining showing that in the context of oncogenic β-catenin (Hesx1 Cre/+; Ctnnb1 lox(ex3)/+ pituitary, top panel), large numbers of EMCN + ve cells also co-express SOX9 and interact closely with the senescent clusters (arrows). However, in the context of wild type β-catenin (loss of tumour suppressor Apc in Hesx1 Cre/+; Apc fl/fl pituitaries), senescent clusters are smaller and show an attenuated SASP that fail to induce changes in the TME. Of note, Hesx1 Cre/+; Apc fl/fl do not develop tumours (see text). Scale bar 50 μm. The figure is
The function of senescent cluster cells in ACP tumourigenesis has been investigated in mice using two genetic approaches. First, in the inducible model, it has been shown that the expression of oncogenic β-catenin in SOX2 + pituitary stem cells at different ages results in a significant reduction in tumour formation when comparing induction in aged (6–9 months old) vs. young (4–6 weeks old) mice. This reduction in tumour burden is associated with a significant decrease in the senescence/SASP response in the clusters of the aged mice. Specifically, pituitaries from older mice contain smaller clusters and reduced expression of SASP factors, while reduced numbers of proliferating cells are also observed surrounding the clusters [42].
A second approach has taken advantage of the fact that deletions or inactivating mutations in the genes and proteins of the β-catenin destruction complex, such as Apc (adenomatous polyposis coli) (Fig. 2), also lead to the over-activation of the WNT/β-catenin pathway and tumour formation (e.g. in intestinal cancers in mouse and humans) [87, 88]. However, the deletion of Apc in both Hesx1-expressing embryonic precursors, or SOX2 + adult stem cells, completely fails to generate ACP-like tumours. Detailed histological and molecular analyses have revealed that deletion of Apc also leads to the formation of β-catenin clusters that activate downstream targets of the WNT pathway and express markers of senescence. However, these clusters are smaller in size in comparison to those carrying oncogenic β-catenin and, importantly, show a drastically reduced expression of SASP factors [42]. In addition to an absence of tumour induction, Apc-null pituitaries do not display the microenvironmental alterations that usually precede tumour growth, such as the aberrant expression of ECM markers or the recruitment of a population of proliferating non-oncogene targeted (YFP-negative) endothelial-like cells expressing SOX9 (Fig. 5c). Therefore, the attenuation of the paracrine activities of the senescent cluster cells in the aged inducible model and in Apc-deficient mice results in reduced tumour induction [42].
In human ACP, the location of the senescent clusters within the finger-like tumour protrusions invading the brain suggests that SASP activities may promote epithelial cell proliferation and invasion [74]. This is further supported by the molecular similarities between human clusters and the enamel knot, a signalling hub controlling epithelial bending and proliferation during tooth formation [67]. In addition, the inhibition of the MAPK pathway in human ACP explant cultures results in reduced proliferation and increased apoptosis, while several ligands capable of activating the MAPK pathway are expressed in both human and murine senescent clusters (e.g. FGFs and EGF) [67, 90]. Together, evidence from mouse and human studies strongly suggests that senescence plays an essential role in tumour initiation in the murine models and in tumour growth, invasion and inflammation in human ACP (Fig. 6).

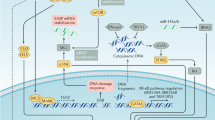
Models depicting the role of senescent cells in mouse and human ACP. a Oncogenic β-catenin expression in either in SOX2 + adult pituitary stem cells or Hesx1 pituitary embryonic progenitors leads to the formation of nucleocytoplasmic β-catenin-accumulating clusters and SASP activation with expression of several cytokines, chemokines and growth factors. Persistent and robust SASP promotes cell transformation of a non-targeted cell (i.e. not expressing oncogenic β-catenin or YFP) in a paracrine manner. In this model SASP-mediated activities of the clusters are required for either tumour initiation, progression or both. b Senescent clusters in human ACP (green cells) are usually found at the base of finger-like tumour projections that invade the brain. The factors secreted by the clusters are proposed to promote tumour cell proliferation of the palisaded epithelium and epithelial bending resulting in tumour invasion. Signals may also promote inflammation in the glial reactive tissue. Created with BioRender.com
Paracrine tumourigenesis and senescence-mediated paracrine tumourigenesis mechanisms have not yet been described in other pituitary tumours. In the case of pituitary adenomas, cellular senescence has been shown to restrict tumour cell proliferation and is thought to underlie the almost invariable benign nature of pituitary tumours [92,93,94,95,96,97]. There are, however, interesting findings suggesting that paracrine SASP signalling might also contribute to pituitary adenoma pathogenesis. For example, it has been shown that senescent cells in somatotroph adenomas also secrete growth hormone (GH) as part of their SASP and that GH and growth hormone releasing hormone (GHRH) are inducers of DNA damage and genomic instability in normal pituitary cells [98,99,100]. Additionally, IL6 is a crucial SASP factor that is normally secreted by pituitary folliculostellate cells, and while it has been shown to induce cellular senescence in adenoma cells, it has also been shown to promote pituitary cell proliferation and to be required for tumour induction in a somatotroph adenoma transplant model [101, 102]. These findings suggest that senescence and the SASP could play a dichotomous role in pituitary adenomas by restricting cell proliferation and preventing the onset of malignancy in established tumours, while promoting the acquisition of genomic instability and the formation of tumour permissive microenvironments.
Paracrine and senescence-induced tumorigenesis
Evidence for paracrine tumorigenesis
Current and widely supported theories of carcinogenesis such as the cancer stem cell/hierarchical model, the genetic/stochastic evolution model and more recent unifying propositions imply that a tumour (or each subclonal population within a tumour) is formed by cells that are descendants of the cell originally targeted by an initiating oncogenic insult [47,48,49]. On the other hand, the observation that tumours can arise cell non-autonomously, as in mouse models of ACP, stands in stark contrast to traditional theories of the origin of cancer. There are, however, several experimental examples showing evidence that cell transformation and tumour initiation mechanisms do not necessarily follow traditional paradigms (Table 1).
Around 3 decades ago, in vitro experiments had already shown that paracrine or autocrine exposure of naïve cells to secreted developmental growth factors of the WNT and FGF families could initiate pre-malignant phenotypical changes, including: (1) ability to grow without attachment in agar plates (a sign of cell transformation); (2) alteration of cell morphologies; (3) increased proliferation and higher cell culture densities; (4) loss of contact inhibition of proliferation [103,104,105,106,107,108,109]. However, these initial studies did not address the capacity of the paracrinally “transformed cells” to generate tumours in vivo. Although research conducted over the last decades has established the importance of mutations and deregulation of these signalling pathways in several cancers [110,111,112,113], the exact relationships between a tumour’s cell-of-origin (also known as tumour-initiating cell), oncogenic driver mutations and the source of paracrine protumourigenic signals have not been widely explored until recently.
In vivo, combinations of different methods such as lineage tracing, cell ablation, DNA sequencing, laser-capture microdissection, chimera aggregations and cell transplantation have been instrumental in providing evidence of paracrine tumourigenesis; while it is important to note that these have also been used to support the validity of cell autonomous mechanisms in some in vivo cancer models [114,115,116]. For example, two notable studies used bone marrow replacement experiments in genetically engineered mouse models to show that the loss of Dicer1 or the activation of oncogenic β-catenin, specifically in osteoprogenitors or osteoblasts, leads to the development of myelodysplastic syndromes and acute myeloid leukaemias in a paracrine manner. Interestingly, they show that the tumour cell of origin does not carry the targeted mutations (i.e. Dicer1 or β-catenin) and instead display novel mutations and genomic aberrations [117, 118].
In breast cancer studies, most experiments so far have used in vitro cocultures to show the capacity of paracrine WNT signalling to transform mammary epithelial cell lines [104,105,106,107]. However, a study has shown that xenotransplanted mammary epithelial cell organoids required HGF and TGB1 secreted by irradiated stromal cells in order to form tumours in nude mice [119]. Likewise, another study has shown the requirement of mammary stroma exposed to the chemical carcinogen N-nitrosomethylurea (NMU) for the induction of mammary epithelial tumours [120].
