
Abstract
Cardiovascular disease (CVD) is a major cause of death in patients with chronic kidney disease (CKD). Both conditions are rising in incidence as well as prevalence, creating poor outcomes for patients and high healthcare costs. Recent data suggests CKD to be an independent risk factor for CVD. Accumulation of uremic toxins, chronic inflammation, and oxidative stress have been identified to act as CKD-specific alterations that increase cardiovascular risk. The association between CKD and cardiovascular mortality is markedly influenced through vascular alterations, in particular atherosclerosis and vascular calcification (VC). While numerous risk factors promote atherosclerosis by inducing endothelial dysfunction and its progress to vascular structural damage, CKD affects the medial layer of blood vessels primarily through VC. Ongoing research has identified VC to be a multifactorial, cell-mediated process in which numerous abnormalities like mineral dysregulation and especially hyperphosphatemia induce a phenotype switch of vascular smooth muscle cells to osteoblast-like cells. A combination of pro-calcifying stimuli and an impairment of inhibiting mechanisms like fetuin A and vitamin K-dependent proteins like matrix Gla protein and Gla-rich protein leads to mineralization of the extracellular matrix. In view of recent studies, intercellular communication pathways via extracellular vesicles and microRNAs represent key mechanisms in VC and thereby a promising field to a deeper understanding of the involved pathomechanisms. In this review, we provide an overview about pathophysiological mechanisms connecting CKD and CVD. Special emphasis is laid on vascular alterations and more recently discovered molecular pathways which present possible new therapeutic targets.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Chronic kidney disease (CKD) is defined as abnormality of kidney structure or function, present for more than 3 months. It is classified and staged based on cause, glomerular filtration rate (GFR G1-G5), and albuminuria category (A1-A3) [1]. Both albuminuria and reduced GFR have been shown to be associated with an increase in all-cause mortality which is especially driven by cardiovascular events [2, 3, 5]. Large meta-analyses have demonstrated that patients with impaired renal function have a 40–50% increased risk of developing coronary artery disease (CAD) compared to patients with normal renal function [4, 6]. This may, at least in part, be mediated by the fact that two of the most common causes for CKD, hypertension and diabetes mellitus, have also been identified as cardiovascular risk factors. However, even after adjustment for classic cardiovascular risk factors, CKD is still associated with an increased risk of coronary events, suggesting CKD to be an independent risk factor for CVD (Table 1) [6,7,8]. Furthermore, renal insufficiency correlates with the severity of coronary atherosclerosis and incidence of coronary events as well as mortality after myocardial infarction [7, 9, 10]. Rates of sudden cardiac deaths are increasing with declining renal function illustrated by a rate of 7 cardiac arrests per 100.000 hemodialysis sessions in the USA [11].
Vascular calcification (VC) is a common finding among CKD patients and even present in young adults with end stage renal disease (ESRD) lacking typical cardiovascular risk factors such as hypertension or dyslipidemia [12,13,14]. VC manifesting in the coronary arteries impairs coronary flow reserve and is associated with a marked increase in adverse cardiac events and cardiovascular mortality [13, 15, 16]. Interestingly, CKD affects CVD in a wider spectrum than ischemic heart disease alone. ESRD is also associated with aortic- and mitral-valve calcification, leading to a faster progression of valve stenosis and thus worse outcome for patients [17,18,19]. Furthermore, left ventricular hypertrophy, diastolic dysfunction, and cardiac fibrosis are known cardiac alterations which are strongly influenced by CKD [20]. Also, VC occurs more frequently in CKD patients, with a reported prevalence in dialysis patients greater than 80% [12, 21]. The underlying pathophysiological mechanisms for these multiple cardiovascular changes associated with CKD are not completely understood and therefore subject of ongoing research.
Cardiorenal syndrome
An early step in attempting to establish a solid definition for the combination of CKD and CVD was taken in 2004 by the working Group of the National Heart, Lung, and Blood Institute in the USA. They proposed a first definition of “cardiorenal syndrome” (CRS) as an endpoint of cardiorenal dysregulation leading to an exacerbation of heart failure symptoms by an increased circulatory volume induced by kidneys and other circulatory compartments [22]. In 2008, this definition was extended by the consensus conference of the Acute Dialysis Quality Initiative into “disorders of the heart and kidneys whereby acute or chronic dysfunction in one organ may induce acute or chronic dysfunction of the other” [23]. They identified 5 subtypes of cardiorenal syndrome characterized by the order of the failing organ (cardiorenal versus reno-cardiac) and the temporal pattern (Table 2).
This sub-classification of the cardiorenal syndrome represents a first important step in creating a clinically applicable definition in the common finding of combined cardiovascular and renal disease. However, it should be acknowledged that the pathophysiological background of this syndrome is more complex and extends beyond the heart and kidney alone. Recent investigations rather suggest a complex interaction of neurohormonal dysregulation, uremia, anemia and inflammatory pathways, endothelial dysfunction, atherosclerosis, and vascular calcification which make it challenging to precisely determine the sequence of pathophysiological events involved [24].
Pathophysiology and therapeutic targets
Chronic inflammation
Atherosclerosis and its manifestation in the coronary arteries, CAD, is supposed to be a key connector between CKD and cardiovascular morbidity and mortality. Atherosclerosis is characterized by a chronic inflammatory process of the vessel wall which is initiated through endothelial dysfunction [28]. CKD strongly correlates with CAD prevalence and progression and is believed to play a role in its pathogenesis as an independent risk factor [6,7,8]. CKD triggers vascular inflammatory processes, which is mirrored by an augmentation of inflammatory markers in the blood of CKD patients [25,26,27]. Elevated levels of inflammatory biomarkers such as C-reactive protein (CRP), interleukin-6 (IL-6), or tumor necrosis factor (TNF) have been shown to be associated with an increased risk of myocardial infarction and mortality [28,29,30]. While patients undergoing renal replacement therapy are exposed to inflammatory triggers owing to the invasive nature of the procedure, available evidence suggests that elevated inflammatory markers can be found in patients prior to initiation of dialysis [31,32,33]. In animal models, it was shown that oxidative stress, which is frequently observed in CKD patients, correlates significantly with an increase in inflammatory markers [34, 35]. Oxidative stress in chronic kidney disease is a multifactorial process which can be caused by an impairment of antioxidant mechanisms as well as an increased production of reactive oxygen species (ROS) [36, 37]. It was shown in rats that CKD is associated with a depressed superoxide dismutase activity parallel to an increase in NADPH oxidase expression [35]. Nrf2, a translation factor which controls expression of antioxidant genes, is a key player in resistance to oxidative stress and may represent a potential target in this process [38]. Thus, in rats with CKD, Nrf2 activity was markedly reduced beside an increase in biomarkers of oxidative stress and inflammation [34].
Recent controlled randomized clinical trials have demonstrated a beneficial effect of SGLT2-inhibitors in heart failure patients with and without diabetes regarding the occurrence of major cardiovascular endpoints [39,40,41]. Of note, SGLT2-inhibition was additionally associated with a slower progression of CKD in patients with and without diabetes [42, 43]. Aside from an improved glycemic control, the discussion about the beneficial cardiovascular and renal effects of SGLT2-inhibitors is widely focused on volume control via the induction of natriuresis and osmotic diuresis [44]. On a molecular level, SGLT2-inhibitors have been shown to reduce oxidative stress, which may suggest a benefit of these substances regarding vascular alterations in patients with or without CKD [45, 46]. Although the molecular mechanisms of SGLT2-inibitiors mediating the protective effects in vascular and kidney disease are still vastly unknown, they represent a promising novel target to treat patients with CAD und CKD.
Recently, interleukin 1 (IL-1) has emerged as a potential therapeutic target to contain inflammation in CKD. IL-1 is a cytokine which is activated by the NLRP3 inflammasome and that induces IL-6 which is independently associated with an increase in mortality in hemodialysis and pre-dialysis patients [47, 48]. Malnutrition is another common finding among CKD patients and was shown to influence circulating inflammation markers in a bidirectional way [49, 50]. In this context, it is of interest that on the basis of the available evidence, a “malnutrition inflammation atherosclerotic syndrome” has been proposed [50].
Chronic inflammation in CKD is a multifactorial condition and specific pharmacological targets to improve outcomes for patients are rare. IL-1β inhibition has shown promising results in animal models with different renal disorders [47, 51, 52]. In a randomized, double-blind placebo-controlled trial with over 10,000 patients, administration of canakinumab, a human monoclonal antibody that targets IL-1β, was associated with a significantly lower rate of recurrent cardiovascular events than placebo [53]. In a subgroup analysis, it was shown that inhibition of IL-1β is particularly promising regarding cardiovascular outcomes in CKD patients [47]. At present, however, canakinumab is not approved for this indication in the USA or Europe. Other anti-inflammatory substances with a broader mechanism of action have also shown promising results. In a recent placebo-controlled trial, colchicine was shown to significantly reduce the risk of ischemic cardiovascular events in patients with myocardial infarction [54]. To date, no such studies have been performed specifically in patients with CKD. In this patient population, safety concerns are of special importance since colchicine is partly eliminated by the kidney and is not removed through hemodialysis [55].
Endothelial dysfunction
Accumulating evidence suggests that CKD promotes atherosclerosis and CAD by inducing damage to endothelial cells. Albuminuria as a consequence of glomerular damage was shown in several studies to correlate with elevated levels of von Willebrand factor, an indicator of endothelial dysfunction [56,57,58]. Furthermore, albuminuria and CKD were shown to be associated with microvascular endothelial dysfunction [59]. In order to control vascular tone, the endothelium synthesizes and releases the vasodilator nitric oxide (NO) from the amino acid L-arginine [60]. In addition, NO plays an important role in regulating vascular permeability, leukocyte adhesion and smooth muscle cell proliferation [61]. Therefore, a hallmark in endothelial dysfunction is a decrease in NO synthesis or bioavailability which is frequently observed in CKD patients [62, 63]. Amador-Martinez et al. were recently able to show in a rat model that cardiac alterations in CKD are partly promoted by an inactivation of endothelial nitric oxide synthase (eNOS) leading to reduced synthesis of NO [64]. L-arginine deficiency is a known factor leading to a decreased activity of eNOS and substitution of L-arginine was shown to positively affect cardiac alterations in rats with CKD [64].
