
Abstract
Breeding for malting quality is an important goal of malting barley breeding programs. Malting quality is a complex phenotype that combines a large number of interrelated components, each of which shows complex inheritance. Currently, only a few genes involved in determining malting quality have been characterized. We combined transcript profiling with phenotypic correlations to identify candidate genes for malting quality. The Barley1 GeneChip® array containing 22,792 probe sets was used to conduct transcript profiling of genes expressed in several different stages of malting of four malting cultivars. Genes that were differentially expressed in comparisons between different malting stages relative to ungerminated seed, as well as in comparisons between malting cultivars in the same malting stage were identified. Correlation analysis of 723 differentially expressed genes with malting quality phenotypes showed that 11–102 of these genes correlated with six malting quality phenotypes. Genes involved in carbohydrate metabolism were among the positively correlated genes. Genes for protein and lipid metabolism, cell wall organization and biogenesis, and genes involved in stress and defense response also correlated with malting quality phenotypes. Expressed sequence tags (ESTs) were generated from a ‘malting-gene enriched’ cDNA library made by suppression subtractive hybridization between malted and ungerminated seeds of ‘Morex’. Eleven percent of the ESTs had no significant homology with sequences in the databases, suggesting that there may be other malting-related genes not represented in the barley gene chip array. The results provide candidate genes for malting quality phenotypes that need to be functionally validated.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Barley (Hordeum vulgare L.) is the fifth most important cereal crop in terms of total world production (FAOSTAT 2006; http://faostat.fao.org/site/370/default.aspx), and it is the major grain used for malting and brewing. Malting is seed germination under strictly controlled environmental conditions. During malting, the stored biopolymers in barley grains are converted into low molecular weight molecules that can be utilized by yeast during brewing (Jones 2005a). The malting process involves three stages: steeping, germination, and kilning (Briggs 1998). During steeping, the barley seed is soaked in water to reach predefined moisture content. During the germination phase, hydrolytic enzymes are synthesized by the aleurone cells (Bamforth and Barclay 1993) and scutellum (Macfadden et al. 1988; Ranki 1990). These hydrolytic enzymes are secreted into the starchy endosperm where they convert proteins and carbohydrates into partially degraded biopolymers (Fincher 1989). In kilning, the malted seed is dried by forcing air at 40–60°C and then the temperature is gradually increased up to 85–95°C (Briggs 1998). Other than the low molecular weight biopolymers, the malt also contains heat-stable hydrolytic enzymes required for further breakdown of carbohydrates in the subsequent mashing process (Potokina et al. 2004).
Understanding the genetic basis of malting quality is important in malt barley breeding. Malting quality is a complex phenotype that combines a large number of interrelated components, each of which shows complex inheritance (Hayes and Jones 2000). Genetic mapping of malting quality phenotypes resulted in more than 150 QTLs associated with 19 traits used to measure malting quality (Gao et al. 2004; Zale et al. 2000). Currently, only a few genes involved in determining malting quality have been characterized. These include genes coding for the starch degrading enzymes α-amylase (1,4-α-d-glucanohydrolase, EC 3.2.1.1), β-amylase (1,4-α–d-glucan malto-hydrolase, EC 3.2.1.2), α-glucosidase, and limit dextrinase (LD, EC 3.2.1.41) (for review, see Fincher 1989). However, the large number of QTLs associated with malting quality traits suggest that many other genes play important roles in malting.
Analysis of gene expression profiles using cDNA-based microarrays has proven useful for deciphering biochemical pathways involved in plant responses to stress and pathogens (Caillaud et al. 2007; Caldo et al. 2004; Venu et al. 2007). Differentially expressed genes during germination in barley were also studied using cDNA arrays (Potokina et al. 2002; Watson and Henry 2005). Using serial analysis of gene expression (SAGE) to conduct transcript profiling during malting in barley, the most abundant transcripts identified by (White et al. 2006) overlapped with most genes identified in cDNA array-based studies (Potokina et al. 2002; Watson and Henry 2005).
The use of cDNA-based arrays for identifying genes that are involved in malting was previously demonstrated (Potokina et al. 2004, 2006). Ten malting barley genotypes showing variation in six malting quality characteristics were compared for gene expression during malting using a cDNA array containing 1400 barley ESTs. The differentially expressed genes were then correlated with malting quality phenotypes (Potokina et al. 2004) resulting in the identification of 17–30 candidate genes for each of the malting parameters. A subsequent study further analyzed a candidate gene, serine carboxypeptidase I (Cxp1), by mapping the relative level of Cxp1 expression in a population made from a cross between the malting barley cultivar Morex and a feed barley cultivar, Steptoe (Potokina et al. 2006). An expression QTL mapped to the same region on chromosome 3H containing a QTL for a malting quality phenotype, diastatic power. A single nucleotide polymorphism (SNP) for Cxp1 was further correlated with phenotypic variation in diastatic power.
This study was conducted to identify genes that are involved in determining malting quality phenotypes. A barley microarray containing more than 22,792 probe sets representing approximately 21,439 barley genes has been developed (Close et al. 2004). We used the barley gene chip array to conduct transcript profiling of genes that are differentially expressed during malting in barley and then correlated differentially expressed genes with malting quality phenotypes. Furthermore, we generated ESTs from a suppression-subtractive hybridization cDNA library made from malted seeds of the malting cultivar ‘Morex’.