In regard to brain cancer, lineage tracing and transplantation experiments have demonstrated an important role for paracrine PDGFB signalling in the recruitment of untargeted oligodendrocyte host cells to induce the formation of gliomas [121, 122]. Similarly, a combination of lineage tracing with chimera aggregations has shown the recruitment of untargeted cells during the formation of polyclonal intestinal tumours [123]. Likewise, combining lineage tracing and bone marrow transplantations has revealed the requirement of macrophage-secreted TNF for the initiation of gastric tumours [124] and that of FGF10 paracrine signalling between the urogenital mesenchyme and prostatic epithelia for the formation of prostatic neoplasias [125,126,127]. FGF signalling has also been implicated in cell non-autonomous tumourigenesis in the liver, as virally-delivered FGF19 or skeletal muscle-derived FGF19 were shown to be required for the induction of hepatocellular carcinomas in mice [128, 129].
In skin models of cancer, it is known that certain oncogenic driver mutations require wounding-derived inflammatory signals to initiate tumourigenesis [130, 131]. Interestingly, lineage tracing in a transgenic skin papilloma model has shown that activating constitutive MAPK signalling in suprabasal epidermal cells, in addition to skin wounding, can induce IL1A-driven hyperproliferation of non-targeted epidermal basal cells, which then formed the bulk of the papilloma [132]. A similar strategy has also demonstrated the cell non-autonomous recruitment of untargeted cells in skin cancer models driven by the carcinogens 7,12-dimethylbenz(a)anthracene (DMBA) and 2-O-tetradecanoylphorbol-13-acetate (TPA) [133, 134]. Finally, the expression of oncogenic β-catenin in K19 + and Lgr5 + stem cells of the hair follicle has been shown to induce outgrowths and benign tumours which are mostly composed of untargeted cells as shown by lineage tracing [135,136,137]. Therefore, plenty of evidence exists supporting the notion that paracrine signalling can initiate tumour formation and fuel progression.
Evidence for senescence-induced tumorigenesis
There is ample in vitro and in vivo evidence demonstrating the tumour promoting activities of cellular senescence on cells carrying oncogenic mutations (see reviews cited above). On the other hand, the requirement of senescence and SASP signalling for the cell non-autonomous initiation of tumours (as it occurs in mouse ACP models) has been less explored, possibly due to the inherent difficulty in studying the early stages of cell transformation and tumour initiation in vivo.
Although a considerable number of in vitro experiments have shown that conditioned media from senescent cell cultures can enhance the cancerous properties of preneoplastic or neoplastic cells (see the reviews mentioned above), it has also been shown that some cells require exposure to senescent cell-conditioned media to acquire cancer stem cell-like phenotypes and the ability to form tumours when transplanted into nude mice [138,139,140]. In vivo, senescence and the SASP-driven paracrine tumourigenesis have been convincingly demonstrated to occur in several tissues of D. melanogaster. In this model, elegant use of gene targeting, genetic lineage tracing and cell ablation approaches have shown that activating oncogenic Ras induces senescence-like phenotypes and SASP activation in targeted cells, which lead to tumour growth in neighbouring wild type cells through the activation of developmental pathways and JNK signalling [141,142,143,144]. In a zebrafish model of melanoma, researchers showed that telomere shortening promotes the accumulation of senescent cells which produces an inflammatory environment that significantly fosters the formation of melanomas [145]. This, taken together with the fact that senescent cells accumulate during aging in several tissues and that their elimination in mice causes a significant delay in cancer deaths [146,147,148,149], implicates senescent cells in age-related tumourigenesis.
In mice, the injection of senescent HRasV12-expressing keratinocytes can induce the formation of skin papillomas that are very similar to those in DMBA/TPA models. Strikingly, a considerable portion of cells within the papillomas contain non-targeted host cells, suggesting an important role for senescence-driven paracrine signalling in tumour formation [150]. Notably, a recent study revealed that p16INK4A-expressing senescent keratinocytes and their SASP induce hyperproliferation and activation of WNT signalling target genes in neighbouring naïve cells, leading to the formation of papillomas when DMBA/TPA is applied [151]. These findings are remarkably similar to the previously discussed studies involving the paracrine recruitment of untargeted cells in skin tumours, thus raising interesting questions about the contributions of senescence and the SASP in those models [132,133,134,135,136,137].
There is also strong evidence suggesting that senescence and the SASP can drive paracrine tumourigenesis in the liver. For example, it has been shown that obese mice develop higher levels of the microbial metabolite deoxycholic acid (DCA) which leads to higher number of senescent hepatic stellate cells (HSCs) and increased SASP expression. Interestingly, depletion of either the senescent HSCs or knockout of IL1B signalling (a common SASP component) significantly prevents the growth of DMBA-induced hepatocellular carcinomas [152]. Moreover, other studies have shown the requirement of senescent fibroblasts and their SASP for the induction of liver tumours from transplanted cancer cell lines [39, 153]. In particular, it has been shown that the pro-tumourigenic effects of the SASP can be inhibited by knockdown of PTBP-1, a factor controlling the alternative splicing of genes involved in intracellular trafficking [39]. The tumour-initiating role of senescence has further been supported by a recent study showing that carcinogen-induced liver tumour formation requires the SASP from senescent HSCs and that it can be prevented by chemically ablating senescent cells [154].
The evidence discussed above suggests three conceivable mechanisms, acting alone or in combination, that could mediate senescence-mediated paracrine tumourigenesis: (1) induction of proliferation in neighbouring cells, therefore, promoting genomic instability and the appearance of novel mutations [155, 156]; (2) promotion of cell reprogramming and plasticity, which has already been shown to occur in the contexts of cancer, aging and repair during injury both in vivo and in vitro [21, 150, 157,158,159]; (3) activation of developmental pathways (such as WNT signalling) related to the induction, maintenance and survival of cancer stem cell phenotypes [160, 161]. Our findings from both humans and mouse models suggest all 3 mechanisms are involved in ACP pathogenesis. First, proliferating cells are often found in close proximity to the senescent clusters, while the expression of the stem cell factor SOX9 is increased in these neighbouring cells. Importantly, the presence of both proliferating and SOX9 + cells is diminished in the context of an attenuated SASP [42, 75]. Second, mouse ACP pituitaries contain more and faster growing pituitary progenitors/stem cells as determined by colony-forming assays [75]. Finally, the senescent clusters in both mouse and human possess a transcriptional profile that is analogous to the enamel knot, while their SASP contains many developmental signalling factors from the WNT, SHH, BMP and FGF families that are critical for tooth morphogenesis [67].
New therapeutic opportunities: senolytics and SASP-modulating drugs
The discovery that senescent cells, through the SASP, are determinant factors of age-related pathogenesis and drivers of organismal ageing has led to the identification and development of drugs able to counteract their damaging effects. Senotherapies aim mostly to either kill senescent cells selectively, by targeting key survival critical pathways, or to regulate their paracrine activities using SASP-modulating drugs. In a couple of milestone studies, Baker and colleagues have demonstrated that the genetic ablation of p16-expressing senescent cells is able to delay ageing-associated disorders [146, 162]. Subsequently, three independent studies have shown that senescent cells can specifically be killed using certain senolytic compounds, resulting in the rejuvenation of tissue stem cells [163,164,165]. These initial studies have been followed by more investigations targeting senescent cells in different disease contexts that have together provided solid evidence for a critical contribution of senescence in several age-related diseases [166,167,168,169,170]. Senolytic therapies have thoroughly been reviewed elsewhere [27, 171, 172]
The previously discussed cell autonomous and non-autonomous functions of senescent cells in tumourigenesis have provided a strong rationale to explore the potential of senotherapies to prevent or delay tumour formation. Senotherapies could improve anticancer treatments, as many currently used therapies may lead to the induction and accumulation of senescent cells in the tumour bed, which could be detrimental for the patients and result in recurrence. In support of this, it has been shown that genetic or chemical ablation of senescent cells that are induced after anti-cancer therapy significantly reduces tumour recurrence in breast and liver cancer models [25, 173].