Important mediators of endothelial dysfunction in CKD are uremic toxins such as asymmetric dimethylarginine (ADMA) and indoxyl sulfate (IS), which accumulate in CKD patients in parallel with declining renal function. ADMA disturbs endothelial function by competitive inhibition of eNOS and is closely associated with the presence and functional significance of CAD in CKD [65, 66]. Similarly, IS is involved in the pathogenesis of CRS through impairment of endothelial NO synthesis and, consecutively, endothelial proliferation in vivo and in vitro [67, 68]. Thus, the role of these uremic toxins may also extend into participating in the complex interrelationship of the heart and the kidney as a mediator of endothelial dysfunction.
Another recently described mechanism of endothelial dysfunction in CKD is driven by a change in the functional properties of LDL-cholesterol. Thus, carbamylation of lysine residues of the LDL-particle is primarily observed in CKD patients promoting endothelial dysfunction by increasing reactive oxygen species (ROS) production and eNOS uncoupling [69].
Hyperphosphatemia as a frequently observed finding among patients with CKD is known to be a major factor involved in the development of medial calcification in CKD (see below). In vitro and in vivo data available suggest a direct influence of elevated phosphate on endothelial function [70, 71]. An hyperphosphatemic milieu results in an impairment of angiogenesis, endothelial migration, and survival; a possible molecular mechanism for this finding could be the downregulation of annexin II [70].
Vascular calcification in CKD
While atherosclerosis is characterized through vascular endothelial dysfunction progressing to vascular structural damage, the medial layer of blood vessels is affected differently in CKD [72]. In this layer of the vessel wall, the primary abnormality is VC [73]. VC is highly prevalent in CKD patients and even present in young adults with ESRD lacking typical cardiovascular risk factors such as hypertension or dyslipidemia [12,13,14]. VC manifesting in the coronary arteries impairs coronary flow reserve and is associated with a marked increase in adverse cardiac events and cardiovascular mortality [13, 15, 16]. VC occurs in two different phenotypes, medial and intimal calcification, differing in their pathogenesis [74, 75]. While intimal calcification is mainly inflammation-driven and closely associated to atherosclerotic plaques, medial calcification is considered to be the major form of VC in CKD [74, 76]. As a result of intimal VC, uremic patients present with heavily calcified atherosclerotic plaques whereas in the absence of CKD, plaques are more fibroatheromatous [77, 78]. Furthermore, increased deposition of CRP in plaques of CKD patients has been reported which may be an indicator for a higher inflammatory component [77, 78].
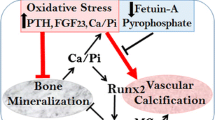
VC is a cell-mediated process similar to skeletal bone formation (Fig. 1). A crucial event is a phenotype switch of vascular smooth muscle cells (VSMC) to osteoblast-like cells which is induced through a number of different stimuli like hyperphosphatemia and hypercalcemia [27, 75, 79]. During the process of transdifferentiation of VSMC, a “reprogramming” is observed with a loss of SMC markers like SM22α and newly expression of osteochondrogenic markers like Runt-related transcription factor 2 (RUNX2), alkaline phosphatase (ALP), osteopontin and osteocalcin [27, 80, 81]. On a cellular level, VC shows similarities with cellular processes observed in senescence which underlines the theory that CKD is a risk factor for premature vascular aging [82, 83]. In the process of VC, VSMC-derived extracellular vesicles (EV) (formerly matrix vesicles) containing calcium phosphate crystals cause mineralization in the extracellular matrix (ECM) [84, 85, 86] .

Intimal and medial calcification in CKD. CKD-specific risk factors are adding up to traditional cardiovascular risk factors and result in endothelial dysfunction and intimal calcification. A phenotype switch of vascular smooth muscle cells to osteoblast-like cells with a loss of smooth muscle cell markers like SM22a and expression of osteochondrogenic markers like Runt-related transcription factor 2 (RUNX2) and alkaline phosphatase (ALP) represents a crucial event in medial calcification. In this process, levels of miRs are altered which act as regulators of calcification in CKD. MGP matrix Gla protein, GRP Gla-rich protein, EV extracellular vesicles, CV cardiovascular, miR microRNA, CCP calciprotein particle
In 2006, a work group of the Kidney Disease: Improving Global Outcomes (KDIGO) recommended the introduction of the term “chronic kidney disease-mineral and bone disorder” (CKD-MBD) to describe a syndrome consisting of abnormalities in bone and mineral metabolism as well as extra-skeletal calcifications including VC [87]. Mineral dysregulation and especially phosphate accumulation play a key role, since inorganic phosphate, known to mediate vascular calcification in a time- and dose-dependent manner, is frequently elevated in CKD patients [79]. Phosphate accumulation as a result of a declining renal clearance and secondary hyperparathyroidism were shown to induce an upregulation of osteogenic gene expression in vitro [76]. In this context, the activity of specific enzymes that regulate biomineralization such as tissue-nonspecific alkaline phosphatase (TNAP) becomes of interest. TNAP regulates extracellular pyrophosphate, an inhibitor of calcification and, accordingly, TNAP overexpressing mice showed a significant increase in medial calcification [88, 89]. Alterations in bone metabolism of CKD patients, or renal osteodystrophy, are a multifaceted disorder with adynamic bone disease as the most frequently observed form among patients on dialysis [90]. Adynamic bone disease is characterized by a low bone turnover and is known to be associated with VC in nondialysis CKD patients as well as in patients on hemodialysis [90,91,92]. A common finding among CKD patients is a deficiency of vitamin D which can lead to hypocalcemia. Low circulating levels of 25-hydroxyvitamin D are known to be associated with increased mortality in CKD patients as well as the development of VC [93, 94]. Vitamin D substitution may have beneficial effects on VC development in CKD patients, a potential mechanism for this finding is that vitamin D known suppresses calcification by downregulation of RUNX2 [95, 96]. However, dosage and indication for vitamin D substitution should be chosen wisely. Elevated concentrations of calcium and the calcium-phosphate product may markedly influence VSMC calcification [27, 97]. Excessive substitution of vitamin D or the administration of calcium containing phosphate binders can be associated with intermittent increases in serum calcium concentration [98]. Moreover, apoptotic or necrotic cells in the vessel wall can also mediate elevated levels of calcium [27]. In the presence of elevated phosphate levels, even small elevations of calcium were shown to act synergistically to promote vascular calcification [97]. Under these calcifying stimuli, calcium phosphate crystals are loaded onto EV by VSMC which nucleate in the ECM as hydroxyapatite [86].
Despite the fact that elevated phosphate levels are known to be associated with adverse cardiovascular outcomes in CKD patients, only few studies have examined clinical endpoints of interventions regarding phosphate control [99]. Meta-analyses of available randomized controlled trials regarding phosphate binding agents have demonstrated a lower all-cause mortality of patients receiving the non-calcium-based agent sevelamer compared to calcium-based binders while there was no significant difference regarding cardiovascular death [100, 101]. According to available data from clinical trials comparing phosphate binders to placebo in nondialysis CKD patients, the relevance of the administration of phosphate binders has not been proven in this population[102,103,103]. The “IMpact of Phosphate Reduction On Vascular End-points in Chronic Kidney Disease” (IMPROVE-CKD) study, a multicenter, randomized parallel-group trial, examined the impact of the non-calcium-based phosphate binder lanthanum carbonate on surrogate markers of cardiovascular disease compared with placebo [99]. Experimental background of that study was, among others, an in vitro study in rat aortic tissue which showed that the administration of iron citrate resulted in an anti-calcific effect by preventing or partially reverting high phosphate induced osteo-chondrocytic shift of ECM [105]. Treatment of CKD patients with lanthanum over 96 weeks however showed no significant difference to placebo regarding arterial stiffness and aortic calcification [106]. However, it is worth mentioning that in this nondialysis CKD population, the patients presented with normophosphatemia at baseline. The influence of a positive phosphate balance on this therapeutic approach needs to be further investigated [106].
In contrast to calcium and phosphate, magnesium has emerged to be a possible protector against VC. Clinical data suggest an inverse association between serum magnesium levels and the expression of VC in peritoneal- and hemodialysis patients [107, 108]. In vitro studies performed on VSMC were able to show a decreased formation of hydroxyapatite formation after exposing the cells to increased magnesium concentrations, a potential molecular mechanism for this finding is suggested to be an impairment of β-glycerophosphate induced ALP activity [109, 110]. In addition, data from experimental studies suggests magnesium to counteract VSMC transdifferentiation by impairing the expression of osteogenic transcription factors [111, 112]. Therefore, magnesium supplementation might present a potential therapeutic approach to battle VC and needs to be investigated in future clinical studies.
More recently, impairment of calcification inhibiting mechanisms, such as fetuin A, klotho, and the vitamin K-dependent matrix Gla protein (MGP) and Gla-rich protein (GRP) have emerged as participants in the complex multifactorial process of VC [113, 114]. These proteins act as inhibitors for the precipitation of calcium/phosphate crystals by forming the calciprotein particle (CPP). CPP exist in two different phenotypes, primary and secondary CPP, which differ in shape, function, and diameter [115]. In vitro, secondary CPP are capable to induce VSMC calcification, expression, and release of TNF-α and may thereby represent a promising new biomarker for VC and potential therapeutic target [115]. In addition to their suggested influence on atherosclerosis, both systemic inflammation and malnutrition are associated with progression of VC [116]. A potential mechanism for this finding is supposed to be mediated through decreases in fetuin A levels, which were shown in several studies to be associated with an increase in vascular calcification and cardiovascular mortality [117, 118].