Materials and methods
Plant material, experimental design, and micromalting conditions
Four North American spring type malting barley cultivars that have different malting quality phenotypes and represent 2-row and 6-row varieties were used. The 2-row varieties were ‘Harrington’ (Klages/7211) and ‘Merit’ (Manley/2B80-350), and the 6-row varieties were ‘Legacy’ (6B86-3517/ Excel), and ‘Morex’ (Cree/Bonanza). All four lines were grown in Idaho Falls during the summer of 2003 in standard plots (1.5 × 4 m) consisting of three replicates in randomized complete block trials optimized for nearest neighbor analysis. Harrington and Merit were grown in one block with several other two-row spring barleys while Morex and Legacy were grown in an adjacent block with several other six-row barleys. The plots were harvested with a plot combine and a sub-sample of 300 g of grain was retained from each plot. Sub-samples of each replicate of each variety were blended together in equal proportions to form a representative sample of about 900 g. The blended grain samples were subsequently cleaned over an A/S Rationel Kornservice model SN4 cleaner to remove thin kernels (by passing through a 6/64th slotted screen). The cleaned blended samples were used for the replicate experiments in this study. By blending samples from replicate plots, field effects should therefore be minimal. Fifty to 100 g seeds of each variety were placed inside a steel container and arranged at random inside the cabinet of a Joe White micromalter located at Busch Agricultural Resources, LLC, Fort Collins, CO. The micromalting conditions were as follows: steeping at 12°C for 37 h, germination at 12°C for 5 h, 17°C for 65 h, 18°C for 23 h, and kilning stages consisting of 55°C for 10 h, 60°C for 4 h, 68°C for 3 h, 80°C for 2 h and 90°C for 3 h. Twenty gram samples of micromalted seeds were collected at four stages for Morex and two stages for Legacy, Harrington, and Merit (Table 1). The four malting stages for Morex were: steeping, 24 h germination (day 1), 93 h germination (day 4), and finished malt after kilning was completed (Fig. 1). Two 20 g samples of un-germinated dry seeds of Morex from the same batch of seeds were used as control. For the three other cultivars, collection was done at day 1 and day 4. Tissues were immediately frozen in liquid nitrogen for RNA extraction.

Barley seeds of cultivar ‘Morex’ at different malting stages analyzed in this study: dry seed, steeped for 37 h, day 1 (germination for 24 h), day 4 (germination for 93 h), and kilning (see text for details of malting conditions)
RNA extraction, microarray experiments and data analysis
Approximately 100 mg of frozen, ground tissue from each sample was used for RNA extraction. Total RNA was extracted using Qiagen RNeasy Plant Mini Kit with RNase Free/DNase (Qiagen, Valencia, CA) following the manufacturer’s instructions. Integrity and quantity of the RNA was tested using Bio-Rad Experion RNA StdSen Chips (Bio-Rad, Hercules, CA). The RNA samples were sent to the University of California at Irvine or the Virginia Bioinformatics Institute Core facility, where additional quality control was performed. These facilities then performed RNA labeling and hybridization to the Affymetrix 22 K Barley1 GeneChip according to the manufacturer’s protocols (http://www.affymetrix.com).
Following hybridization to the gene chip arrays, different quality control checks were performed including inspection of hybridized images, boxplots and histograms of log2(PM) values, and examination of hybridization and PolyA controls. GeneChips with poor quality were discarded from the analysis. Twenty-two arrays were analyzed with two biological replications per cultivar per time point, with the exception of Merit day 4 with three replications and Harrington day 1, where one of the replicates was discarded (Table 1). All microarray data have been deposited in NCBI’s Gene Expression Omnibus (Edgar et al. 2002) and are accessible through GEO Series accession number GSE11200 (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE11200) (Table 1).
Data analysis was carried out using Bioconductor in R (Gentleman et al. 2004). Data preprocessing and summarization were performed using Robust Multichip Average (RMA) (Irizarry et al. 2003). Statistical tests of differential expression were conducted using the moderated t test through the limma (Linear Models for Microarrays) package in Bioconductor. The Benjamini-Hochberg multiple testing adjustment was applied in order to control the comparison-wise false discovery rate (Benjamini and Hochberg 1995).
Two sets of analyses were performed (Table 1). First, expression values in four malting stages in Morex cultivar (steeping, day 1, day 4, and kilned) were compared to the ungerminated, dry seed (non-malting control) stage. Second, expression values were compared during day 1 and day 4 among the four cultivars Morex, Legacy, Harrington, and Merit.
For the Morex time course experiment, 5,579 probe sets that were called “Absent” on every Morex array were removed from further analysis. For the cultivars comparison, a comparison of 2-row cultivars versus 6-row cultivars and all pair-wise cultivar comparisons were performed for day 1 and day 4 samples separately. We note that since there was only a single Harrington replicate for day 1 we did not consider any pair-wise cultivar comparisons involving Harrington for day 1. Five thousand two hundred sixty probe sets that were called “Absent” on every array (in the cultivars experiment) were removed from further analysis. For both analyses, genes were considered to be differentially expressed if the probe sets had an absolute value of log2 fold change |log2(FC)| > 1 and adjusted P-values of ≤0.05.
The probe sets identified as differentially expressed were annotated using HarvEST:Barley (Version 1.65) assembly 25 (http://www.harvest-web.org/). For gene ontology (GO), we used the Arabidopsis gene model for each probe set and used the program PLAN (He et al. 2007) to obtain functional categories. GO annotation was obtained for the top significant hit (using an e-value cutoff of 1e-4) for each probe set.
Malting quality phenotypes
To mitigate bias of environmental effects from a single location, we used data from a relatively large number of field experiments. Nine malting quality traits routinely used for screening breeding materials at Busch Agricultural Resources were initially considered. The data for malting quality phenotypes included phenotypic measurements of individual malting traits from as many as 49 independent field experiments (Table 2). These trials were conducted from 2001 to 2006 over a wide range of geography and different farming practices, such as irrigated versus dry land. For a given trait, the analysis was restricted to those field experiments for which all four cultivars were represented. A two-way analysis of variance (ANOVA) including a random experiment effect was calculated for each of the nine traits (Table 2). Only wort protein did not have a statistically significant cultivar effect (at P ≤ 0.05 level) and it was excluded from further analysis.
Broad-sense heritability (H2) estimates for some of these traits have been reported (Foster et al. 1967; Hockett and Nilan 1985; Therrien 2006) and there is a wide range of values obtained in different studies, as well as different sets of environments within a study. Because we used a relatively large set of paired phenotypic data over a range of locations and years as the best unbiased estimate of the true phenotypic response of the four varieties for the respective malting traits the distorting effects of individual locations and years should have been minimized in our analysis.