In ACP models, initial experiments have shown that the genetic attenuation of the senescence/SASP response is able to reduce significantly the tumour-inducing potential of the senescent cell clusters [42]. In human ACP, the inhibition of IL6 (a SASP factor expressed in mouse and human ACP) results in reduced motility of ACP epithelial tumour cells, suggesting that SASP attenuation may reduce tumour burden in vivo [174]. A number of studies have shown that senescent cluster cells in the embryonic mouse ACP model can be ablated chemically using senolytic agents in vitro. Treatment of pre-tumoural mouse pituitaries with ABT-263 and ABT-737, two well-studied senolytics that target the anti-apoptotic proteins Bcl2, Bcl-xL and Bcl-w [164, 165], results in smaller clusters due to cell apoptosis in ex vivo explants cultures [42, 175]. Likewise, ouabain and digoxin, two members of the cardiac glycoside family of organic compounds, can induce apoptosis in senescent cluster cells [175]. In a recent report, a galactose-modified cytotoxic prodrug has been used to preferentially kill the cluster senescent cells in vitro based on the higher levels of β-galactosidase in these cells [176]. Therefore, mouse ACP models can potentially be used to study novel senolytic compounds in the context of senescence-mediated paracrine tumourigenesis in vivo. Preclinical studies are still required to establish proof-of-principle evidence that senotherapies may be relevant against craniopharyngioma and therefore mouse ACP models are ideal tools to test these novel treatments.
Conclusions
Early studies showing a negative correlation between senescent cell burden and tumour progression towards malignancy helped establish cellular senescence as a tumour-suppressive mechanism. However, when viewed under the precepts of widely accepted cell autonomous paradigms of tumorigenesis, this inverse senescence-cancer progression relationship implies that cells carrying oncogenic driver mutations have to either bypass or escape the senescent phenotype for tumours to progress. Although there are some convincing experiments in vitro [161, 177], conclusive evidence demonstrating the occurrence of either senescence bypass or escape has yet to be produced in vivo [178]. Of relevance, a genome-wide methylation analysis has shown that the methylation signature of transformed cells is acquired stochastically and independently of the senescent epigenetic state, which argues against the senescence escape hypothesis [179]. Likewise, although NOTCH1 is the most commonly mutated gene in the physiologically aged human oesophagus (followed by TP53), NOTCH1 mutations are considerably underrepresented in oesophageal cancers, suggesting these are more likely to evolve from epithelial cells without NOTCH1 mutations. In contrast, TP53 mutations, which are several-fold less frequent in the aged oesophagus, are almost universally present in oesophageal cancers, suggesting that these cancers originate from the small fraction of TP53 mutant cells [180, 181]. These findings also argue against senescence escape as an underlying mechanism of oesophageal cancer development, as it would be expected for mutations in NOTCH1 to be as abundant as TP53. More recently, the mechanisms of action of known carcinogens have been challenged. By analysing the mutational signatures of tumours generated in mice exposed to one of 20 carcinogens, it has been found that most of these agents are not directly mutagenic on the genome (i.e. they do not increase mutation burden in the tumours), with most mutations, including driver mutations, resulting from tissue-specific endogenous processes. This suggests that these carcinogens promote tumour initiation in a different manner, possibly by creating a permissive environment that allows tumour growth rather than, as initially believed, by increasing mutation rate [182]. Therefore, a combination of techniques such as genetic lineage-tracing, transplantation and sequencing strategies will be required to demonstrate that oncogene-targeted cells can escape or avoid the senescent phenotype in animal models of cancer.
The ACP models have revealed that the relationship between oncogenic driver-mutations, tumour cells-of-origin and the tumour microenvironment (TME) can be much more complex than what can be explained by conventional models of carcinogenesis. Lineage tracing has shown the otherwise counterintuitive observation that tumours can be formed from cells that are not related to the original oncogenic insult. Molecular studies in these models indicate that senescent cells bring about a pro-tumourigenic TME that induces tumour initiation in a paracrine manner through the SASP, while in human ACP, the evidence strongly supports a role of senescent cells in tumour growth and invasion. Further preclinical research using senolytics in the ACP mouse models may pave the way to clinical trials to evaluate the clinical relevance of senotherapies, alone or in combination, against human ACP.
It remains to be seen how common the occurrence of senescence-induced paracrine tumourigenesis in other tumours and cancers is, but it will be particularly interesting to assess the role of senescent cells and the SASP in the cancer models for which cell non-autonomous initiating mechanisms have already been described (Sect. 4.1 and Table 1). Evidence of the prevalence of such mechanisms in the origin or establishment of some human cancers will also have to be produced, which may be difficult due to the fact that early stages of tumourigenesis are hard to get hold of in humans. However, within the context of therapy-induced senescence, it may be easier to demonstrate the pro-tumourigenic effects of both senescent tumoural cells and senescent cells within the TME. Therefore, therapies capable of inducing senescent phenotypes such as radiotherapy, chemotherapy or even targeted approaches, in combination with senolytics or SASP-modulators [183], could significantly improve the current standard of care and result in better clinical outcomes.
References
Hayflick L, Moorhead PS (1961) The serial cultivation of human diploid cell strains. Exp Cell Res 25:585–621. https://doi.org/10.1016/0014-4827(61)90192-6
Hayflick L (1965) The limited in vitro lifetime of human diploid cell strains. Exp Cell Res 37:614–636. https://doi.org/10.1016/0014-4827(65)90211-9
Orgel LE (1963) The maintenance of the accuracy of protein synthesis and its relevance to ageing. Proc Natl Acad Sci 49:517–521. https://doi.org/10.1073/PNAS.49.4.517
Hernandez-Segura A, Nehme J, Demaria M (2018) Hallmarks of cellular senescence. Trends Cell Biol 28:436–453. https://doi.org/10.1016/j.tcb.2018.02.001
Gorgoulis V, Adams PD, Alimonti A et al (2019) Cellular senescence: defining a path forward. Cell 179:813–827. https://doi.org/10.1016/j.cell.2019.10.005
Storer M, Mas A, Robert-Moreno A et al (2013) Senescence is a developmental mechanism that contributes to embryonic growth and patterning. Cell 155:1119–1130. https://doi.org/10.1016/j.cell.2013.10.041
Muñoz-Espín D, Cañamero M, Maraver A et al (2013) Programmed cell senescence during mammalian embryonic development. Cell 155:1104. https://doi.org/10.1016/j.cell.2013.10.019