Another key calcification inhibitor which is impaired in CKD patients is the klotho/FGF23 axis. The klotho gene was first described as an aging suppressor and encodes for a single-pass transmembrane protein which functions as a coreceptor for fibroblast growth factor 23 (FGF-23) [119, 120]. CKD is characterized by klotho deficiency and low levels of circulating klotho were shown to be associated with adverse renal outcome [120, 121]. In addition to being a biomarker for CKD, klotho plays a pathogenic role in CKD and especially in VC. Evidence suggests that klotho improves the phosphate metabolism by inducing phosphaturia and supresses thereby VSMC calcification [120]. In line with these findings, animal models of klotho substitution have shown positive effects regarding kidney injury and phosphaturia [122, 123]. Klotho substitution may represent a promising therapeutic approach to influence VC in CKD patients. FGF23 in contrast is known to be elevated in CKD patients and is a potent predictor of adverse cardiovascular outcomes [124]. However, FGF23 was shown to be independently associated with VC, its relevance as an therapeutic target remains subject of ongoing research [125]. In a rat model of CKD, FGF23 neutralization was shown to positively influence secondary hyperparathyroidism but accelerated hyperphosphatemia, vascular calcification, and mortality [126].
Interestingly, hyperaldosteronism, which is a frequent finding among patients with CKD, is also observed in klotho-deficient mice and is supposed to participate in VC [127, 128]. VSMC express the mineralcorticoid receptor which is sensitive for aldosterone and can be blocked by the competitive antagonist spironolactone [129]. Experimental data provide evidence that aldosterone leads to the expression of the type III sodium-dependent phosphate transporter PIT1, which leads to an increase of ALP activity and expression of ostegenic transcription factors [130]. In an animal model with klotho-deficient mice, this effect was ameliorated through the administration of spironolactone-associated with an enhanced survival of the animals [130]. However, until now, no data are available regarding the positive influence of spironolactone on VC in patients.
Subclinical vitamin K deficiency is frequently observed in CKD patients and can be due to malnutrition, dietary restrictions, or anticoagulant therapies with vitamin K antagonists [131, 132]. This leads to a reduced bioavailability of vitamin K-dependent proteins. MGP and GRP are proteins synthesized by vascular smooth muscle cells and known as inhibitors of tissue calcification, while GRP may also exhibit anti-inflammatory efficacy [114, 133, 134]. To be fully active, both require vitamin K-dependent posttranslational modification (gamma carboxylation) [135]. Both MGP or GRP deficiency are associated with early development of VC in vitro and in vivo and thus represent a molecular mechanism connecting vitamin K deficiency in CKD with VC [136, 137]. The pathophysiological importance of an impairment of calcifying inhibitors is impressively highlighted by the example of calciphylaxis. Calciphylaxis is a rare syndrome with a high mortality characterized by calcification and thrombotic occlusion of microvessels in subcutaneous adipose tissue which results in painful, ischemic skin lesions [138]. It shares a number of pathophysiological features with VC such as a reduction of carboxylated MGP, fetuin A deficiency, and the use of vitamin K antagonists as a risk factor [138]. Despite its synonym “calcific uremic arteriolopathy,” calciphylaxis is also reported in nonuremic patients without advanced renal impairment. In contrast to VC, which typically affects the aorta, coronary, and femoral arteries, calciphylaxis occurs in the microcirculation of subcutaneous adipose tissue [138]. Therefore, calciphylaxis represents a distinct entity of VC rather than a continuum of VC [138].
Since vitamin K deficiency represents a key feature in the development of VC, the effects of vitamin K substitution have been investigated. A recently published study comparing oral vitamin K2 substitution versus placebo in CKD patients showed no improvement in vascular stiffness or other measures of vascular health [139]. Vitamin K1 supplementation however is a promising and uncomplicated way to positively influence VC in CKD patients and, therefore, its effect on CV risk is under investigation in current clinical trials (Table 3) [135, 140]. Further support for an essential role of vitamin K in vascular calcification is provided by experimental and clinical studies with vitamin K antagonists suggesting adverse effects on cardiovascular outcomes[141,142,143].
Extracellular vesicles as intercellular messengers in CKD and CAD
In the past years, circulating EV have emerged to play a key role in cardiovascular health and disease [72]. EV are membrane shed particles that consist of a lipid bilayer and are categorized into exosomes (30–100 nm), microvesicles (200–1000 nm), and apoptotic bodies (1–4 μm) with each category having its own formation process [144, 145]. EV play an important role in intercellular communication by transferring their bioactive cargos including proteins, lipids, and nucleic acids. This mechanism of signal transfer has evolved as an important regulator of cardiovascular health and disease [146, 147]. Circulating EV are released from their origin cell in consequence of certain stimuli. Circulating EV are known to mediate many physiological and pathophysiological mechanisms like inflammation and coagulation[148,149,150]. Endothelial cell damage and apoptosis are crucial steps in the pathophysiology of CVD and endothelial microvesicles have been shown to be elevated in patients with CVD compared to healthy control subjects. This has been described in patients with hypertension, CAD, acute coronary syndrome, and myocardial infarction[151,152,153,154]. We have previously shown that endothelial-derived EV promote endothelial cell repair by delivering functional miR-126 into recipient cells. This mechanism was shown to be altered under pathological hyperglycemic conditions [155]. Furthermore, elevated levels of endothelial- and platelet-derived EV have been reported in CKD patients compared to healthy controls [156, 157]. Endothelial cells incubated with IS show an increase in EV release and a significantly reduced angiogenesis in endothelial progenitor cells [158]. Increased levels of endothelial EV were shown to be an independent predictor of CV mortality in patients with ESRD [159]. In vitro, endothelial EV from plasma of patients with ESRD led to reduced endothelium-dependent relaxations and cyclic guanosine monophosphate (cGMP) generation [160]. In addition to these findings, endothelial-derived EV correlate with an impaired arterial function in patients with ESRD, demonstrated by an accelerated aortic pulse wave velocity and loss of flow-mediated dilation [160].
VSMC-derived EV play a crucial role in the early stages of development of VC [84,85,86]. EV produced by VSMC under physiological conditions do not contain calcium phosphate crystals and moreover transport calcification inhibitory proteins such as vitamin K-dependent MGP and fetuin-A [84, 161]. Under calcifying stimuli such as elevated phosphate levels as they are present in CKD patients, calcium phosphate crystals are loaded onto the EV [86]. It is important to mention that EV secretion by VSMC is not a pathological process itself. Thus, removal of exosomes from healthy serum promoted VC which underlines the importance of exosomes as calcifying inhibitors under certain circumstances [162]. In vitro, VSMC that were incubated with serum of CKD patients demonstrated increased calcification compared with controls. Serum of CKD from which EV had been removed showed a marked decrease in VSMC calcification [162]. Taken together, these findings indicate that, on a molecular level, EV act as mediators between CKD and CVD. Influencing the phenotype and cargo of EV derived from VSMC could therefore represent an interesting therapeutic target for VC in CKD patients.
microRNAs as gene regulators of vascular alteration in CKD
MicroRNAs (miRNAs) play an outstanding role in post transcriptional gene regulation and can be transferred intercellularly through EV [163]. MiRNAs are small, noncoding nucleotides of about 20 basepaires that modulate different biological pathways in angiogenesis and apoptosis as well as diseases such as atherosclerosis and CKD [164, 165]. We have previously shown that atherosclerotic conditions promote the packaging of endothelial miR-92a-3p into endothelial microvesicles which regulates angiogenesis and may act as a potential regenerative messenger in intercellular communication [166]. Patients with diabetic nephropathy express a different exosomal miRNA profile than healthy subjects and upregulated miRNAs closely correlate with the degree of albuminuria [167]. A pilot study, comparing urinary and plasmatic miRNA-profiles of CKD patients with eGFR < 30 ml·min−1·1.73 cm−2 and patients with eGFR ≥ 30 ml·min−1·1.73 cm−2 showed 266 circulatory and 384 urinary miRNA that were differently expressed [168]. A number of upregulated miRNA in this study, including miR-130a-3p and miR-1825, target the TGFβ pathway [168]. Own data reveal that CKD patients exhibit significantly lower vesicular levels of vascular protective miR-126-3p and miR-130a-3p [169]. In vitro treatment with the model uremia toxin IS leads to a decreased packaging of the two miRNAs into EV through a hnRNPU-dependent sorting mechanism. This altered miRNA packaging was shown to be functionally relevant as it influences endothelial cell migratory capacity [169]. Another miRNA, miR-92a, which is relevant for endothelial function circulates in augmented levels in the peripheral blood of CKD patients. It correlates with levels of IS and is transported by endothelial microvesicles that originate from uremia-damaged endothelial cells [170]. In cultured endothelial cells, miR-92a induced inflammasome activation and thus mediated endothelial cell damage [170]. MiRNAs have been investigated as therapeutic targets to reduce CKD-associated atherosclerosis. Thus, in mice with renal injury, increased levels of miR-92a-3p were observed and inhibition of miR-92a-3p with a single injection miRNA inhibitors complexed to HDL significantly reduced atherosclerotic lesions [171]. Inhibition of these miRNAs significantly altered the TGFβ pathway and STAT3 trancriptional activity [171]. Furthermore, recent data support the concept that substitution of cardiovascular protective miRNAs may represent a potential treatment strategy. One such target investigated is miR-142-3p which is inversely correlated with carotid-femoral pulse wave velocity in humans as an indicator of vascular stiffness and is significantly decreased in patients with ESRD [172]. Also, in uremic mice with markedly reduced acetylcholine-induced relaxation, intravenous injection of synthetic mimic syn-mmu-mir-142-3p in order to restore bioavailability of miR-142-3p 2 days before sacrifice increased aortic relaxation of aortic rings to control levels [172].