A short description of the nine malting quality phenotypes follows. α-Amylase is a measure of the units of enzyme activity to break down a known standardized branched dextrin into smaller linear pieces by internal cleavage in a given amount of time at a constant temperature and pH 9. Beta glucan is a measure of the degree of cell wall modification and indicates the enzyme activity level of beta-glucanase, as measured by the ability of a cold water extract of malt to digest a known standardized beta glucan substrate using a Skalar instrument. Barley protein is a measure of the percent protein found in the grain. It is measured indirectly using near-infrared technology and is corrected to a standard dry moisture (13%) basis. Diastatic power refers to the starch-degrading capacity of malt, and represents the complementary actions of α-amylase and β-amylase on starch to produce sugars and low-molecular weight dextrins. Diastatic power is measured like α-amylase by incubating malt extract with a starch substrate, but the malt extract used as enzyme source is not heated leaving the more thermolabile β-amylase active. Percent fine extract refers to the concentration of solids when finely ground malt is mixed with water and the stirred mash is subjected to a time/temperature program that facilitates amylolysis, proteolysis and other enzyme reactions. Free amino nitrogen is a measure of the concentration of individual wort amino acids and small peptides which can be utilized by yeast for cell growth and proliferation. Malt protein is a measurement of total nitrogen on kilned finished malt. Soluble/total protein ratio, also known as Kolbach index, represents the ratio of soluble to total nitrogen. Wort protein, also called soluble protein, is a measure of percent soluble protein in the wort after hot water extraction of the malt. It is measured spectrophotometrically as set by a wet chemistry standard curve. Measurements for these malting quality traits are based on standard procedures described by the American Society of Brewing Chemists (ASBC 1992) and are discussed in previous reviews (Bamforth and Barclay 1993; Burger and LaBerge 1985).
Correlation between gene expression and malting quality phenotypes
A subset of genes that were differentially expressed in at least one malting stage versus dry seed and in at least one comparison between cultivars in day 1 and day 4 was correlated with the four malting quality phenotypes. The correlation between gene expression (taking day 1 and day 4 separately) and average phenotype (for each of the nine traits averaged of field experiments) was calculated. Correlation was calculated based on the four pairs of observations representing the expression and phenotypic data across the four cultivars. Calculation of correlation between gene expression and phenotype average, however, ignores experiment and environmental effects which are known to be present in the data. In order to take the variability due to field experiment into account, we performed an additional analysis where the correlation was calculated by gene and by field experiment. Genes that showed statistically significant correlation to average phenotype (P ≤ 0.01) and strong correlation (absolute value of correlation ≥0.7) for at least half of the individual field experiments for a phenotype were identified.
Quantitative reverse transcription-PCR (qRT-PCR)
Primer pairs for each target gene were based on specific barley sequences from the 22 K Barley1 Affymetrix GeneChip probe array (Affymetrix, Inc., Santa Clara, CA) and were designed online using Primer3 (Rozen and Skaletsky 1998). Four genes were selected for these experiments: α-amylase, α-glucosidase, limit dextrinase, and β-ketoacyl synthase. Primer sequences were as follows: α-amylase (contig3953) L: 5′-CGGCAATGACTATGCCGTAT-3′, R: 5′-GCATGTCCCTCATCCTCACT-3′; α-glucosidase (contig7937) L: 5′-AGCGATTGGTGCATGGTAAG-3′; R: 5′-AGTCCTCCTGCGTTCTGGTA-3′; limit dextrinase (contig11648) L: 5′-CGGTTTCAACACGAGGATCT-3′; R: 5′-ACTAGCAGCTTGGG CACTA-3′; and β-ketoacyl synthase: (contig6642_at) L: 5′-GAGGACACAACTCGGTGGTT-3′, R: 5′-GCAATGGATCTTGGATCCTC-3′. qRT- PCR was performed using the qPCR MasterMix Plus for SYBR(R) Green I with fluorescein (Eurogentec, Inc., San Diego, CA, USA) using 100 ng of cDNA and 10 pmol of both forward and reverse primers per 25 μl reaction. All qPCR reactions were carried out in triplicate. Amplification and fluorescence measurements were carried out on iCycler iQ Real-Time PCR Detection Instrument (Bio-Rad Laboratories, Hercules, CA) with the following thermocycling parameters: 2 min at 50°C and 10 min at 95°C, followed by 60 amplification cycles of 15 s at 95°C and 1 min at 58°C. A melting curve was constructed for the amplification product by increasing the temperature from 55°C by 0.5°C per cycle for 80 cycles. Standard curves were generated using a 10-fold dilution series of the Merit cDNA sample and used to determine the PCR efficiency for each target transcript amplification. The unregulated 18S rRNA transcript was selected as suitable endogenous control and used for normalization during relative quantification of target genes (Pfaffl 2001).
Construction of subtracted cDNA library from malted barley seeds and DNA sequencing
A cDNA library was constructed from malted seeds of cv. Morex using suppression subtractive hybridization (SSH) technique (Diatchenko et al. 1996). Total RNA was extracted from dry seed (control) and seed at day 4 micromalting (96 h at 20°C) as described above. cDNA populations were derived from the two RNA samples using the SMART cDNA synthesis system (BD Biosciences-Clontech, Palo Alto, CA). SSH was performed between these two cDNA populations designating the one derived from dry seed as “Driver” and the population derived from malted seed as “Tester”. Forward PCR-based subtraction was performed (Tester minus Driver) and the amplification products ligated into the pTAdvantage PCR cloning vector (BD Biosciences-Clontech, Palo Alto, CA). E. coli was transformed with this ligation reaction by electroporation. Aliquots of the plasmid library were plated on Luria Broth plates containing the appropriate antibiotic and grown overnight at 37°C. Seven hundred sixty-eight white clones were picked onto 96-well plates. DNA sequences for ~650 clones were determined by Sanger sequencing (Macrogen Inc., Maryland, USA and Washington University Genome Sequencing Center, St. Louis, MO).