Davaapil H, Brockes JP, Yun MH (2017) Conserved and novel functions of programmed cellular senescence during vertebrate development. Development 144:106–114. https://doi.org/10.1242/dev.138222
Gibaja A, Aburto MR, Pulido S et al (2019) TGFβ2-induced senescence during early inner ear development. Sci Rep 9:1–13. https://doi.org/10.1038/s41598-019-42040-0
Feng T, Meng J, Kou S et al (2019) CCN1-induced cellular senescence promotes heart regeneration. Circulation 139:2495–2498. https://doi.org/10.1161/CIRCULATIONAHA.119.039530
Sarig R, Rimmer R, Bassat E et al (2019) Transient p53-mediated regenerative senescence in the injured heart. Circulation 139:2491–2494. https://doi.org/10.1161/CIRCULATIONAHA.119.040125
Da Silva-Álvarez S, Guerra-Varela J, Sobrido-Cameán D et al (2020) Cell senescence contributes to tissue regeneration in zebrafish. Aging Cell 19:1–5. https://doi.org/10.1111/acel.13052
Krizhanovsky V, Yon M, Dickins RA et al (2008) Senescence of activated stellate cells limits liver fibrosis. Cell 134:657–667. https://doi.org/10.1016/j.cell.2008.06.049
Chiche A, Le Roux I, von Joest M et al (2017) Injury-induced senescence enables in vivo reprogramming in skeletal muscle. Cell Stem Cell 20:407–414. https://doi.org/10.1016/j.stem.2016.11.020
Hickson LTJ, Langhi Prata LGP, Bobart SA et al (2019) Senolytics decrease senescent cells in humans: preliminary report from a clinical trial of Dasatinib plus Quercetin in individuals with diabetic kidney disease. EBioMedicine 47:446–456. https://doi.org/10.1016/j.ebiom.2019.08.069
Demaria M, Ohtani N, Youssef SAA et al (2014) An essential role for senescent cells in optimal wound healing through secretion of PDGF-AA. Dev Cell 31:722–733. https://doi.org/10.1016/j.devcel.2014.11.012
Lewis-McDougall FC, Ruchaya PJ, Domenjo-Vila E et al (2019) Aged-senescent cells contribute to impaired heart regeneration. Aging Cell 18:1–15. https://doi.org/10.1111/acel.12931
Yun MH, Davaapil H, Brockes JP (2015) Recurrent turnover of senescent cells during regeneration of a complex structure. Elife 4:e05505. https://doi.org/10.7554/eLife.05505
Wang H, Wang Z, Huang Y et al (2020) Senolytics (DQ) mitigates radiation ulcers by removing senescent cells. Front Oncol 9:1–13. https://doi.org/10.3389/fonc.2019.01576
Wang Z, Chen Z, Jiang Z et al (2019) Cordycepin prevents radiation ulcer by inhibiting cell senescence via NRF2 and AMPK in rodents. Nat Commun. https://doi.org/10.1038/s41467-019-10386-8
Rhinn M, Ritschka B, Keyes WM (2019) Cellular senescence in development, regeneration and disease. Developments. https://doi.org/10.1242/dev.151837
Walters HE, Yun MH (2020) Rising from the ashes: cellular senescence in regeneration. Curr Opin Genet Dev 64:94–100. https://doi.org/10.1016/j.gde.2020.06.002
Ogrodnik M, Zhu Y, Langhi LGP et al (2019) Obesity-induced cellular senescence drives anxiety and impairs neurogenesis. Cell Metab 29:1061–1077. https://doi.org/10.1016/j.cmet.2018.12.008
Palmer AK, Xu M, Zhu Y et al (2019) Targeting senescent cells alleviates obesity-induced metabolic dysfunction. Aging Cell 18:1–15. https://doi.org/10.1111/acel.12950
Demaria M, O’Leary MN, Chang J et al (2017) Cellular senescence promotes adverse effects of chemotherapy and cancer relapse. Cancer Discov 7:165–176. https://doi.org/10.1158/2159-8290.CD-16-0241
Wiley CD, Liu S, Limbad C et al (2019) SILAC analysis reveals increased secretion of hemostasis-related factors by senescent cells. Cell Rep 28:3329–3337. https://doi.org/10.1016/j.celrep.2019.08.049
Childs BG, Gluscevic M, Baker DJ et al (2017) Senescent cells: an emerging target for diseases of ageing. Nat Rev Drug Discov 16:718–735. https://doi.org/10.1038/nrd.2017.116
He S, Sharpless NE (2017) Senescence in health and disease. Cell 169:1000–1011. https://doi.org/10.1016/j.cell.2017.05.015
Muñoz-Espín D, Serrano M, Munoz-Espin D, Serrano M (2014) Cellular senescence: from physiology to pathology. Nat Rev Mol Cell Biol 15:482–496. https://doi.org/10.1038/nrm3823
Calcinotto A, Kohli J, Zagato E et al (2019) Cellular senescence: aging, cancer, and injury. Physiol Rev 99:1047–1078. https://doi.org/10.1152/physrev.00020.2018
Brenner E, Schörg BF, Ahmetlić F et al (2020) Cancer immune control needs senescence induction by interferon-dependent cell cycle regulator pathways in tumours. Nat Commun. https://doi.org/10.1038/s41467-020-14987-6
Prieur A, Peeper DS (2008) Cellular senescence in vivo: a barrier to tumorigenesis. Curr Opin Cell Biol 20:150–155. https://doi.org/10.1016/j.ceb.2008.01.007
Collado M, Serrano M (2010) Senescence in tumours: evidence from mice and humans. Nat Rev Cancer 10:51–57. https://doi.org/10.1038/nrc2772
Bavik C, Coleman I, Dean JP et al (2006) The gene expression program of prostate fibroblast senescence modulates neoplastic epithelial cell proliferation through paracrine mechanisms. Cancer Res 66:794–802. https://doi.org/10.1158/0008-5472.CAN-05-1716
Guo Y, Ayers JL, Carter KT et al (2019) Senescence-associated tissue microenvironment promotes colon cancer formation through the secretory factor GDF15. Aging Cell 18:1–12. https://doi.org/10.1111/acel.13013
Hwang HJ, Lee Y-R, Kang D et al (2020) Endothelial cells under therapy-induced senescence secrete CXCL11, which increases aggressiveness of breast cancer cells. Cancer Lett. https://doi.org/10.1016/j.canlet.2020.06.019
Han L, Long Q, Li S et al (2020) Senescent stromal cells promote cancer resistance through SIRT1 loss-potentiated overproduction of small extracellular vesicles. Cancer Res. https://doi.org/10.1158/0008-5472.CAN-20-0506
Farsam V, Basu A, Gatzka M et al (2016) Senescent fibroblast-derived chemerin promotes squamous cell carcinoma migration. Oncotarget 7:83554–83569. https://doi.org/10.18632/oncotarget.13446
Georgilis A, Klotz S, Hanley CJ et al (2018) PTBP1-mediated alternative splicing regulates the inflammatory secretome and the pro-tumorigenic effects of senescent cells. Cancer Cell 34:85–102. https://doi.org/10.1016/j.ccell.2018.06.007
Kim YH, Choi YW, Lee J et al (2017) Senescent tumor cells lead the collective invasion in thyroid cancer. Nat Commun 8:15208. https://doi.org/10.1038/ncomms15208
Alimirah F, Pulido T, Valdovinos A et al (2020) Cellular senescence promotes skin carcinogenesis through p38MAPK and p44/p42MAPK signaling. Cancer Res. https://doi.org/10.1158/0008-5472.CAN-20-0108
Gonzalez-Meljem JM, Haston S, Carreno G et al (2017) Stem cell senescence drives age-attenuated induction of pituitary tumours in mouse models of paediatric craniopharyngioma. Nat Commun 8:1819. https://doi.org/10.1038/s41467-017-01992-5
Andoniadou CL, Matsushima D, Mousavy-gharavy SN et al (2013) The Sox2 + population of the adult murine pituitary includes progenitor/stem cells with tumour-inducing potential. Cell Stem Cell 13:433
Gonzalez-Meljem JM, Apps JR, Fraser HC, Martinez-Barbera JP (2018) Paracrine roles of cellular senescence in promoting tumourigenesis. Br J Cancer. https://doi.org/10.1038/s41416-018-0066-1
Coppé J-P, Desprez P-Y, Krtolica A, Campisi J (2010) The senescence-associated secretory phenotype: the dark side of tumor suppression. Annu Rev Pathol Mech Dis 5:99–118. https://doi.org/10.1146/annurev-pathol-121808-102144
Faget DV, Ren Q, Stewart SA (2019) Unmasking senescence: context-dependent effects of SASP in cancer. Nat Rev Cancer 19:439–453
Visvader JE, Lindeman GJ (2012) Cancer stem cells: current status and evolving complexities. Cell Stem Cell 10:717–728. https://doi.org/10.1016/j.stem.2012.05.007