VC is a cell-mediated process that requires genetic alterations which may at least in part be modulated through miRNAs [173]. RUNX2, a transcription factor which regulates osteoblast differentiation and VSMC calcification, represents a key target for miRs influencing VC. Recent data show protective miR like miR-133a, miR-204, and miR-205, as well as miR triggering VC like miR-32 influence RUNX2 by decreasing or inducing its expression [174,175,176,177]. Furthermore, miR-30b and miR-125b have recently been identified to protect against VC. Bone morphogenetic protein-2 (BMP-2) may promote vascular calcification by decreasing miR-30b and miR-30c to induce RUNX2 expression whereas upregulation of miR-30b attenuated VC in vivo and in vitro [178, 179]. In vitro, downregulation of endogenous miR-125b led to osteogenic transdifferentiation of VSMC and increased activity of alkaline phosphatase activity and matrix mineralization which was confirmed in ApoE knockout mice [180]. Osterix, an osteoblast transcription factor, may be a molecular target of miR-125b in this process [180]. In an in vitro biomineralization model, VSMC that have been transfected with miR-125b showed significantly lower osteocalcin expression than control VSMC [181]. These data are supported by the observation that decreased circulating levels of miR-125b in CKD patients is associated with a decline in renal function [182]. Recent studies have elucidated the potential use of miR-125b as a biomarker for VC in uremic patients. MiR-125b levels correlated significantly with VC severity as well as levels of fetuin-A and mediators of mineral bone disorder like osteoprotegerin and FGF-23 [181, 183]. In one study, high levels of serum osteoprotegerin and low serum miR-125b levels were able to synergistically enhance VC risk estimating ability [183]. Furthermore, miR-135a, miR-762, miR-714, and miR-712 targeting Ca2+-transporters in VSMC were shown in vitro and in vivo in a rat model to be overexpressed in the aortic media under calcifying conditions [184]. Recently, Fakhry et al. investigated potential miRs involved in VC by exposing rat aortic explants to high concentrations of phosphate and characterized the miR expression profile versus control samples [80]. In that study, five miRs were expressed differently compared to controls (miR-155, -200c, -322, -331, -708) 3 days after exposition to phosphate and five miRs (miR-328, -546, -301a, -409, -542) 6 days after exposition [80]. It is of interest that some of the altered miRs (miR-322, miR-155, miR-200c) are known to act by participating in inflammatory and osteoblastic processes [80]. Finally, Pan et al. were able to show that miR profiles in EV from mouse VSMC were significantly influenced through VC [185]. Taken together, miRs represent important regulators of VC in CKD and further understanding of single pathways will be crucial to develop molecular treatment targets.
In summary, cardiorenal syndrome, the combination of cardiovascular and chronic kidney disease, is a crucial challenge for modern medicine with rising numbers in incidence and prevalence. Vascular calcification represents an important link between CKD and cardiovascular mortality which is the leading cause of death among patients with impaired renal function. While numerous potential pathophysiological mechanisms have been uncovered in recent years, many details remain unknown, and as a consequence, therapeutic options to provide better outcomes for patients with VC are limited. A further understanding of molecular mechanisms and genetic targets involved in the complex process of VC is pivotal to develop novel therapeutic targets. However, more research is required to transfer promising experimental results into routine clinical practice. With a better understanding of the pathophysiological mechanisms involved in VC, in the future, additional therapeutic targets with a chance for further improvements in therapy may be identified.
Data availability
Not applicable.
Abbreviations
- ADMA:
-
Asymmetric dimethylarginine
- ALP:
-
Alkaline phosphatase
- BMP-2:
-
Bone morphogenetic protein-2
- CAD:
-
Coronary artery disease
- cGMP:
-
Cyclic guanosine monophosphate
- CKD:
-
Chronic kidney disease
- CRP:
-
C-reactive protein
- CPP:
-
Calciprotein particle
- CRS:
-
Cardiorenal syndrome
- CVD:
-
Cardiovascular disease
- ECM:
-
Extracellular matrix
- eNOS:
-
Nitric oxide synthase
- ESRD:
-
End stage renal disease
- EV:
-
Extracellular vesicles
- GRP:
-
Gla-rich protein
- IL-1:
-
Interleukin-1
- IL-6:
-
Interleukin-6
- IS:
-
Indoxyl sulfate
- MGP:
-
Matrix Gla protein
- miRNA:
-
MicroRNAs
- NO:
-
Nitric oxide
- ROS:
-
Reactive oxygen species
- RUNX2:
-
Runt-related transcription factor 2
- TNAP:
-
Tissue-nonspecific alkaline phosphatase
- TNF:
-
Tumor necrosis factor
- VC:
-
Vascular calcification
- VSMC:
-
Vascular smooth muscle cells
References
KDIGO Board Members (2013) KDIGO 2012 Clinical Practice Guideline for the Evaluation and Managment of Chronic Kidney Disease, Summary of Recommendation Statements. Kidney Int Suppl 3:5–14. https://doi.org/10.1038/kisup.2012.77
Thompson S, James M, Wiebe N et al (2015) Cause of death in patients with reduced kidney function. J Am Soc Nephrol 26:2504–2511
Matsushita K, van der Welde M, Astor BC et al (2010) Association of estimated glomerular filtration rate and albuminuria with all-cause and cardiovascular mortality in general population cohorts: a collaborative meta-analysis. Lancet 375:2073–2081
Di Angelantonio E, Danesh J, Eiriksdottir G, Gudnason V (2007) Renal function and risk of coronary heart disease in general populations: new prospective study and systematic review. PLoS Med 4:e270. https://doi.org/10.1371/journal.pmed.0040270
Webster AC, Nagler EV, Morton RL, Masson P (2017) Chronic kidney disease. Lancet 389:1238–1252
Perkovic V, Verdon C, Ninomiya T, Barzi F, Cass A, Patel A, Jardine M, Gallagher M, Turnbull F, Chalmers J, Craig J, Huxley R (2008) The relationship between proteinuria and coronary risk: a systematic review and meta-analysis. PLoS Med 5:e207. https://doi.org/10.1371/journal.pmed.0050207
Tonelli M, Muntner P, Lloyd A et al (2012) Risk of coronary events in people with chronic kidney disease compared with those with diabetes: a population-level cohort study. Lancet 380:807–814
Sarnak MJ, Levey AS, Schoolwerth AC, Coresh J, Culleton B, Hamm LL, McCullough P, Kasiske BL, Kelepouris E, Klag MJ, Parfrey P, Pfeffer M, Raij L, Spinosa DJ, Wilson PW, American Heart Association Councils on Kidney in Cardiovascular Disease, High Blood Pressure Research, Clinical Cardiology, and Epidemiology and Prevention (2003) Kidney disease as a risk factor for development of cardiovascular disease: a statement from the American Heart Association Councils on Kidney in Cardiovascular Disease, High Blood Pressure Research, Clinical Cardiology, and Epidemiology and Prevention. Circulation 108:2154–2169
Bae EH, Lim SY, Cho KH et al (2012) GFR and cardiovascular outcomes after acute myocardial infarction: results from the Korea Acute Myocardial Infarction Registry. Am J Kidney Dis 59:795–802
Nakano T, Ninomiya T, Sumiyoshi S, Fujii H, Doi Y, Hirakata H, Tsuruya K, Iida M, Kiyohara Y, Sueishi K (2010) Association of kidney function with coronary atherosclerosis and calcification in autopsy samples from Japanese elders: the Hisayama Study. Am J Kidney Dis 55:21–30
Shamseddin MK, Parfrey PS (2011) Sudden cardiac death in chronic kidney disease: epidemiology and prevention. Nat Rev Nephrol 7:145–154
Goodman WG, Goldin J, Kuizon BD et al (2000) Coronary-artery calcification in young adults with end-stage renal disease who are undergoing dialysis. N Engl J Med 342:1478–1483
London GM, Guérin AP, Marchais SJ et al (2003) Arterial media calcification in end-stage renal disease: impact on all-cause and cardiovascular mortality. Nephrol Dial Transplant 18:1731–1740
Toussaint ND, Kerr PG (2007) Vascular calcification and arterial stiffness in chronic kidney disease: implications and management. Nephrology 12:500–509
Kim J, Bravo PE, Gholamrezanezhad A et al (2013) Coronary artery and thoracic aorta calcification is inversely related to coronary flow reserve as measured by 82Rb PET/CT in intermediate risk patients. J Nucl Cardiol 20:375–384
Raggi P (2017) Coronary artery calcification predicts risk of CVD in patients with CKD. Nat Rev Nephrol 13:324–326
Perkovic V, Hunt D, Griffin SV et al (2004) Accelerated progression of calcific aortic stenosis in dialysis patients. Nephron Clin Pract 94:c40–c45