Annotation of ESTs
Vector sequences, polyA and polyT stretches, were trimmed from the DNA sequences. Sequences less than 100 bp were discarded. Annotation of individual ESTs was performed using Basic Local Alignment Search Tool (BlastN and tBlastX) (Altschul et al. 1990) searches against the non-redundant protein sequences at NCBI (http://www.ncbi.nlm.nih.gov/blast). Best matches (E-value < 10−10) were used for putative gene assignments. Nucleotide BLAST searches against the EST database at NCBI was conducted for ESTs showing no significant hits with protein sequences. Functional categories were assigned on the basis of biological, functional and molecular annotations available from the gene ontology (GO) database (http://www.geneontology.org/). The sequences were deposited into the NCBI dbEST resource using NCBI sequence-submission protocols.
Results
Transcript profiles of different malting stages
The number of differentially expressed genes (i.e., probe sets with absolute value of log2 fold change >1 and adjusted P-values of ≤0.05) increased as barley seeds went from steeping to 24 h germination (day 1), 93 h germination (day 4), and kilning (Table 3). During steeping, when seeds imbibed water, 966 probe sets were up-regulated in comparison to dry seed. Categories of the genes based on biological processes assigned using the PLAN program (He et al. 2007) are shown in Table 3 (see Supplemental Tables S1 for expression data and S2 for list of genes under each category). Altogether, genes involved in metabolism of carbohydrates, nucleotides, amino acids, and proteins, made up 15% of the genes induced during this stage. Another large group of genes (~15%) were those involved in response to stimulus, including pathogens, chemicals, and abiotic stress. Following steeping, extensive reprogramming occurred as shown by the dramatic increase in numbers of induced genes in day 1 and day 4 malting (1,432 and 1,997 genes, respectively). Genes involved in metabolism of macromolecules (i.e., proteins, amino acids, nucleotides, carbohydrates, and lipids) continued to increase from steeping to day 4. Genes involved in transport more than doubled from steeping to day 4. The number of up-regulated genes decreased slightly during kilning (1466). At this stage when the seed was subjected to heat, the number of genes involved in metabolism of macromolecules decreased. Genes involved in cell organization and development decreased by more than half. In comparison, the percentage of genes involved in stress response slightly increased.
The number of down-regulated genes increased from steeping to kilning (Table 3). While up-regulated genes decreased from day 4 to kilning, down-regulated genes continued to increase during this transition. The kilning stage contained the largest number of down-regulated genes (2135). Down-regulated genes involved in metabolism of macromolecules increased from day 4 to kilning, reflecting the necessity to decrease or shut down these processes. The exception is carbohydrate metabolism where down-regulated genes did not increase from day 4 to kilning. The number of down regulated genes for stress response decreased from day 4 to kilning, indicating the possible need for induction of these genes. Genes for which there were no significant homology with DNA sequences in GenBank made up approximately 40–50% of the differentially expressed genes during the different stages of malting.
Differentially expressed genes among four malting barley cultivars
To determine the best stages to use for comparing gene expression during malting in different barley cultivars, we investigated the expression patterns of barley genes that had been previously associated with one or more malting quality phenotypes. The list included genes encoding starch degradation enzymes, cell wall degrading enzymes, protein hydrolytic enzymes, and proteinase inhibitors (Jones 2005a, b; Potokina et al. 2004). Seventy probe sets representing these genes were identified and hierarchical clustering was performed according to expression patterns in dry seed and the four malting stages of Morex (Supplemental Fig. S1). A majority of the genes were down-regulated in the dry seed and steeping stage. More than 90% of the probe sets were highly expressed during either or both day 1 and day 4 germination stages. Day 4 contained the largest number of up-regulated genes, consistent with the pattern for all differentially expressed genes in the different malting stages. Based on these results, we decided to use day 1 and day 4 germination stages to investigate expression patterns in three additional cultivars.
Pairwise comparisons of expression patterns between cultivars show that the largest numbers of differentially expressed genes were in comparisons between 2-row and 6-row cultivars (Fig. 2). In day 1, the largest number of differentially up-regulated genes was in the combined 2-row versus 6-row comparison, followed by Legacy (6-row) versus Merit (2-row), then Merit versus Morex (6-row) (see Supplemental Table S3 for list of genes). A small number of genes (~30) were differentially up-regulated between Legacy and Morex. A similar trend was observed for differentially down-regulated genes in day 1, with the exception that a greater number of differentially expressed genes were found between Merit and Morex compared to Legacy and Merit. Pairwise comparisons involving Harrington were not available in day 1 since there was only one replication. For day 4, the largest numbers of differentially up-regulated genes were found in comparisons between Harrington and the 6-row cultivars, Legacy and Morex (see Supplemental Table S4 for list of genes). This was followed by combined 2 row versus 6 row, and Harrington versus Merit and Merit versus Morex. The Legacy versus Morex comparison showed the least number of differentially expressed genes (~30). The pattern for down-regulated genes for day 4 was similar to that of up-regulated genes. In general, the lowest numbers of differentially expressed genes were found in comparisons between same row-class cultivars (Merit vs. Harrington and Legacy vs. Morex).

Number of differentially expressed genes in all possible pairwise comparisons between the four malting barley cultivars
Correlation of differentially expressed genes with malting quality phenotypes
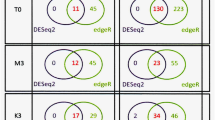
Genes that were differentially expressed compared to dry seed from the Morex time course experiment are potentially important in determining malting quality traits. However, many genes expressed during these stages are involved with growth and development and may not contribute directly to malting quality parameters. Similarly, many genes that were differentially expressed between the cultivars may potentially underlie malting quality trait differences among the cultivars. However, since the cultivars have different genetic backgrounds, many genes identified in these comparisons may not be directly related to the malting quality differences among the cultivars. In an attempt to identify genes that may be the most relevant to malting quality traits, we set the following criteria: the genes had to be differentially expressed in at least one of the four malting stages in the Morex time course experiment, as well as differentially expressed in the cultivars comparison for day 1 or day 4 (Fig. 3). There were a total of 723 genes that overlapped between Morex malting stages and at least one or both of the two malting stages for all cultivar comparisons (see Supplemental Table S5 for list of genes).