Kreso A, Dick JE (2014) Evolution of the cancer stem cell model. Cell Stem Cell 14:275–291. https://doi.org/10.1016/j.stem.2014.02.006
Song Y, Wang Y, Tong C et al (2017) A unified model of the hierarchical and stochastic theories of gastric cancer. Br J Cancer 116:973–989. https://doi.org/10.1038/bjc.2017.54
Bunin GR, Surawicz TS, Witman PA et al (1998) The descriptive epidemiology of craniopharyngioma. J Neurosurg 89:547–551. https://doi.org/10.3171/jns.1998.89.4.0547
Zacharia BE, Bruce SS, Goldstein H et al (2012) Incidence, treatment and survival of patients with craniopharyngioma in the surveillance, epidemiology and end results program. Neuro Oncol. https://doi.org/10.1093/neuonc/nos142
Martinez-Barbera JP, Andoniadou CL (2020) Biological behaviour of craniopharyngiomas. Neuroendocrinology. https://doi.org/10.1159/000506904
Müller HL, Merchant TE, Puget S, Martinez-Barbera J-PP (2017) New outlook on the diagnosis, treatment and follow-up of childhood-onset craniopharyngioma. Nat Rev Endocrinol 13:299–312. https://doi.org/10.1038/nrendo.2016.217
Hoffmann A, Warmth-Metz M, Gebhardt U, Pietsch T, Pohl F, Kortmann R-D, Calaminus G, Müller H (2014) Childhood Craniopharyngioma – Changes of Treatment Strategies in the Trials KRANIOPHARYNGEOM 2000/2007. Klin Pädiatrie 226(03):161–168. https://doi.org/10.1055/s-0034-1368785
Prieto R, Rosdolsky M, Hofecker V et al (2020) Craniopharyngioma treatment: an updated summary of important clinicopathological concepts. Expert Rev Endocrinol Metab. https://doi.org/10.1080/17446651.2020.1770081
Hengartner AC, Prince E, Vijmasi T, Hankinson TC (2020) Adamantinomatous craniopharyngioma: moving toward targeted therapies. Neurosurg Focus. https://doi.org/10.3171/2019.10.FOCUS19705
Zuccaro G (2016) 44th Annual Meeting of International Society for Pediatric Neurosurgery, Kobe, Japan, Oct 23–27, 2016. Child’s Nerv Syst 32:1957–2040. https://doi.org/10.1007/s00381-016-3209-9
Müller HL, Merchant TE, Warmuth-Metz M et al (2019) Craniopharyngioma. Nat Rev Dis Prim 5:75. https://doi.org/10.1038/s41572-019-0125-9
Karavitaki N, Cudlip S, Adams CBT, Wass JAH (2006) Craniopharyngiomas. Endocr Rev 27(4):371–397. https://doi.org/10.1210/er.2006-0002
Erfurth E-M (2020) Diagnosis, background and treatment of hypothalamic damage in craniopharyngioma. Neuroendocrinology. https://doi.org/10.1159/000509616
Özyurt J, Müller HL, Thiel CM (2015) A systematic review of cognitive performance in patients with childhood craniopharyngioma. J Neurooncol 125(1):9–21. https://doi.org/10.1007/s11060-015-1885-z
Wijnen M, Olsson DS, van den Heuvel-Eibrink MM et al (2018) Excess morbidity and mortality in patients with craniopharyngioma: a hospital-based retrospective cohort study. Eur J Endocrinol 178:95–104. https://doi.org/10.1530/EJE-17-0707
Larkin SJ, Ansorge O (2013) Pathology and pathogenesis of craniopharyngiomas. Pituitary 16:9
Martinez-Barbera JP, Buslei R (2015) Adamantinomatous craniopharyngioma: pathology, molecular genetics and mouse models. J Pediatr Endocrinol Metab. https://doi.org/10.1515/jpem-2014-0442
Johnson LN, Hepler RS, Yee RD et al (1986) Magnetic resonance imaging of craniopharyngioma. Am J Ophthalmol 102:242–244. https://doi.org/10.1016/0002-9394(86)90152-2
Warmuth-Metz M (2016) Imaging and diagnosis in pediatric brain tumor studies
Apps JR, Carreno G, Gonzalez-Meljem JM et al (2018) Tumour compartment transcriptomics demonstrates the activation of inflammatory and odontogenic programmes in human adamantinomatous craniopharyngioma and identifies the MAPK/ERK pathway as a novel therapeutic target. Acta Neuropathol 135:757–777. https://doi.org/10.1007/s00401-018-1830-2
Sekine S, Shibata T, Kokubu A et al (2002) Craniopharyngiomas of adamantinomatous type harbor β-catenin gene mutations. Am J Pathol 161:1997–2001. https://doi.org/10.1016/S0002-9440(10)64477-X
Buslei R, Nolde M, Hofmann B et al (2005) Common mutations of beta-catenin in adamantinomatous craniopharyngiomas but not in other tumours originating from the sellar region. Acta Neuropathol 109:589–597. https://doi.org/10.1007/s00401-005-1004-x
Clevers H, Nusse R (2012) Wnt/β-catenin signaling and disease. Cell 149:1192–1205. https://doi.org/10.1016/j.cell.2012.05.012
Buslei R, Hölsken A, Hofmann B et al (2007) Nuclear beta-catenin accumulation associates with epithelial morphogenesis in craniopharyngiomas. Acta Neuropathol 113:585–590. https://doi.org/10.1007/s00401-006-0184-3
Martinez-Barbera JP (2015) Molecular and cellular pathogenesis of adamantinomatous craniopharyngioma. Neuropathol Appl Neurobiol 41:721–732. https://doi.org/10.1111/nan.12226
Apps JR, Stache C, Gonzalez-Meljem JM et al (2020) CTNNB1 mutations are clonal in adamantinomatous craniopharyngioma. Neuropathol Appl Neurobiol. https://doi.org/10.1111/nan.12613
Apps JR, Hutchinson JC, Arthurs OJ et al (2016) Imaging invasion: micro-CT imaging of adamantinomatous craniopharyngioma highlights cell type specific spatial relationships of tissue invasion. Acta Neuropathol Commun 4:57. https://doi.org/10.1186/s40478-016-0321-8
Gaston-Massuet C, Andoniadou CL, Signore M et al (2011) Increased wingless (Wnt) signaling in pituitary progenitor/stem cells gives rise to pituitary tumors in mice and humans. Proc Natl Acad Sci USA 108:11482–11487. https://doi.org/10.1073/pnas.1101553108
Kato K, Nakatani Y, Kanno H et al (2004) Possible linkage between specific histological structures and aberrant reactivation of the Wnt pathway in adamantinomatous craniopharyngioma. J Pathol 203:814–821. https://doi.org/10.1002/path.1562
Tateyama H, Tada T, Okabe M et al (2001) Different keratin profiles in craniopharyngioma subtypes and ameloblastomas. Pathol Res Pract 197:735–742. https://doi.org/10.1078/0344-0338-00152
Apps JR, Martinez-Barbera JP (2017) Genetically engineered mouse models of craniopharyngioma: an opportunity for therapy development and understanding of tumor biology. Brain Pathol 27:364–369. https://doi.org/10.1111/bpa.12501
Jayakody SA, Andoniadou CL, Gaston-Massuet C et al (2012) SOX2 regulates the hypothalamic-pituitary axis at multiple levels. J Clin Investig 122:3635–3646. https://doi.org/10.1172/JCI64311
Carreno G, Boult JK, Apps J et al (2019) SHH pathway inhibition is protumourigenic in adamantinomatous craniopharyngioma. Endocr Relat Cancer 26:355–366. https://doi.org/10.1530/ERC-18-0538
Boult JKR, Apps JR, Hölsken A et al (2017) Preclinical transgenic and patient-derived xenograft models recapitulate the radiological features of human adamantinomatous craniopharyngioma. Brain Pathol 28:475–483. https://doi.org/10.1111/bpa.12525
Jurkiewicz E, Bekiesińska-Figatowska M, Duczkowski M et al (2010) Antenatal diagnosis of the congenital craniopharyngioma. Pol J Radiol 75:98–102
Scagliotti V, Avagliano L, Gualtieri A et al (2016) Histopathology and molecular characterisation of intrauterine-diagnosed congenital craniopharyngioma. Pituitary 19:50–56. https://doi.org/10.1007/s11102-015-0682-1
Kostadinov S, Hanley CL, Lertsburapa T et al (2014) Fetal craniopharyngioma: management, postmortem diagnosis, and literature review of an intracranial tumor detected in utero. Pediatr Dev Pathol 17:409–412. https://doi.org/10.2350/14-06-1506-CR.1