Rattazzi M, Bertacco E, Del Vecchio A et al (2013) Aortic valve calcification in chronic kidney disease. Nephrol Dial Transplant 28:2968–2976
Patel KK, Shah SY, Arrigain S, Jolly S, Schold JD, Navaneethan SD, Griffin BP, Nally JV, Desai MY (2019) Characteristics and outcomes of patients with aortic stenosis and chronic kidney disease. J Am Heart Assoc 8:e009980
Tumlin JA, Costanzo MR, Chawla LS et al (2013) Cardiorenal syndrome type 4: insights on clinical presentation and pathophysiology from the Eleventh Consensus Conference of the Acute dialysis Quality Initiative (ADQI). In: McCullough PA, Kellum JA, Mehta RL et al (eds) Contributions to nephrology. S. KARGER AG, Basel, pp 158–173
Oh J, Wunsch R, Turzer M, Bahner M, Raggi P, Querfeld U, Mehls O, Schaefer F (2002) Advanced coronary and carotid arteriopathy in young adults with childhood-onset chronic renal failure. Circulation 106:100–105
Rangaswami J, Bhalla V, Blair JEA et al (2019) Cardiorenal syndrome: classification, pathophysiology, diagnosis, and treatment strategies: a scientific statement from the American Heart Association. Circulation 139. https://doi.org/10.1161/CIR.0000000000000664
Ronco C, McCullough P, Anker SD, Anand I, Aspromonte N, Bagshaw SM, Bellomo R, Berl T, Bobek I, Cruz DN, Daliento L, Davenport A, Haapio M, Hillege H, House AA, Katz N, Maisel A, Mankad S, Zanco P, Mebazaa A, Palazzuoli A, Ronco F, Shaw A, Sheinfeld G, Soni S, Vescovo G, Zamperetti N, Ponikowski P, for the Acute Dialysis Quality Initiative (ADQI) consensus group (2010) Cardio-renal syndromes: report from the consensus conference of the acute dialysis quality initiative. Eur Heart J 31:703–711
Hatamizadeh P, Fonarow GC, Budoff MJ et al (2012) Cardiorenal syndrome: pathophysiology and potential targets for clinical management. Nat Rev Nephrol 9:99
Zimmermann J, Herrlinger S, Pruy A, Metzger T, Wanner C (1999) Inflammation enhances cardiovascular risk and mortality in hemodialysis patients. Kidney Int 55:648–658
Schiffrin EL, Lipman ML, Mann JFE (2007) Chronic kidney disease: effects on the cardiovascular system. Circulation 116:85–97
Shanahan CM, Crouthamel MH, Kapustin A, Giachelli CM (2011) Arterial calcification in chronic kidney disease: key roles for calcium and phosphate. Circ Res 109:697–711
Ridker PM, Cushman M, Stampfer MJ, Tracy RP, Hennekens CH (1997) Inflammation, aspirin, and the risk of cardiovascular disease in apparently healthy men. N Engl J Med 336:973–979
Bologa R, Levine D, Parker T, Cheigh JS, Serur D, Stenzel KH, Rubin AL (1998) Interleukin-6 predicts hypoalbuminemia, hypocholesterolemia, and mortality in hemodialysis patients. Am J Kidney Dis 32:107–114
Zoccali C, Vanholder R, Massy ZA et al (2017) The systemic nature of CKD. Nat Rev Nephrol 13:344–358
Pastan S, Soucie JM, McClellan WM (2002) Vascular access and increased risk of death among hemodialysis patients. Kidney Int 62:620–626
Parker TF, Wingard RL, Husni L et al (1996) Effect of the membrane biocompatibility on nutritional parameters in chronic hemodialysis patients. Kidney Int 49:551–556
Stenvinkel P, Heimbürger O, Paultre F, Diczfalusy U, Wang T, Berglund L, Jogestrand T (1999) Strong association between malnutrition, inflammation, and atherosclerosis in chronic renal failure. Kidney Int 55:1899–1911
Kim HJ, Vaziri ND (2010) Contribution of impaired Nrf2-Keap1 pathway to oxidative stress and inflammation in chronic renal failure. Am J Physiol-Ren Physiol 298:F662–F671
Vaziri ND, Dicus M, Ho ND, Boroujerdi-Rad L, Sindhu RK (2003) Oxidative stress and dysregulation of superoxide dismutase and NADPH oxidase in renal insufficiency. Kidney Int 63:179–185
Manning RD Jr, Tian N, Meng S (2005) Oxidative stress and antioxidant treatment in hypertension and the associated renal damage. Am J Nephrol 25:311–317
Moradi H, Pahl MV, Elahimehr R, Vaziri ND (2009) Impaired antioxidant activity of high-density lipoprotein in chronic kidney disease. Transl Res 153:77–85
Ma Q (2013) Role of Nrf2 in oxidative stress and toxicity. Annu Rev Pharmacol Toxicol 53:401–426
Zinman B, Wanner C, Lachin JM et al (2015) Empagliflozin, cardiovascular outcomes, and mortality in type 2 diabetes. N Engl J Med 373:2117–2128
McMurray JJV, Solomon SD, Inzucchi SE et al (2019) Dapagliflozin in patients with heart failure and reduced ejection fraction. N Engl J Med 381:1995–2008
Bhatt DL, Szarek M, Pitt B et al (2020) Sotagliflozin in patients with diabetes and chronic kidney disease. N Engl J Med NEJMoa2030186. https://doi.org/10.1056/NEJMoa2030186
Wanner C, Inzucchi SE, Lachin JM et al (2016) Empagliflozin and progression of kidney disease in type 2 diabetes. N Engl J Med 375:1799–1802. https://doi.org/10.1056/NEJMc1611290
Heerspink HJL, Stefánsson BV, Correa-Rotter R et al (2020) Dapagliflozin in patients with chronic kidney disease. N Engl J Med 383:1436–1446
Lytvyn Y, Bjornstad P, Udell JA et al (2017) Sodium glucose cotransporter-2 inhibition in heart failure: potential mechanisms, clinical applications, and summary of clinical trials. Circulation 136:1643–1658
Li C, Zhang J, Xue M et al (2019) SGLT2 inhibition with empagliflozin attenuates myocardial oxidative stress and fibrosis in diabetic mice heart. Cardiovasc Diabetol 18:15
Chen Y-Y, Wu T-T, Ho C-Y et al (2019) Dapagliflozin prevents NOX- and SGLT2-dependent oxidative stress in Lens cells exposed to fructose-induced diabetes mellitus. Int J Mol Sci 20:4357
Ridker PM, MacFadyen JG, Glynn RJ et al (2018) Inhibition of interleukin-1β by canakinumab and cardiovascular outcomes in patients with chronic kidney disease. J Am Coll Cardiol 71:2405–2414
Barreto DV, Barreto FC, Liabeuf S, Temmar M, Lemke HD, Tribouilloy C, Choukroun G, Vanholder R, Massy ZA, European Uremic Toxin Work Group (EUTox) (2010) Plasma interleukin-6 is independently associated with mortality in both hemodialysis and pre-dialysis patients with chronic kidney disease. Kidney Int 77:550–556
Qureshi AR, Alvestrand A, Danielsson A et al (1998) Factors predicting malnutrition in hemodialysis patients: a cross-sectional study. Kidney Int 53:773–782
Stenvinkel P, Heimbürger O, Lindholm B et al (2000) Are there two types of malnutrition in chronic renal failure? Evidence for relationships between malnutrition, inflammation and atherosclerosis (MIA syndrome). Nephrol Dial Transplant 15:953–960
Chen A, Lai-Fa S, Wei-Yuan C et al (1997) Interleukin-1 receptor antagonist modulates the progression of a spontaneously occurring IgA nephropathy in mice. Am J Kidney Dis 30:693–702
Shahzad K, Bock F, Dong W et al (2015) Nlrp3-inflammasome activation in non-myeloid-derived cells aggravates diabetic nephropathy. Kidney Int 87:74–84
Ridker PM, Everett BM, Thuren T, MacFadyen JG, Chang WH, Ballantyne C, Fonseca F, Nicolau J, Koenig W, Anker SD, Kastelein JJP, Cornel JH, Pais P, Pella D, Genest J, Cifkova R, Lorenzatti A, Forster T, Kobalava Z, Vida-Simiti L, Flather M, Shimokawa H, Ogawa H, Dellborg M, Rossi PRF, Troquay RPT, Libby P, Glynn RJ (2017) Antiinflammatory therapy with canakinumab for atherosclerotic disease. N Engl J Med 377:1119–1131
Tardif J-C, Kouz S, Waters DD et al (2019) Efficacy and safety of low-dose colchicine after myocardial infarction. N Engl J Med 381:2497–2505
Vargas-Santos AB, Neogi T (2017) Management of gout and hyperuricemia in CKD. Am J Kidney Dis 70:422–439
Huang M-J, Wei R, Zhao J et al (2017) Albuminuria and endothelial dysfunction in patients with non-diabetic chronic kidney disease. Med Sci Monit 23:4447–4453
Stehouwer CDA, Stroes ESG, Hackeng WHL, Mulder PGH, den Ottolander GJH (1991) Von Willebrand factor and development of diabetic nephropathy in IDDM. Diabetes 40:971–976
Stehouwer CA, Zeldenrust GC, den Ottolander GH et al (1992) Urinary albumin excretion, cardiovascular disease, and endothelial dysfunction in non-insulin-dependent diabetes mellitus. Lancet 340:319–323
Seliger SL, Salimi S, Pierre V, Giffuni J, Katzel L, Parsa A (2016) Microvascular endothelial dysfunction is associated with albuminuria and CKD in older adults. BMC Nephrol 17:82
Ignarro LJ, Buga GM, Wood KS, Byrns RE, Chaudhuri G (1987) Endothelium-derived relaxing factor produced and released from artery and vein is nitric oxide. Proc Natl Acad Sci 84:9265–9269
Martens CR, Edwards DG (2011) Peripheral vascular dysfunction in chronic kidney disease. Cardiol Res Pract 2011:1–9
Schmidt RJ, Baylis C (2000) Total nitric oxide production is low in patients with chronic renal disease. Kidney Int 58:1261–1266