Venn diagram showing the overlap between differentially expressed genes in four malting stages of Morex, and differentially expressed genes in the cultivars-comparisons for day 1 and day 4. The broken-lined ellipse shows the 723 genes that overlapped between the Morex malting stages, day 1 cultivars-comparisons, and day 4 cultivars-comparisons. Expression levels of these 723 genes were used to correlate with malting quality phenotypes
We examined the correlation between the expression values of the 723 genes and the malting quality phenotypes using a cut-off value P ≤ 0.05, and requiring more than 50% of the sites to have correlation values ≥0.7. Correlations with six malting quality traits (α-amylase, diastatic power, free amino nitrogen, fine extract, malting protein, and soluble/total protein ratio) were detected (Table 4). Eleven to 72 genes showed positive correlation while 19–102 genes showed negative correlation with these traits. Representative genes correlated with the malting quality phenotypes are listed in Table 5 (see Supplemental Table S6 for complete list). The list includes genes involved in metabolism of carbohydrates, proteins, lipids, and amino acids, genes involved in transport, cell wall organization and biogenesis, as well as genes for stress and defense response. Genes for carbohydrate metabolism include genes that were previously associated with malting quality traits (Clark et al. 2003; Jones 2005a, b; Potokina et al. 2004). β-Glucosidase was positively correlated with diastatic power while limit dextrinase was positively correlated with fine extract. Other genes coding for carbohydrate metabolic enzymes, such as Sucrose synthase I and β-fructofuranidase I, were also positively correlated with diastatic power. On the other hand, many genes coding for enzymes involved in glycolysis negatively correlated with fine extract, diastatic power, soluble/total protein ratio, and free amino nitrogen. Lipid metabolic enzyme genes showing positive correlations with the malting quality traits included Lipoxygenase 2.1, several lipases, and oleosin. Protein inhibitor genes including α-amylase inhibitor, protein synthesis inhibitor I, and lipid transfer protein (LTP) showed negative correlation with many of the malting traits (Table 5). Other genes showing correlation with malting quality phenotypes are those involved in transport of proteins and macromolecules and genes involved in defense and stress response. Genes with unclear functions as well as genes with no significant homology were also identified.
Characterization of barley ESTs from malted seeds
To identify other barley genes that may be important in malting, ESTs were generated from a cDNA library made by suppression-subtractive hybridization between cDNA made from day 4 malted Morex seeds and cDNA from Morex dry seeds. Suppression-subtractive hybridization was performed to enrich for genes expressed during day 4 germination. A total of 509 ESTs were produced and grouped according to functional categories (Fig. 4 and Supplemental Table S7). Fifty-six percent of the genes were of unknown function. Many of these did not produce BLASTX hits with proteins. Significant homologies were only found with barley ESTs and may be barley-specific genes. Other genes in this group showed homology to hypothetical proteins or to ESTs from other plant or animal species. Eleven percent of the ESTs showed no significant homology to sequences in the databases. Among the genes that could be classified into functional categories, the largest group (9%) was involved in defense and stress response. Jasmonate-induced protein genes made up 26% of the genes in this group. Genes involved in lipid biosynthesis made up approximately 6% of all ESTs, although 96% of the ESTs in this group consisted of β-ketoacyl ACP synthase. Surprisingly, this gene was not differentially expressed during malting in any of the four cultivars examined. Genes involved in gene regulation, protein metabolism, and carbohydrate metabolism made up 4.3, 3.9, and 3.1% of the ESTs, respectively. a-Amylase genes were the largest group of genes under carbohydrate metabolism (Supplemental Table S7). Other ESTs identified were involved in metal ion binding (2.2%), membrane biosynthesis (1.4%), amino acid metabolism (1.8%), energy production (1%), cell wall biogenesis (0.6%), and seed storage protein (0.4%).

Functional categories of barley ESTs generated from a cDNA library made by suppression-subtractive hybridization between cDNA from ungerminated seed of Morex as ‘driver’ and cDNA from day 4 malted seed of Morex as tester
Quantitative RT-PCR of selected genes
To validate gene expression observed in microarray experiments, qRT-PCR of four selected genes (α-amylase, α-glucosidase, limit dextrinase, and β-ketoacyl synthase) was conducted on the four malting cultivars using cDNA prepared from day 4 malted seeds (Fig. 5a). cDNA from day 4 malted seed of a non-malting cultivar, Steptoe, was included for comparison. For all four genes, there was a significant difference in expression levels among the five cultivars (Fig. 5a). α-Amylase and α-glucosidase expression levels were significantly different among the cultivars at P ≤ 0.0001. Limit dextrinase and β-ketoacyl synthase expression levels were significantly different among the cultivars at P ≤ 0.001 and 0.01, respectively. In comparison, only α-amylase and limit dextrinase showed differential expression in microarrays at day 4 as defined by |log2(FC)| > 1 and adjusted P ≤ 0.05 (Fig. 5b). There were also differences in expression patterns between qRT-PCR and microarrays for α-amylase expression (Fig. 5a, b). Merit and Legacy showed the highest levels of α-amylase expression in qRT-PCR while Harrington showed the highest level of α-amylase expression in microarrays. For limit dextrinase, Merit and Harrington showed the highest levels of expression in both qRT-PCR and microarrays (Fig. 5a, b). Therefore, there was limited correlation between expression patterns observed with qRT-PCR and microarrays. Including Steptoe in qRT-PCR experiments allowed comparison of gene expression between the four malting cultivars and a non-malting cultivar. Steptoe showed at least three times lower expression levels for α-amylase and α-glucosidase than the lowest expressing malting cultivar, Morex (Fig. 5a). For limit dextrinase, Legacy had lower expression than Steptoe, while for β-ketoacyl synthase, Steptoe, Morex, and Harrington had similar expression levels.