Isaacs H (2009) Fetal brain tumors: a review of 154 cases. Am J Perinatol 26:453–466. https://doi.org/10.1055/s-0029-1214245
Kretzschmar K, Watt FM (2012) Lineage tracing. Cell 148:33–45. https://doi.org/10.1016/j.cell.2012.01.002
Zhu L, Gibson P, Currle DS et al (2009) Prominin 1 marks intestinal stem cells that are susceptible to neoplastic transformation. Nature. https://doi.org/10.1038/nature07589
Barker N, Ridgway RA, Van Es JH et al (2009) Crypt stem cells as the cells-of-origin of intestinal cancer. Nature 457:608–611. https://doi.org/10.1038/nature07602
Lodge EJ, Santambrogio A, Russell JP et al (2019) Homeostatic and tumourigenic activity of SOX2+ pituitary stem cells is controlled by the LATS/YAP/TAZ cascade. Elife. https://doi.org/10.7554/eLife.43996
Andoniadou CL, Gaston-Massuet C, Reddy R et al (2012) Identification of novel pathways involved in the pathogenesis of human adamantinomatous craniopharyngioma. Acta Neuropathol 124:259–271. https://doi.org/10.1007/s00401-012-0957-9
Russell JP, Lim X, Santambrogio A et al (2020) Pituitary stem cells produce paracrine WNT signals to control the expansion of their descendant progenitor cells. bioRxiv. https://doi.org/10.1101/2020.05.22.107045
Sabatino ME, Petiti JP, del Valle Sosa JP et al (2015) Evidence of cellular senescence during the development of estrogen-induced pituitary tumors. Endocr Relat Cancer 22:299–317. https://doi.org/10.1530/ERC-14-0333
Chesnokova V, Zonis S, Kovacs K et al (2008) p21Cip1 restrains pituitary tumor growth. Proc Natl Acad Sci USA 105:17498–17503. https://doi.org/10.1073/pnas.0804810105
Mongi-Bragato B, Grondona E, del Sosa VL et al (2020) Pivotal role of NF-κB in cellular senescence of experimental pituitary tumours. J Endocrinol 245:179–191. https://doi.org/10.1530/joe-19-0506
Chesnokova V, Zonis S, Rubinek T et al (2007) Senescence mediates pituitary hypoplasia and restrains pituitary tumor growth. Cancer Res 67:10564–10572. https://doi.org/10.1158/0008-5472.CAN-07-0974
Manojlovic-Gacic E, Skender-Gazibara M, Popovic V et al (2016) Oncogene-induced senescence in pituitary adenomas—an immunohistochemical study. Endocr Pathol 27:1–11. https://doi.org/10.1007/s12022-015-9405-4
Alexandraki KI, Munayem Khan M, Chahal HS et al (2012) Oncogene-induced senescence in pituitary adenomas and carcinomas. Hormones (Athens) 11:297–307
Ben-Shlomo A, Deng N, Ding E et al (2020) DNA damage and growth hormone hypersecretion in pituitary somatotroph adenomas. J Clin Investig. https://doi.org/10.1172/jci138540
Chesnokova V, Zhou C, Ben-Shlomo A et al (2013) Growth hormone is a cellular senescence target in pituitary and nonpituitary cells. Proc Natl Acad Sci USA 110:3331–3339. https://doi.org/10.1073/pnas.1310589110
Chesnokova V, Melmed S (2020) Peptide hormone regulation of DNA damage responses. Endocr Rev 41:519–537. https://doi.org/10.1210/endrev/bnaa009
Sapochnik M, Haedo MR, Fuertes M et al (2017) Autocrine IL-6 mediates pituitary tumor senescence. Oncotarget 8:4690–4702. https://doi.org/10.18632/oncotarget.13577
Sapochnik M, Fuertes M, Arzt E (2017) Programmed cell senescence: role of IL-6 in the pituitary. J Mol Endocrinol 58:R241–R253. https://doi.org/10.1530/JME-17-0026
Rogelj S, Weinberg RA, Fanning P, Klagsbrun M (1988) Basic fibroblast growth factor fused to a signal peptide transforms cells. Nature 331:173–175. https://doi.org/10.1038/331173a0
Jue SF, Bradley RS, Rudnicki JA et al (1992) The mouse Wnt-1 gene can act via a paracrine mechanism in transformation of mammary epithelial cells. Mol Cell Biol 12:321–328. https://doi.org/10.1128/mcb.12.1.321
Mason JO, Kitajewski J, Varmus HE (1992) Mutational analysis of mouse wnt-1 identifies two temperature-sensitive alleles and attributes of Wnt-1 protein essential for transformation of a mammary cell line. Mol Biol Cell 3:521–533. https://doi.org/10.1091/mbc.3.5.521
Christiansen JH, Monkley SJ, Wainwright BJ (1996) Murine WNT11 is a secreted glycoprotein that morphologically transforms mammary epithelial cells. Oncogene
Shimizu H, Julius MA, Giarré M et al (1997) Transformation by wnt family proteins correlates with regulation of β-catenin. Cell Growth Differ 8:1349–1358
Forough R, Zhan X, MacPhee M et al (1993) Differential transforming abilities of non-secreted and secreted forms of human fibroblast growth factor-1. J Biol Chem 268:2960–2968
Li Y, Basilico C, Mansukhani A (1994) Cell transformation by fibroblast growth factors can be suppressed by truncated fibroblast growth factor receptors. Mol Cell Biol 14:7660–7669. https://doi.org/10.1128/mcb.14.11.7660
Anastas JN, Moon RT (2013) WNT signalling pathways as therapeutic targets in cancer. Nat Rev Cancer 13:11–26. https://doi.org/10.1038/nrc3419
Klaus A, Birchmeier W (2008) Wnt signalling and its impact on development and cancer. Nat Rev Cancer 8:387–398. https://doi.org/10.1038/nrc2389
Babina IS, Turner NC (2017) Advances and challenges in targeting FGFR signalling in cancer. Nat Rev Cancer 17:318–332. https://doi.org/10.1038/nrc.2017.8
Turner N, Grose R (2010) Fibroblast growth factor signalling: from development to cancer. Nat Rev Cancer 10:116–129. https://doi.org/10.1038/nrc2780
Schepers AG, Snippert HJ, Stange DE et al (2012) Lineage tracing reveals Lgr5+ stem cell activity in mouse intestinal adenomas. Science (80-) 337:730–735. https://doi.org/10.1126/science.1224676
Rycaj K, Tang DG (2015) Cell-of-origin of cancer versus cancer stem cells: assays and interpretations. Cancer Res 75:4003–4011. https://doi.org/10.1158/0008-5472.CAN-15-0798
Batlle E, Clevers H (2017) Cancer stem cells revisited. Nat Med 23:1124–1134. https://doi.org/10.1038/nm.4409
Raaijmakers MHGP, Mukherjee S, Guo S et al (2010) Bone progenitor dysfunction induces myelodysplasia and secondary leukaemia. Nature 464:852–857. https://doi.org/10.1038/nature08851
Kode A, Manavalan JS, Mosialou I et al (2014) Leukaemogenesis induced by an activating β-catenin mutation in osteoblasts. Nature 506:240–244. https://doi.org/10.1038/nature12883
Kuperwasser C, Chavarria T, Wu M et al (2004) Reconstruction of functionally normal and malignant human breast tissues in mice. Proc Natl Acad Sci USA 101:4966–4971. https://doi.org/10.1073/pnas.0401064101
Maffini MV, Soto AM, Calabro JM et al (2004) The stroma as a crucial target in rat mammary gland carcinogenesis. J Cell Sci 117:1495–1502. https://doi.org/10.1242/jcs.01000
Fomchenko EI, Dougherty JD, Helmy KY et al (2011) Recruited cells can become transformed and overtake PDGF-induced murine gliomas in vivo during tumor progression. PLoS ONE 6:e20605. https://doi.org/10.1371/journal.pone.0020605
Chen Y, Wang ZZ, Dai X et al (2015) Glioma initiating cells contribute to malignant transformation of host glial cells during tumor tissue remodeling via PDGF signaling. Cancer Lett 365:174–181. https://doi.org/10.1016/j.canlet.2015.05.026
Thliveris AT, Schwefel B, Clipson L et al (2013) Transformation of epithelial cells through recruitment leads to polyclonal intestinal tumors. Proc Natl Acad Sci USA 110:11523–11528. https://doi.org/10.1073/pnas.1303064110
Oshima H, Ishikawa T, Yoshida GJ et al (2014) TNF-α/TNFR1 signaling promotes gastric tumorigenesis through induction of Noxo1 and Gna14 in tumor cells. Oncogene 33:3820–3829. https://doi.org/10.1038/onc.2013.356