Schmidt RJ, Yokota S, Tracy TS et al (1999) Nitric oxide production is low in end-stage renal disease patients on peritoneal dialysis. Am J Physiol-Ren Physiol 276:F794–F797
Amador-Martínez I, Pérez-Villalva R, Uribe N et al (2019) Reduced endothelial nitric oxide synthase activation contributes to cardiovascular injury during chronic kidney disease progression. Am J Physiol-Ren Physiol 317:F275–F285
Fujii H, Takiuchi S, Kawano Y, Fukagawa M (2008) Putative role of asymmetric dimethylarginine in microvascular disease of kidney and heart in hypertensive patients. Am J Hypertens 21:650–656
Mangiacapra F, Conte M, Demartini C, Muller O, Delrue L, Dierickx K, di Sciascio G, Trimarco B, de Bruyne B, Wijns W, Bartunek J, Barbato E (2016) Relationship of asymmetric dimethylarginine (ADMA) with extent and functional severity of coronary atherosclerosis. Int J Cardiol 220:629–633
Tumur Z, Niwa T (2009) Indoxyl sulfate inhibits nitric oxide production and cell viability by inducing oxidative stress in vascular endothelial cells. Am J Nephrol 29:551–557
Dou L, Bertrand E, Cerini C, Faure V, Sampol J, Vanholder R, Berland Y, Brunet P (2004) The uremic solutes p-cresol and indoxyl sulfate inhibit endothelial proliferation and wound repair. Kidney Int 65:442–451
Speer T, Owala FO, Holy EW, Zewinger S, Frenzel FL, Stähli BE, Razavi M, Triem S, Cvija H, Rohrer L, Seiler S, Heine GH, Jankowski V, Jankowski J, Camici GG, Akhmedov A, Fliser D, Lüscher TF, Tanner FC (2014) Carbamylated low-density lipoprotein induces endothelial dysfunction. Eur Heart J 35:3021–3032
Di Marco GS, König M, Stock C et al (2013) High phosphate directly affects endothelial function by downregulating annexin II. Kidney Int 83:213–222
Shuto E, Taketani Y, Tanaka R, Harada N, Isshiki M, Sato M, Nashiki K, Amo K, Yamamoto H, Higashi Y, Nakaya Y, Takeda E (2009) Dietary phosphorus acutely impairs endothelial function. J Am Soc Nephrol 20:1504–1512
Jansen F, Li Q, Pfeifer A, Werner N (2017) Endothelial- and immune cell-derived extracellular vesicles in the regulation of cardiovascular health and disease. JACC Basic Transl Sci 2:790–807
Ketteler M, Rothe H, Krüger T et al (2011) Mechanisms and treatment of extraosseous calcification in chronic kidney disease. Nat Rev Nephrol 7:509–516
Jablonski KL, Chonchol M (2013) Vascular calcification in end-stage renal disease: vascular calcification in ESRD. Hemodial Int 17:S17–S21
Vervloet M, Cozzolino M (2017) Vascular calcification in chronic kidney disease: different bricks in the wall? Kidney Int 91:808–817
Hutcheson JD, Blaser MC, Aikawa E (2017) Giving calcification its due: recognition of a diverse disease: a first attempt to standardize the field. Circ Res 120:270–273
Schwarz U, Buzello M, Ritz E et al (2000) Morphology of coronary atherosclerotic lesions in patients with end-stage renal failure. Nephrol Dial Transplant 15:218–223
Gross M-L, Meyer H-P, Ziebart H et al (2007) Calcification of coronary intima and media: immunohistochemistry, backscatter imaging, and X-ray analysis in renal and nonrenal patients. Clin J Am Soc Nephrol 2:121–134
Jono S, McKee MD, Murry CE et al (2000) Phosphate regulation of vascular smooth muscle cell calcification. Circ Res 87:E10–E17
Fakhry M, Skafi N, Fayyad-Kazan M, Kobeissy F, Hamade E, Mebarek S, Habib A, Borghol N, Zeidan A, Magne D, Fayyad-Kazan H, Badran B (2018) Characterization and assessment of potential microRNAs involved in phosphate-induced aortic calcification. J Cell Physiol 233:4056–4067
Durham AL, Speer MY, Scatena M, Giachelli CM, Shanahan CM (2018) Role of smooth muscle cells in vascular calcification: implications in atherosclerosis and arterial stiffness. Cardiovasc Res 114:590–600
Stojanović SD, Fiedler J, Bauersachs J, Thum T, Sedding DG (2020) Senescence-induced inflammation: an important player and key therapeutic target in atherosclerosis. Eur Heart J 41:2983–2996
Dai L, Schurgers LJ, Shiels PG, Stenvinkel P (2020) Early vascular ageing in chronic kidney disease: impact of inflammation, vitamin K, senescence and genomic damage. Nephrol Dial Transplant 35:ii31–ii37
Cui L, Houston DA, Farquharson C, MacRae VE (2016) Characterisation of matrix vesicles in skeletal and soft tissue mineralisation. Bone 87:147–158
Aikawa E (2016) Extracellular vesicles in cardiovascular disease: focus on vascular calcification: editorial. J Physiol 594:2877–2880
Kapustin AN, Chatrou MLL, Drozdov I, Zheng Y, Davidson SM, Soong D, Furmanik M, Sanchis P, de Rosales RTM, Alvarez-Hernandez D, Shroff R, Yin X, Muller K, Skepper JN, Mayr M, Reutelingsperger CP, Chester A, Bertazzo S, Schurgers LJ, Shanahan CM (2015) Vascular smooth muscle cell calcification is mediated by regulated exosome secretion. Circ Res 116:1312–1323
Moe S, Drüeke T, Cunningham J et al (2006) Definition, evaluation, and classification of renal osteodystrophy: a position statement from Kidney Disease: Improving Global Outcomes (KDIGO). Kidney Int 69:1945–1953
Sheen CR, Kuss P, Narisawa S, Yadav MC, Nigro J, Wang W, Chhea TN, Sergienko EA, Kapoor K, Jackson MR, Hoylaerts MF, Pinkerton AB, O'Neill WC, Millán JL (2015) Pathophysiological role of vascular smooth muscle alkaline phosphatase in medial artery calcification: role of TNAP in medial vascular calcification. J Bone Miner Res 30:824–836
Savinov AY, Salehi M, Yadav MC et al (2015) Transgenic overexpression of tissue-nonspecific alkaline phosphatase (TNAP) in vascular endothelium results in generalized arterial calcification. J Am Heart Assoc 4. https://doi.org/10.1161/JAHA.115.002499
Rocha LA, Higa A, Barreto FC et al (2006) Variant of adynamic bone disease in hemodialysis patients: fact or fiction? Am J Kidney Dis 48:430–436
London GM (2004) Arterial calcifications and bone histomorphometry in end-stage renal disease. J Am Soc Nephrol 15:1943–1951
Tomiyama C, Carvalho AB, Higa A et al (2010) Coronary calcification is associated with lower bone formation rate in CKD patients not yet in dialysis treatment. J Bone Miner Res 25:499–504
Ravani P, Malberti F, Tripepi G et al (2009) Vitamin D levels and patient outcome in chronic kidney disease. Kidney Int 75:88–95
de Boer IH, Kestenbaum B, Shoben AB, Michos ED, Sarnak MJ, Siscovick DS (2009) 25-Hydroxyvitamin D levels inversely associate with risk for developing coronary artery calcification. J Am Soc Nephrol 20:1805–1812
Hou Y-C, Liu W-C, Zheng C-M et al (2017) Role of vitamin D in uremic vascular calcification. Biomed Res Int 2017:1–13
Drissi H (2002) 1,25-(OH)2-vitamin D3 suppresses the bone-related Runx2/Cbfa1 gene promoter. Exp Cell Res 274:323–333
Reynolds JL (2004) Human vascular smooth muscle cells undergo vesicle-mediated calcification in response to changes in extracellular calcium and phosphate concentrations: a potential mechanism for accelerated vascular calcification in ESRD. J Am Soc Nephrol 15:2857–2867
Peacock M (2010) Calcium metabolism in health and disease. Clin J Am Soc Nephrol 5:S23–S30
Lioufas N, Toussaint ND, Pedagogos E, Elder G, Badve SV, Pascoe E, Valks A, Hawley C, IMPROVE-CKD Writing Committee (2019) Can we IMPROVE cardiovascular outcomes through phosphate lowering in CKD? Rationale and protocol for the IMpact of Phosphate Reduction On Vascular End-points in Chronic Kidney Disease (IMPROVE-CKD) study. BMJ Open 9:e024382. https://doi.org/10.1136/bmjopen-2018-024382
Patel L, Bernard LM, Elder GJ (2016) Sevelamer versus calcium-based binders for treatment of hyperphosphatemia in CKD: a meta-analysis of randomized controlled trials. Clin J Am Soc Nephrol 11:232–244
Ruospo M, Palmer SC, Natale P et al (2018) Phosphate binders for preventing and treating chronic kidney disease-mineral and bone disorder (CKD-MBD). Cochrane Database Syst Rev. https://doi.org/10.1002/14651858.CD006023.pub3
Block GA, Wheeler DC, Persky MS et al (2012) Effects of phosphate binders in moderate CKD. J Am Soc Nephrol 23:1407–1415
Seifert ME, de las Fuentes L, Rothstein M et al (2013) Effects of phosphate binder therapy on vascular stiffness in early-stage chronic kidney disease. Am J Nephrol 38:158–167
Ix JH, Isakova T, Larive B et al (2019) Effects of nicotinamide and lanthanum carbonate on serum phosphate and fibroblast growth factor-23 in CKD: the COMBINE trial. J Am Soc Nephrol 30:1096–1108
Ciceri P, Falleni M, Tosi D, Martinelli C, Bulfamante G, Block GA, Messa P, Cozzolino M (2019) High-phosphate induced vascular calcification is reduced by iron citrate through inhibition of extracellular matrix osteo-chondrogenic shift in VSMCs. Int J CardiolS0167527319323873 297:94–103