Bar charts showing expression levels of α-amylase (Alpha-Amy), α-glucosidase (Alpha-Gluc), limit dextrinase (L-Dex), and β-ketoacyl synthase (Beta-KAS) based on (a) qRT-PCR, and (b) microarrays during day 4 germination in Morex, Merit, Harrington, and Legacy. For A, a non-malting cultivar, Steptoe, was included in the qRT-PCR experiments. Relative quantification of transcript abundance was performed using Steptoe as calibrator; this sample therefore had a baseline relative fold change of one. Asterisks denote P-values based on ANOVA: **** ≤0.0001, *** ≤0.001, ** ≤0.01, * ≤0.05. Differential expression in B was based on absolute log2 fold change >1 and P ≤ 0.05
Discussion
Transcript profiles of germinating barley seeds
The use of the Barley1 GeneChip® array in this study provided a comprehensive view of global changes in gene expression during germination in barley. Up-regulation of genes involved in metabolism of carbohydrates, nucleotides, amino acids, and proteins during steeping indicates that a reactivation of metabolism was occurring. Twenty-four hours following steeping (day 1), extensive reprogramming of gene expression occurred, similar to what others have previously reported (Watson and Henry 2005; White et al. 2006). Every functional category included in the analysis increased in the number of induced genes during the malting stages. Doubling in number of induced genes involved in transport from steeping to day 4 may reflect the importance of protein movement from embryo and scutellum to the endosperm (Fincher 1989). Hydrolytic enzymes synthesized in the embryo are transported to the endosperm, where they break down the carbohydrate and protein reserve (Bamforth and Barclay 1993; Macfadden et al. 1988; Ranki 1990). In addition, transcripts for hydrolytic enzymes are also transcribed or stored in the endosperm as recently reported (Sreenivasulu et al. 2008). In this recent study, the authors investigated the transcriptome of barley during maturation, dessication, and germination, separating the endosperm plus aleurone from the embryo. They showed that transcriptional activation of storage reserve mobilization events occurs at an early stage during germination, well before protrusion.
Genes involved in stress and defense response made up a major portion of differentially expressed transcripts in all four malting stages studied. Previous studies of germinating barley seeds have reported similar findings (Potokina et al. 2002; Watson and Henry 2005; White et al. 2006). This is not surprising since malting is not performed under aseptic conditions and barley seeds contain an inherent microbial flora consisting of fungi, bacteria, and yeasts (Flannigan 2003). It has been shown that the molds and bacteria in barley increase during malting, reaching a peak during the germination stage (Flannigan 2003), consistent with the observation that defense and stress response genes increased from steeping to day 4. Analysis of the proteome of malt and beer also showed the predominance of proteins involved in protection against pathogens and insects (Perrocheau et al. 2005). In addition, seed germination may be considered a critical time point in a plant’s life cycle and it is not surprising that a large number of defense-related genes are normally upregulated as a protective measure.
While previous studies of gene expression in germinating barley seeds mainly focused on steeping and germination stages, we also looked at kilning. Exposure of the seeds to gradually increasing temperatures resulted in the overall decrease in number of up-regulated genes involved in metabolism of macromolecules. This is to be expected as kilning is done to terminate the germination process. Some genes that remained up-regulated from germination through kilning included those coding for α-amylase, limit dextrinase, β-glucosidase, and β-glucanase, indicating that the enzymes are thermostable. Other genes that were up-regulated from germination through kilning included those coding for peptidases, lipid transfer proteins, and proteinase inhibitors. These proteins were identified as thermostable components of malt and beer (Perrocheau et al. 2005). Other genes that were up-regulated from germination to kilning were those for heat shock proteins, which function to protect enzymes during heat stress (Jinn et al. 1995; Waters et al. 1996).
Genes correlated with malting quality phenotypes
The strategy used to identify barley genes associated with malting quality was to focus on genes that were differentially expressed in both the malting stages in Morex and in the four-cultivar comparisons. Using this approach, we took advantage of differential expression between the malting stages relative to dry seed as well as differential expression among four malting cultivars. There were 723 genes that met the criteria. To address the concern that this approach might be too stringent and might eliminate genes that are important in malting, we determined whether genes that have been previously associated with malting quality phenotypes were represented among the 723 genes. The genes for α-amylase, limit dextrinase, and α-glucosidase were in this group of genes. The endosperm-specific β-amylase gene, βmy1, has also been associated with diastatic power (Clark et al. 2003) but was not included in the list, since βmy1 is transcribed during seed development (Daussant and Corvazier 1970). Other genes previously identified based on differential expression and correlation with malting quality traits including β-1,3 glucanase, Sucrose synthase I, and Lipoxygenase (Potokina et al. 2004), were also among the 723 genes. Therefore, it appears that the approach used was able to identify genes that are potentially important in malting.
Of the 723 genes, 11–102 genes correlated with six malting quality phenotypes. Well-characterized genes for hydrolytic enzymes known to be important in malting (e.g., Limit dextrinase, β-glucanase) showed the expected association with malting quality phenotypes (Table 5). Many other genes that have not been characterized for their roles in determining specific malting quality traits were identified in this study. For example, α-amylase measurements were negatively correlated with genes coding for α-amylase inhibitors and positively correlated with Lipoxygenase 2.1. Diastatic power measurement, which is an indication of α-amylase and β-amylase activities, was correlated with genes for carbohydate metabolism enzymes, as well as genes for lipid metabolism, several defense and stress response genes, and genes of unknown function.
It is noteworthy that many of the genes showing correlation with malting quality traits were also previously shown to be associated with the same or different malting quality traits in this current report (Potokina et al. 2004). The previous study investigated the expression of 1400 ESTs in ten malting barley varieties different from those used in our study. Examples of common genes that correlated with malting quality traits in the two studies are those coding for β-glucosidase, sucrose synthase I, limit dextrinase, cysteine protease, β-glucanase, lipid transfer proteins, lipoxygenase 2.1, lipases, peroxidase, and heat shock proteins.