Memarzadeh S, Xin L, Mulholland DJ et al (2007) Enhanced paracrine FGF10 expression promotes formation of multifocal prostate adenocarcinoma and an increase in epithelial androgen receptor. Cancer Cell 12:572–585. https://doi.org/10.1016/j.ccr.2007.11.002
Memarzadeh S, Cai H, Janzen DM et al (2011) Role of autonomous androgen receptor signaling in prostate cancer initiation is dichotomous and depends on the oncogenic signal. Proc Natl Acad Sci USA 108:7962–7967. https://doi.org/10.1073/pnas.1105243108
Li Q, Ingram L, Kim S et al (2018) Paracrine fibroblast growth factor initiates oncogenic synergy with epithelial FGFR/Src transformation in prostate tumor progression. Neoplasia (United States) 20:233–243. https://doi.org/10.1016/j.neo.2018.01.006
Nicholes K, Guillet S, Tomlinson E et al (2002) A mouse model of hepatocellular carcinoma: ectopic expression of fibroblast growth factor 19 in skeletal muscle of transgenic mice. Am J Pathol 160:2295–2307. https://doi.org/10.1016/S0002-9440(10)61177-7
Zhou M, Yang H, Learned RM et al (2017) Non-cell-autonomous activation of IL-6/STAT3 signaling mediates FGF19-driven hepatocarcinogenesis. Nat Commun 8:1–16. https://doi.org/10.1038/ncomms15433
Arwert EN, Hoste E, Watt FM (2012) Epithelial stem cells, wound healing and cancer. Nat Rev Cancer 12:170–180. https://doi.org/10.1038/nrc3217
Burclaff J, Mills JC (2018) Plasticity of differentiated cells in wound repair and tumorigenesis, part II: skin and intestine. DMM Dis Model Mech. https://doi.org/10.1242/dmm.035071
Arwert EN, Lal R, Quist S et al (2010) Tumor formation initiated by nondividing epidermal cells via an inflammatory infiltrate. Proc Natl Acad Sci USA 107:19903–19908. https://doi.org/10.1073/pnas.1007404107
Demehri S, Turkoz A, Kopan R (2009) Epidermal Notch1 loss promotes skin tumorigenesis by impacting the stromal microenvironment. Cancer Cell 16:55–66. https://doi.org/10.1016/j.ccr.2009.05.016
Reeves MQ, Kandyba E, Harris S et al (2018) Multicolour lineage tracing reveals clonal dynamics of squamous carcinoma evolution from initiation to metastasis. Nat Cell Biol 20:699–709. https://doi.org/10.1038/s41556-018-0109-0
Brown S, Pineda CM, Xin T et al (2017) Correction of aberrant growth preserves tissue homeostasis. Nature 548:334–337. https://doi.org/10.1038/nature23304
Kretzschmar K, Weber C, Driskell RR et al (2016) Compartmentalized epidermal activation of β-catenin differentially affects lineage reprogramming and underlies tumor heterogeneity. Cell Rep 14:269–281. https://doi.org/10.1016/j.celrep.2015.12.041
Deschene ER, Myung P, Rompolas P et al (2014) β-Catenin activation regulates tissue growth non-cell autonomously in the hair stem cell niche. Science (80-) 343:1353–1356. https://doi.org/10.1126/science.1248373
Cahu J, Bustany S, Sola B (2012) Senescence-associated secretory phenotype favors the emergence of cancer stem-like cells. Cell Death Dis 3:e446. https://doi.org/10.1038/cddis.2012.183
Ohanna M, Giulano S, Bonet C et al (2011) Senescent cells develop a PARP-1 and nuclear factor-kB-associated secretome (PNAS). Genes Dev 25:1245–1261
Ohanna M, Cheli Y, Bonet C et al (2013) Secretome from senescent melanoma engages the STAT3 pathway to favor reprogramming of naive melanoma towards a tumor-initiating cell phenotype. Oncotarget 4:2212–2224. https://doi.org/10.18632/oncotarget.1143
Uhlirova M, Jasper H, Bohmann D (2005) Non-cell-autonomous induction of tissue overgrowth by JNK/Ras cooperation in a Drosophila tumor model. Proc Natl Acad Sci USA 102:13123–13128. https://doi.org/10.1073/pnas.0504170102
Cosolo A, Jaiswal J, Csordás G et al (2019) JNK-dependent cell cycle stalling in G2 promotes survival and senescence-like phenotypes in tissue stress. Elife. https://doi.org/10.7554/eLife.41036
Ohsawa S, Sato Y, Enomoto M et al (2012) Mitochondrial defect drives non-autonomous tumour progression through Hippo signalling in Drosophila. Nature 490:547–551. https://doi.org/10.1038/nature11452
Nakamura M, Ohsawa S, Igaki T (2014) Mitochondrial defects trigger proliferation of neighbouring cells via a senescence-associated secretory phenotype in Drosophila. Nat Commun 5:1–11. https://doi.org/10.1038/ncomms6264
Lex K, Maia Gil M, Lopes-Bastos B et al (2020) Telomere shortening produces an inflammatory environment that increases tumor incidence in zebrafish. Proc Natl Acad Sci. https://doi.org/10.1073/pnas.1920049117
Baker DJ, Childs BG, Durik M et al (2016) Naturally occurring p16 Ink4a-positive cells shorten healthy lifespan. Nature 530:184–189. https://doi.org/10.1038/nature16932
Dimri GP, Lee X, Basile G et al (1995) A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc Natl Acad Sci 92:9363–9367. https://doi.org/10.1073/pnas.92.20.9363
Herbig U, Ferreira M, Condel L et al (2006) Cellular senescence in aging primates. Science (80-) 311:1–2. https://doi.org/10.1126/science.1122446
Wang C, Jurk D, Maddick M et al (2009) DNA damage response and cellular senescence in tissues of aging mice.pdf. Aging Cell 8:311–323. https://doi.org/10.1111/j.1474-9726.2009.00481.x
Ritschka B, Storer M, Mas A et al (2017) The senescence-associated secretory phenotype induces cellular plasticity and tissue regeneration. Genes Dev 31:172–183. https://doi.org/10.1101/gad.290635.116
Azazmeh N, Assouline B, Winter E et al (2020) Chronic expression of p16INK4a in the epidermis induces Wnt-mediated hyperplasia and promotes tumor initiation. Nat Commun 11:1–13. https://doi.org/10.1038/s41467-020-16475-3
Yoshimoto S, Loo TM, Atarashi K et al (2013) Obesity-induced gut microbial metabolite promotes liver cancer through senescence secretome. Nature 499:97–101. https://doi.org/10.1038/nature12347
Herranz N, Gallage S, Mellone M et al (2015) mTOR regulates MAPKAPK2 translation to control the senescence-associated secretory phenotype. Nat Cell Biol 17:1205–1217. https://doi.org/10.1038/ncb3225
Li F, Huangyang P, Burrows M et al (2020) FBP1 loss disrupts liver metabolism and promotes tumorigenesis through a hepatic stellate cell senescence secretome. Nat Cell Biol 22:728–739. https://doi.org/10.1038/s41556-020-0511-2
Löbrich M, Jeggo PA (2007) The impact of a negligent G2/M checkpoint on genomic instability and cancer induction. Nat Rev Cancer 7:861–869. https://doi.org/10.1038/nrc2248
Gaillard H, García-Muse T, Aguilera A (2015) Replication stress and cancer. Nat Rev Cancer 15:276–289. https://doi.org/10.1038/nrc3916
Mosteiro L, Pantoja C, Alcazar N et al (2016) Tissue damage and senescence provide critical signals for cellular reprogramming in vivo. Science (80-). https://doi.org/10.1126/science.aaf4445
Ferreirós A, Pedrosa P, Da Silva-Álvarez S et al (2019) Context-dependent impact of RAS oncogene expression on cellular reprogramming to pluripotency. Stem Cell Rep 12:1099–1112. https://doi.org/10.1016/j.stemcr.2019.04.006
Vernot JP (2020) Senescence-associated pro-inflammatory cytokines and tumor cell plasticity. Front Mol Biosci 7:1–15. https://doi.org/10.3389/fmolb.2020.00063
Milanovic M, Yu Y, Schmitt CA (2018) The senescence-stemness alliance—a cancer—hijacked regeneration principle. Trends Cell Biol 28:1049–1061
Milanovic M, Fan DNY, Belenki D et al (2018) Senescence-associated reprogramming promotes cancer stemness. Nature 553:96–100. https://doi.org/10.1038/nature25167
Baker DJ, Wijshake T, Tchkonia T et al (2011) Clearance of p16 Ink4a-positive senescent cells delays ageing-associated disorders. Nature 479:232–236. https://doi.org/10.1038/nature10600