Toussaint ND, Pedagogos E, Lioufas NM, Elder GJ, Pascoe EM, Badve SV, Valks A, Block GA, Boudville N, Cameron JD, Campbell KL, Chen SSM, Faull RJ, Holt SG, Jackson D, Jardine MJ, Johnson DW, Kerr PG, Lau KK, Hooi LS, Narayan O, Perkovic V, Polkinghorne KR, Pollock CA, Reidlinger D, Robison L, Smith ER, Walker RJ, Wang AYM, Hawley CM, IMPROVE-CKD Trial Investigators (2020) A randomized trial on the effect of phosphate reduction on vascular end points in CKD (IMPROVE-CKD). J Am Soc Nephrol 31:2653–2666
Molnar AO, Biyani M, Hammond I et al (2017) Lower serum magnesium is associated with vascular calcification in peritoneal dialysis patients: a cross sectional study. BMC Nephrol 18:129
Ishimura E, Okuno S, Kitatani K, Tsuchida T, Yamakawa T, Shioi A, Inaba M, Nishizawa Y (2007) Significant association between the presence of peripheral vascular calcification and lower serum magnesium in hemodialysis patients. Clin Nephrol 68:222–227
Bai Y, Zhang J, Xu J et al (2015) Magnesium prevents β-glycerophosphate-induced calcification in rat aortic vascular smooth muscle cells. Biomed Rep 3:593–597
ter Braake AD, Tinnemans PT, Shanahan CM et al (2018) Magnesium prevents vascular calcification in vitro by inhibition of hydroxyapatite crystal formation. Sci Rep 8:2069
Montezano AC, Zimmerman D, Yusuf H et al (2010) Vascular smooth muscle cell differentiation to an osteogenic phenotype involves TRPM7 modulation by magnesium. Hypertension 56:453–462
Kircelli F, Peter ME, Sevinc Ok E, Celenk FG, Yilmaz M, Steppan S, Asci G, Ok E, Passlick-Deetjen J (2012) Magnesium reduces calcification in bovine vascular smooth muscle cells in a dose-dependent manner. Nephrol Dial Transplant 27:514–521
Viegas C, Araújo N, Marreiros C, Simes D (2019) The interplay between mineral metabolism, vascular calcification and inflammation in Chronic Kidney Disease (CKD): challenging old concepts with new facts. Aging 11:4274–4299
Schurgers LJ, Uitto J, Reutelingsperger CP (2013) Vitamin K-dependent carboxylation of matrix Gla-protein: a crucial switch to control ectopic mineralization. Trends Mol Med 19:217–226
Aghagolzadeh P, Bachtler M, Bijarnia R et al (2016) Calcification of vascular smooth muscle cells is induced by secondary calciprotein particles and enhanced by tumor necrosis factor-α. Atherosclerosis 251:404–414
Choi SR, Lee Y-K, Cho AJ et al (2019) Malnutrition, inflammation, progression of vascular calcification and survival: inter-relationships in hemodialysis patients. PLoS One 14:e0216415
Ketteler M, Bongartz P, Westenfeld R et al (2003) Association of low fetuin-a (AHSG) concentrations in serum with cardiovascular mortality in patients on dialysis: a cross-sectional study. Lancet 361:827–833
Yamada S, Tokumoto M, Tsuruya K, Tatsumoto N, Noguchi H, Kitazono T, Ooboshi H (2015) Fetuin-A decrease induced by a low-protein diet enhances vascular calcification in uremic rats with hyperphosphatemia. Am J Physiol-Ren Physiol 309:F744–F754
Kuro-o M, Matsumura Y, Aizawa H et al (1997) Mutation of the mouse klotho gene leads to a syndrome resembling ageing. Nature 390:45–51
Hu MC, Shi M, Zhang J et al (2011) Klotho deficiency causes vascular calcification in chronic kidney disease. J Am Soc Nephrol 22:124–136
Kim HR, Nam BY, Kim DW et al (2013) Circulating α-Klotho levels in CKD and relationship to progression. Am J Kidney Dis 61:899–909
Hu M-C, Shi M, Zhang J, Quiñones H, Kuro-o M, Moe OW (2010) Klotho deficiency is an early biomarker of renal ischemia–reperfusion injury and its replacement is protective. Kidney Int 78:1240–1251
Hu MC, Shi M, Zhang J, Pastor J, Nakatani T, Lanske B, Razzaque MS, Rosenblatt KP, Baum MG, Kuro-O M, Moe OW (2010) Klotho: a novel phosphaturic substance acting as an autocrine enzyme in the renal proximal tubule. FASEB J 24:3438–3450
Lu X, Hu MC (2017) Klotho/FGF23 axis in chronic kidney disease and cardiovascular disease. Kidney Dis 3:15–23
on behalf of the European Uremic Toxin (EUTox) Work Group, Desjardins L, Liabeuf S et al (2012) FGF23 is independently associated with vascular calcification but not bone mineral density in patients at various CKD stages. Osteoporos Int 23:2017–2025
Shalhoub V, Shatzen EM, Ward SC, Davis J, Stevens J, Bi V, Renshaw L, Hawkins N, Wang W, Chen C, Tsai MM, Cattley RC, Wronski TJ, Xia X, Li X, Henley C, Eschenberg M, Richards WG (2012) FGF23 neutralization improves chronic kidney disease–associated hyperparathyroidism yet increases mortality. J Clin Invest 122:2543–2553
Fischer SS, Kempe DS, Leibrock CB, Rexhepaj R, Siraskar B, Boini KM, Ackermann TF, Föller M, Hocher B, Rosenblatt KP, Kuro-o M, Lang F (2010) Hyperaldosteronism in Klotho-deficient mice. Am J Physiol-Ren Physiol 299:F1171–F1177
Hené RJ, Boer P, Koomans HA, Dorhout Mees EJ (1982) Plasma aldosterone concentrations in chronic renal disease. Kidney Int 21:98–101
Jaffe IZ, Mendelsohn ME (2005) Angiotensin II and aldosterone regulate gene transcription via functional mineralocortocoid receptors in human coronary artery smooth muscle cells. Circ Res 96:643–650
Voelkl J, Alesutan I, Leibrock CB et al (2013) Spironolactone ameliorates PIT1-dependent vascular osteoinduction in klotho-hypomorphic mice. J Clin Invest:JCI64093. https://doi.org/10.1172/JCI64093
Cozzolino M, Mangano M, Galassi A et al (2019) Vitamin K in chronic kidney disease. Nutrients 11:168
Cranenburg ECM, Schurgers LJ, Uiterwijk HH et al (2012) Vitamin K intake and status are low in hemodialysis patients. Kidney Int 82:605–610
Viegas CSB, Costa RM, Santos L et al (2017) Gla-rich protein function as an anti-inflammatory agent in monocytes/macrophages: implications for calcification-related chronic inflammatory diseases. PLoS One 12:e0177829
Viegas CSB, Rafael MS, Enriquez JL et al (2015) Gla-rich protein acts as a calcification inhibitor in the human cardiovascular system. Arterioscler Thromb Vasc Biol 35:399–408
Krueger T, Schlieper G, Schurgers L et al (2014) Vitamin K1 to slow vascular calcification in haemodialysis patients (VitaVasK trial): a rationale and study protocol. Nephrol Dial Transplant 29:1633–1638
Luo G, Ducy P, McKee MD et al (1997) Spontaneous calcification of arteries and cartilage in mice lacking matrix GLA protein. Nature 386:78–81
Willems BA, Furmanik M, Caron MMJ et al (2018) Ucma/GRP inhibits phosphate-induced vascular smooth muscle cell calcification via SMAD-dependent BMP signalling. Sci Rep 8:4961
Nigwekar SU, Thadhani R, Brandenburg VM (2018) Calciphylaxis. N Engl J Med 378:1704–1714
Witham MD, Lees JS, White M et al (2020) Vitamin K supplementation to improve vascular stiffness in CKD: the K4Kidneys randomized controlled trial. J Am Soc Nephrol 31:2434–2445
Holden RM, Booth SL, Day AG et al (2015) Inhibiting the progression of arterial calcification with vitamin K in HemoDialysis patients (iPACK-HD) trial: rationale and study design for a randomized trial of vitamin K in patients with end stage kidney disease. Can J Kidney Health Dis 2:53
Krüger T, Floege J (2014) Vitamin K antagonists: beyond bleeding. Semin Dial 27:37–41
Price PA, Faus SA, Williamson MK (1998) Warfarin causes rapid calcification of the elastic lamellae in rat arteries and heart valves. Arterioscler Thromb Vasc Biol 18:1400–1407
Krüger T, Oelenberg S, Kaesler N et al (2013) Warfarin induces cardiovascular damage in mice. Arterioscler Thromb Vasc Biol 33:2618–2624
Zappulli V, Friis KP, Fitzpatrick Z et al (2016) Extracellular vesicles and intercellular communication within the nervous system. J Clin Invest 126:1198–1207
Colombo M, Raposo G, Théry C (2014) Biogenesis, secretion, and intercellular interactions of Exosomes and other extracellular vesicles. Annu Rev Cell Dev Biol 30:255–289
Horstman LL, Jy W, Jimenez JJ, Bidot C, Ahn YS (2004) New horizons in the analysis of circulating cell-derived microparticles. Keio J Med 53:210–230
Loyer X, Vion A-C, Tedgui A, Boulanger CM (2014) Microvesicles as cell–cell messengers in cardiovascular diseases. Circ Res 114:345–353
Mesri M, Altieri DC (1998) Endothelial cell activation by leukocyte microparticles. J Immunol 161:4382
Mack M, Kleinschmidt A, Brühl H, Klier C, Nelson PJ, Cihak J, Plachý J, Stangassinger M, Erfle V, Schlöndorff D (2000) Transfer of the chemokine receptor CCR5 between cells by membrane-derived microparticles: a mechanism for cellular human immunodeficiency virus 1 infection. Nat Med 6:769–775
Rectenwald JE, Myers DD, Hawley AE et al (2005) D-dimer, P-selectin, and microparticles: novel markers to predict deep venous thrombosis: a pilot study. Thromb Haemost 94:1312–1317