The finding that several proteinase inhibitor genes correlated with malting quality traits agrees with the suggestion by Jones (2005b) that it is important to define the hydrolytic enzyme inhibitors in malting barley since these proteins can regulate the activity of the endoproteases. It has been recognized that current measurements of malting quality are not precise since they do not identify all enzymes that contribute to a malting quality trait (Bamforth and Barclay 1993). The results presented here may provide candidate genes that are involved in malting quality traits. A caveat to keep in mind is that these results are exploratory and the role of these candidate genes in determining malting quality will need to be validated.
One way to validate candidate genes is by determining whether the gene or the expression level polymorphism maps to a malting quality QTL, as previously demonstrated (Potokina et al. 2006). A candidate gene (CxpI) and the relative expression level polymorphism both mapped to a region containing a QTL for diastatic power on chromosome 3H (Potokina et al. 2006). Another potential approach for validating the function of genes identified in this study would be to knock down gene expression using RNA interference or TILLING (McCallum et al. 2000; Till et al. 2003). TILLING lines developed in malting barley cultivars are available (Caldwell et al. 2004). The use of TILLING lines provides an advantage in that transformation is not required, since barley transformation is still a technical challenge and requires a particle bombardment facility. Identification of SNPs associated with malting quality traits would provide another confirmation for the function of candidate genes in malting as well as markers that can be used by breeders. SNP analysis of the CxpI gene in 90 barley cultivars identified two haplotypes, one of which showed association with diastatic power (Potokina et al. 2006).
ESTs from malted barley seed include novel genes
The majority of ESTs generated from a cDNA library made by suppression-subtractive hybridization between malted and dry seed were in common with the 723 differentially expressed genes identified in the microarray experiments. Furthermore, many of the ESTs corresponded to genes correlated with malting quality phenotypes (Supplemental Table S7). Some examples are limit dextrinase, lipid transfer protein, α-amylase inhibitors, and jasmonate-induced proteins.
Approximately 6% of the ESTs generated showed homology to β-ketoacyl ACP synthase. This gene was not differentially expressed in any of the malting stages of the four cultivars investigated using microarrays. However, qRT-PCR results showed significant differences (P < 0.01) in the expression level of this gene among the four cultivars, with transcript levels in Morex lower than in the non-malting cultivar Steptoe. Whether this gene plays an important role in malting is not clear. β-ketoacyl ACP synthase catalyzes the chain elongation step in fatty acid synthesis leading to palmitoyl-ACP and stearoyl ACP (Kauppinen 1992). Other genes involved in lipid metabolism were found to be correlated with malting quality traits in this study and in previous studies (Potokina et al. 2004).
Conclusion
In conclusion, transcript profiling using microarrays and ESTs identified a common set of genes that were differentially expressed during malting in barley. Correlation of expression profiles with malting quality phenotypes resulted in the identification of 11–102 genes showing correlation with six malting quality traits. These candidate genes will need to be further validated for their importance in malting. A large group of genes with unknown function were identified with both methods, emphasizing the fact that the molecular basis of malting quality traits is not well understood. Finally, ESTs generated in this study with no known homology to sequences in the genome database may be novel genes and these may be important in malting.
References
Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ (1990) Basic local alignment search tool. J Mol Biol 215:403–410
ASBC (1992) Methods of analysis of the American Society of Brewing Chemists, 8th edn. American Society of Brewing Chemists, St. Paul
Bamforth CW, Barclay AHP (1993) Malting technology and the uses of malt. In: MacGregor A, Bhatty RS (eds) Barley: chemistry and technology. American Association of cereal Chemists, St. Paul, pp 297–354
Benjamini Y, Hochberg Y (1995) Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Ser B 57:289–300
Briggs DE (1998) Malts and malting. Blackie Academic and Professional, London
Burger WC, LaBerge DE (1985) Malting and brewing quality. In: Ramusson DG (ed) Barley, Madison, pp 367–401
Caillaud MC, Dubreuil G, Quentin M, Perfus-Barbeoch L, Lecomte P, de Almeida Engler J, Abad P, Rosso MN, Favery B (2007) Root-knot nematodes manipulate plant cell functions during a compatible interaction. J Plant Physiol 165:104–113
Caldo RA, Nettleton D, Wise RP (2004) Interaction-dependent gene expression in Mla-specified response to barley powdery mildew. Plant Cell 16:2514–2528
Caldwell DG, McCallum N, Shaw P, Muehlbauer GJ, Marshall DF, Waugh R (2004) A structured mutant population for forward and reverse genetics in Barley (Hordeum vulgare L.). Plant J 40:143–150
Clark SE, Hayes PM, Henson CA (2003) Effects of single nucleotide polymorphisms in β-amylase 1 alleles from barley on functional properties of the enzymes. Plant Physiol Biochem 41:798–804
Close TJ, Wanamaker SI, Caldo RA, Turner SM, Ashlock DA, Dickerson JA, Wing RA, Muehlbauer GJ, Kleinhofs A, Wise RP (2004) A new resource for cereal genomics: 22 K barley GeneChip comes of age. Plant Physiol 134:960–968