Zhu Y, Tchkonia T, Fuhrmann-Stroissnigg H et al (2016) Identification of a novel senolytic agent, navitoclax, targeting the Bcl-2 family of anti-apoptotic factors. Aging Cell 15:428–435. https://doi.org/10.1111/acel.12445
Yosef R, Pilpel N, Tokarsky-Amiel R et al (2016) Directed elimination of senescent cells by inhibition of BCL-W and BCL-XL. Nat Commun 7:11190. https://doi.org/10.1038/ncomms11190
Chang J, Wang Y, Shao L et al (2016) Clearance of senescent cells by ABT263 rejuvenates aged hematopoietic stem cells in mice. Nat Med 22:78–83. https://doi.org/10.1038/nm.4010
Roos CM, Zhang B, Palmer AK et al (2016) Chronic senolytic treatment alleviates established vasomotor dysfunction in aged or atherosclerotic mice. Aging Cell 15:973–977. https://doi.org/10.1111/acel.12458
Xu M, Pirtskhalava T, Farr JN et al (2018) Senolytics improve physical function and increase lifespan in old age. Nat Med 24:1246–1256. https://doi.org/10.1038/s41591-018-0092-9
Amor C, Feucht J, Leibold J et al (2020) Senolytic CAR T cells reverse senescence-associated pathologies. Nature. https://doi.org/10.1038/s41586-020-2403-9
Farr JN, Xu M, Weivoda MM et al (2017) Targeting cellular senescence prevents age-related bone loss in mice. Nat Med 23:1072–1079. https://doi.org/10.1038/nm.4385
Zhang P, Kishimoto Y, Grammatikakis I et al (2019) Senolytic therapy alleviates Aβ-associated oligodendrocyte progenitor cell senescence and cognitive deficits in an Alzheimer’s disease model. Nat Neurosci. https://doi.org/10.1038/s41593-019-0372-9
Paez-Ribes M, González-Gualda E, Doherty GJ, Muñoz-Espín D (2019) Targeting senescent cells in translational medicine. EMBO Mol Med 11:1–19. https://doi.org/10.15252/emmm.201810234
Song S, Lam EWF, Tchkonia T et al (2020) Senescent cells: emerging targets for human aging and age-related diseases. Trends Biochem Sci 45:578–592. https://doi.org/10.1016/j.tibs.2020.03.008
Wang C, Vegna S, Jin H et al (2019) Inducing and exploiting vulnerabilities for the treatment of liver cancer. Nature. https://doi.org/10.1038/s41586-019-1607-3
Zhou J, Zhang C, Pan J et al (2017) Interleukin-6 induces an epithelial–mesenchymal transition phenotype in human adamantinomatous craniopharyngioma cells and promotes tumor cell migration. Mol Med Rep. https://doi.org/10.3892/mmr.2017.6538
Guerrero A, Herranz N, Sun B et al (2019) Cardiac glycosides are broad-spectrum senolytics. Nat Metab. https://doi.org/10.1038/s42255-019-0122-z
Guerrero A, Guiho R, Herranz N et al (2020) Galactose-modified duocarmycin prodrugs as senolytics. Aging Cell. https://doi.org/10.1111/acel.13133
Saleh T, Tyutyunyk-Massey L, Gewirtz DA (2019) Tumor cell escape from therapy-induced senescence as a model of disease recurrence after dormancy. Cancer Res. https://doi.org/10.1158/0008-5472.CAN-18-3437
Lee S, Schmitt CA (2019) The dynamic nature of senescence in cancer. Nat Cell Biol 21:94–101. https://doi.org/10.1038/s41556-018-0249-2
Xie W, Kagiampakis I, Pan L et al (2018) DNA methylation patterns separate senescence from transformation potential and indicate cancer risk. Cancer Cell 33:309–321. https://doi.org/10.1016/j.ccell.2018.01.008
Yokoyama A, Kakiuchi N, Yoshizato T et al (2019) Age-related remodelling of oesophageal epithelia by mutated cancer drivers. Nature 565:312–317. https://doi.org/10.1038/s41586-018-0811-x
Martincorena I, Fowler JC, Wabik A et al (2018) Somatic mutant clones colonize the human esophagus with age. Science (80-) 362:911–917. https://doi.org/10.1126/science.aau3879
Riva L, Pandiri AR, Li YR et al (2020) The mutational signature profile of known and suspected human carcinogens in mice. Nat Genet 52:1189–1197. https://doi.org/10.1038/s41588-020-0692-4
Wang B, Kohli J, Demaria M (2020) Senescent cells in cancer therapy: friends or foes? Trends Cancer. https://doi.org/10.1016/j.trecan.2020.05.004
Rupec RA, Jundt F, Rebholz B et al (2005) Stroma-mediated dysregulation of myelopoiesis in mice lacking IκBα. Immunity 22:479–491. https://doi.org/10.1016/j.immuni.2005.02.009
Walkley CR, Olsen GH, Dworkin S et al (2007) A microenvironment-induced myeloproliferative syndrome caused by retinoic acid receptor γ deficiency. Cell 129:1097–1110. https://doi.org/10.1016/j.cell.2007.05.014
Kim YW, Koo BK, Jeong HW et al (2008) Defective Notch activation in microenvironment leads tomyeloproliferative disease. Blood 112:4628–4638. https://doi.org/10.1182/blood-2008-03-148999
Bao L, Al-Assar O, Drynan LF et al (2017) A non-cell autonomous mouse model of CNS haemangioblastoma mediated by mutant KRAS. Sci Rep 7:44899. https://doi.org/10.1038/srep44899
Dai X, Wang Y, Dong X et al (2020) Downregulation of miRNA-146a-5p promotes malignanttransformation of mesenchymal stromal/stem cells by glioma stem-like cells. Aging (Albany NY) 12:9151–9172. https://doi.org/10.18632/aging.103185
Bohm AK, DePetro J, Binding CE et al (2020) In vitro modeling of glioblastoma initiation using PDGF-AA and p53-null neural progenitors. Neuro Oncol. https://doi.org/10.1093/neuonc/noaa093
Echizen K, Horiuchi K, Aoki Y et al (2019) NF-κB-induced NOX1 activation promotes gastric tumorigenesis through the expansion of SOX2-positive epithelial cells. Oncogene 38:4250–4263. https://doi.org/10.1038/s41388-019-0702-0
Bhowmick NA, Chytil A, Plieth D, et al (2004) {TGF}-beta signaling in fibroblasts modulates the oncogenic potential of adjacent epithelia. Science 303:848–851. https://doi.org/10.1126/science.1090922
Weber TJ, Siegel RW, Markillie LM et al (2005) A paracrine signal mediates the cell transformation response to low dose gamma radiation in JB6 cells. Mol Carcinog 43:31–37. https://doi.org/10.1002/mc.20092
Martin D, Nguyen Q, Molinolo A, Gutkind JS (2014) Accumulation of dephosphorylated 4EBP after mTOR inhibition with rapamycin is sufficient to disrupt paracrine transformation by the KSHV vGPCR oncogene. Oncogene. https://doi.org/10.1038/onc.2013.193
Hu B, Castillo E, Harewood L et al (2012) Multifocal epithelial tumors and field cancerization from loss of mesenchymal CSL signaling. Cell 149:1207–1220. https://doi.org/10.1016/j.cell.2012.03.048
Acknowledgements
J.M.G.-M. is part of the Bioengineering Department at Tecnologico de Monterrey, Mexico City Campus, and belongs to the Translational Omics Strategic Focus Research Group. J.P.M.-B. is a Great Ormond Street Hospital for Children’s Charity Principal Investigator.
Funding
J.P.M.-B. laboratory is funded by Cancer Research UK, Children’s Cancer and Leukaemia Group, Children with Cancer UK, The Brain Tumour Charity (SIGNAL and EVEREST), Great Ormond Street Hospital Children’s Charity, the Morgan Adams Foundation and National Institute of Health Research Biomedical Research Centre at the Great Ormond Street Hospital for Children NHS Foundation Trust, and the University College London.
Author information
Authors and Affiliations
Contributions
Both authors performed the literature search as well as drafted, revised and approved this manuscript.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Gonzalez-Meljem, J.M., Martinez-Barbera, J.P. Adamantinomatous craniopharyngioma as a model to understand paracrine and senescence-induced tumourigenesis. Cell. Mol. Life Sci. 78, 4521–4544 (2021). https://doi.org/10.1007/s00018-021-03798-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00018-021-03798-7