Burnier L, Fontana P, Kwak BR, Angelillo-Scherrer A (2009) Cell-derived microparticles in haemostasis and vascular medicine. Thromb Haemost 101:439–451
Diamant M, Tushuizen ME, Sturk A, Nieuwland R (2004) Cellular microparticles: new players in the field of vascular disease? Eur J Clin Investig 34:392–401
Bernal-Mizrachi L, Jy W, Fierro C, Macdonough R, Velazques HA, Purow J, Jimenez JJ, Horstman LL, Ferreira A, de Marchena E, Ahn YS (2004) Endothelial microparticles correlate with high-risk angiographic lesions in acute coronary syndromes. Int J Cardiol 97:439–446
Boulanger CM, Amabile N, Guérin AP et al (2007) In vivo shear stress determines circulating levels of endothelial microparticles in end-stage renal disease. Hypertension 49:902–908
Jansen F, Yang X, Hoelscher M, Cattelan A, Schmitz T, Proebsting S, Wenzel D, Vosen S, Franklin BS, Fleischmann BK, Nickenig G, Werner N (2013) Endothelial microparticle–mediated transfer of MicroRNA-126 promotes vascular endothelial cell repair via SPRED1 and is abrogated in glucose-damaged endothelial microparticles. Circulation 128:2026–2038
Faure V, Dou L, Sabatier F et al (2006) Elevation of circulating endothelial microparticles in patients with chronic renal failure. J Thromb Haemost 4:566–573
Burton JO, Hamali HA, Singh R et al (2013) Elevated levels of procoagulant plasma microvesicles in dialysis patients. PLoS One 8:e72663
Carmona A, Guerrero F, Buendia P et al (2017) Microvesicles derived from indoxyl sulfate treated endothelial cells induce endothelial progenitor cells dysfunction. Front Physiol 8:666
Amabile N, Guerin AP, Tedgui A, Boulanger CM, London GM (2012) Predictive value of circulating endothelial microparticles for cardiovascular mortality in end-stage renal failure: a pilot study. Nephrol Dial Transplant 27:1873–1880
Amabile N, Guérin AP, Leroyer A, Mallat Z, Nguyen C, Boddaert J, London GM, Tedgui A, Boulanger CM (2005) Circulating endothelial microparticles are associated with vascular dysfunction in patients with end-stage renal failure. J Am Soc Nephrol 16:3381–3388
Kapustin AN, Shanahan CM (2012) Calcium regulation of vascular smooth muscle cell–derived matrix vesicles. Trends Cardiovasc Med 22:133–137
Viegas CSB, Santos L, Macedo AL, Matos AA, Silva AP, Neves PL, Staes A, Gevaert K, Morais R, Vermeer C, Schurgers L, Simes DC (2018) Chronic kidney disease circulating calciprotein particles and extracellular vesicles promote vascular calcification: a role for GRP (Gla-rich protein). Arterioscler Thromb Vasc Biol 38:575–587
EL Andaloussi S, Mäger I, Breakefield XO, Wood MJA (2013) Extracellular vesicles: biology and emerging therapeutic opportunities. Nat Rev Drug Discov 12:347–357
Yang D, Tan S, Yang Z, Jiang P, Qin C, Yuan Q, Dang R, Yao X, Qu J, Lu Q, Xu P, Zhang B, Xiang D, Chen L (2018) Dihydromyricetin attenuates TNF- α -induced endothelial dysfunction through miR-21-mediated DDAH1/ADMA/NO signal pathway. Biomed Res Int 2018:1–12
Abbasian N, Herbert KE, Pawluczyk I et al (2018) Vesicles bearing gifts: the functional importance of micro-RNA transfer in extracellular vesicles in chronic kidney disease. Am J Physiol-Ren Physiol 315:F1430–F1443
Liu Y, Li Q, Hosen MR, Zietzer A, Flender A, Levermann P, Schmitz T, Frühwald D, Goody P, Nickenig G, Werner N, Jansen F (2019) Atherosclerotic conditions promote the packaging of functional MicroRNA-92a-3p into endothelial microvesicles. Circ Res 124:575–587
Kim H, Bae Y-U, Jeon JS, Noh H, Park HK, Byun DW, Han DC, Ryu S, Kwon SH (2019) The circulating exosomal microRNAs related to albuminuria in patients with diabetic nephropathy. J Transl Med 17:236
Muralidharan J, Ramezani A, Hubal M et al (2017) Extracellular microRNA signature in chronic kidney disease. Am J Physiol-Ren Physiol 312:F982–F991
Zietzer A, Steffen E, Niepmann S et al (2020) MicroRNA-mediated vascular intercellular communication is altered in chronic kidney disease. Cardiovasc Res:cvaa322. https://doi.org/10.1093/cvr/cvaa322
Shang F, Wang S-C, Hsu C-Y, Miao Y, Martin M, Yin Y, Wu CC, Wang YT, Wu G, Chien S, Huang HD, Tarng DC, Shiu YT, Cheung AK, Huang PH, Chen Z, Shyy JYJ (2017) MicroRNA-92a mediates endothelial dysfunction in CKD. J Am Soc Nephrol 28:3251–3261
Wiese CB, Zhong J, Xu Z-Q, Zhang Y, Ramirez Solano MA, Zhu W, Linton MRF, Sheng Q, Kon V, Vickers KC (2019) Dual inhibition of endothelial miR-92a-3p and miR-489-3p reduces renal injury-associated atherosclerosis. Atherosclerosis 282:121–131
Kétszeri M, Kirsch A, Frauscher B, Moschovaki-Filippidou F, Mooslechner AA, Kirsch AH, Schabhuettl C, Aringer I, Artinger K, Pregartner G, Ekart R, Breznik S, Hojs R, Goessler W, Schilcher I, Müller H, Obermayer-Pietsch B, Frank S, Rosenkranz AR, Eller P, Eller K (2019) MicroRNA-142-3p improves vascular relaxation in uremia. Atherosclerosis 280:28–36
Leopold J (2014) MicroRNAs regulate vascular medial calcification. Cells 3:963–980
Liao X-B, Zhang Z-Y, Yuan K et al (2013) MiR-133a modulates osteogenic differentiation of vascular smooth muscle cells. Endocrinology 154:3344–3352
Cui R-R, Li S-J, Liu L-J et al (2012) MicroRNA-204 regulates vascular smooth muscle cell calcification in vitro and in vivo. Cardiovasc Res 96:320–329
Qiao W, Chen L, Zhang M (2014) MicroRNA-205 regulates the calcification and osteoblastic differentiation of vascular smooth muscle cells. Cell Physiol Biochem 33:1945–1953
Liu J, Xiao X, Shen Y et al (2017) MicroRNA-32 promotes calcification in vascular smooth muscle cells: implications as a novel marker for coronary artery calcification. PLoS One 12:e0174138
Balderman JAF, Lee H, Mahoney CE et al (2012) Bone morphogenetic protein-2 decreases microRNA-30b and microRNA-30c to promote vascular smooth muscle cell calcification. J Am Heart Assoc 1:e003905
Xu T, Qiu X, Sheng Z et al (2019) Restoration of microRNA-30b expression alleviates vascular calcification through the mTOR signaling pathway and autophagy. J Cell Physiol 234:14306–14318
Goettsch C, Rauner M, Pacyna N, Hempel U, Bornstein SR, Hofbauer LC (2011) miR-125b regulates calcification of vascular smooth muscle cells. Am J Pathol 179:1594–1600
Chao C-T, Liu Y-P, Su S-F, Yeh HY, Chen HY, Lee PJ, Chen WJ, Lee YM, Huang JW, Chiang CK, Hung KY, Chen HW (2017) Circulating microRNA-125b predicts the presence and progression of uremic vascular calcification. Arterioscler Thromb Vasc Biol 37:1402–1414
Chen NX, Kiattisunthorn K, O’Neill KD et al (2013) Decreased microRNA is involved in the vascular remodeling abnormalities in chronic kidney disease (CKD). PLoS One 8:e64558. https://doi.org/10.1371/journal.pone.0064558
Chao C, Yuan T, Yeh H et al (2019) Risk factors associated with altered circulating microRNA-125b and their influences on uremic vascular calcification among patients with end-stage renal disease. J Am Heart Assoc 8. https://doi.org/10.1161/JAHA.118.010805
Gui T, Zhou G, Sun Y, Shimokado A, Itoh S, Oikawa K, Muragaki Y (2012) MicroRNAs that target Ca2+ transporters are involved in vascular smooth muscle cell calcification. Lab Investig 92:1250–1259
Pan W, Liang J, Tang H, Fang X, Wang F, Ding Y, Huang H, Zhang H (2020) Differentially expressed microRNA profiles in exosomes from vascular smooth muscle cells associated with coronary artery calcification. Int J Biochem Cell Biol 118:105645
Code availability
Not applicable.
Funding
Open Access funding enabled and organized by Projekt DEAL. This work was supported by the medical faculty of the University of Bonn [BONFOR to P.D. and A.Z., by the German Cardiac Society [DGK16/2018 to A.Z.], the Deutsche Forschungsgemeinschaft [Project-ID 397484323 – TRR 259 to F.J. and G.N.). FJ was further supported by the DFG (JA-2351/2-1) and the Corona foundation. C.K. was supported by the Deutsche Forschungsgemeinschaft (SFBTRR259 A01, SFBTR57 P10, SFB1192 A8, GRK2168 272482170, EXC2151 390873048).
Author information
Authors and Affiliations
Contributions
P.D., A.Z., and F.J. wrote the manuscript. F.J. and A.Z. had the idea for the article. P.D. performed the literature search. G.N., C.K., M.R.H, and P.R.G critically revised the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Düsing, P., Zietzer, A., Goody, P.R. et al. Vascular pathologies in chronic kidney disease: pathophysiological mechanisms and novel therapeutic approaches. J Mol Med 99, 335–348 (2021). https://doi.org/10.1007/s00109-021-02037-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00109-021-02037-7