Daussant J, Corvazier P (1970) Biosynthesis and modifications of alpha- and beta-amylases in germinating wheat seeds. FEBS Lett 7:191–194
Diatchenko L, Lau YF, Campbell AP, Chenchik A, Moqadam F, Huang B, Lukyanov S, Lukyanov K, Gurskaya N, Sverdlov ED, Siebert PD (1996) Suppression subtractive hybridization: a method for generating differentially regulated or tissue-specific cDNA probes and libraries. Proc Nat Acad Sci USA 93:6025–6030
Edgar R, Domrachev M, Lash AE (2002) Gene expression omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res 30:207–210
Fincher GB (1989) Molecular and cellular biology associated with endosperm mobilization in germinating cereal grains. Annu Rev Plant Physiol Plant Mol Biol 40:305–346
Flannigan B (2003) Microbiota of barley and malt. In: Priest FG, Campbell I (eds) Brewing microbiology, 3rd edn. Kluwer Academic/Plenum, New York, pp 113–180
Foster AE, Peterson GA, Banasik OJ (1967) Heritability of factors affecting malting quality of barley, Hordeum vulgare L., emend. Lam Crop Sci 7:611–613
Gao W, Clancy JA, Han F, Jones BL, Budde A, Wesenberg DM, Kleinhofs A, Ullrich SE, Project NABG (2004) Fine mapping of a malting-quality QTL complex near the chromosome 4HS telomere in barley. Theor Appl Genet 109:750–760
Gentleman RC, Carey VJ, Bates DM, Bolstad B, Dettling M, Dudoit S, Ellis B, Gautier L, Ge Y, Gentry J, Hornik K, Hothorn T, Huber W, Iacus S, Irizarry R, Leisch F, Li C, Maechler M, Rossini AJ, Sawitzki G, Smith C, Smyth G, Tierney L, Yang JY, Zhang J (2004) Bioconductor: open software development for computational biology and bioinformatics. Genome Biol 5:R80
Hayes P, Jones BL (2000) Malting quality from a QTL perspective. 8th international Barley genetics symposium 8:99–105
He J, Dai X, Zhao X (2007) PLAN: a web platform for automating high-throughput BLAST searches and for managing and mining results. BMC Bioinformatics 8:53
Hockett EA, Nilan RA (1985) Genetics. In: Rasmussen DC (ed) Barley. American Society of Agronomy, Madison, p 195
Irizarry RA, Bolstad BM, Collin F, Cope LM, Hobbs B, Speed TP (2003) Summaries of Affymetrix GeneChip probe level data. Nucleic Acids Res 31:e15
Jinn TL, Chen YM, Lin CY (1995) Characterization and physiological function of class I low-molecular-mass, heat-shock protein complex in soybean. Plant Physiol 108:693–701
Jones BL (2005a) Endoproteases of barley and malt. J Cereal Sci 42:139–156
Jones BL (2005b) The endogenous endoprotease inhibitors of barley and malt and their roles in malting and brewing. J Cereal Sci 42:271–280
Kauppinen S (1992) Structure and expression of the Kas12 gene encoding a beta-ketoacyl-acyl carrier protein synthase I isozyme from barley. J Biol Chem 267:23999–24006
Macfadden GI, Ahluwalia B, Clarke AE, Fincher GB (1988) Expression sites and developmental regulation of genes encoding (1–3, 1–4)-beta-glucanases in germinated barley. Planta 173:500–508
McCallum CM, Comai L, Greene A, Henikoff S (2000) Targeting induced local lesions in genomes (TILLING) for plant functional genomics. Plant Physiol 123:439–442
Perrocheau L, Rogniaux H, Boivin P, Marion D (2005) Probing heat-stable water-soluble proteins from barley to malt and beer. Proteomics 5:2849–2858
Pfaffl MW (2001) A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res 29:e45
Potokina E, Sreenivasulu N, Altschmied L, Michalek W, Graner A (2002) Differential gene expression during seed germination in barley (Hordeum vulgare L.). Funct Integr Genomics 2:28–39
Potokina E, Caspers M, Prasad M, Kota R, Zhang H, Sreenivasulu N, Wang M, Graner A (2004) Functional association between malting quality trait components and cDNA arrays based on expression patterns in barley (Hordeum vulgare L.). Mol Breed 14:153–170
Potokina E, Prasad M, Malysheva L, Roder MS, Graner A (2006) Expression genetics and haplotype analysis reveal cis regulation of serine carboxypeptidase I (Cxp1), a candidate gene for malting quality in barley (Hordeum vulgare L.). Funct Integr Genomics 6:25–35
Ranki H (1990) Secretion of α-amylase by the epihelium of barley scutellum. F Inst Brew 96:307–309
Rozen S, Skaletsky HJ (1998) Primer3 on the WWW for general users and for biologist programmers. In: Krawetz SMS (ed) Bioinformatics methods and protocols: methods in molecular biology. Humana Press, Totowa, pp 365–386
Sreenivasulu N, Usadel B, Winter A, Radchuk V, Scholz U, Stein N, Weschke W, Strickert M, Close TJ, Stitt M, Graner A, Wobus U (2008) Barley grain maturation and germination: metabolic pathway and regulatory network commonalities and differences highlighted by new MapMan/PageMan profiling tools. Plant Physiol 146:1738–1758
Therrien MC (2006) Estimates of heritability of major malting quality traits in Canadian barley. Barley Genet Newsl 36:10–11
Till BJ, Reynolds SH, Greene EA, Codomo CA, Enns LC, Johnson JE, Burtner C, Odden AR, Young K, Taylor NE, Henikoff JG, Comai L, Henikoff S (2003) Large-scale discovery of induced point mutations with high-throughput TILLING. Genome Res 13:524–530
Venu RC, Jia Y, Gowda M, Jia MH, Jantasuriyarat C, Stahlberg E, Li H, Rhineheart A, Boddhireddy P, Singh P, Rutger N, Kudrna D, Wing R, Nelson JC, Wang GL (2007) RL-SAGE and microarray analysis of the rice transcriptome after Rhizoctonia solani infection. Mol Genet Genomics 278:421–431
Waters ER, Lee GJ, Vierling E (1996) Evolution, structure and function of the small heat shock proteins in plants. J Exp Bot 47:325–338
Watson L, Henry RJ (2005) Microarray analysis of gene expression in germinating barley embryos (Hordeum vulgare L.). Funct Integr Genomics 5:155–162
White J, Pacey-Miller T, Crawford A, Cordeiro G, Barbary D, Bundock P, Henry R (2006) Abundant transcripts of malting barley identified by serial analysis of gene expression (SAGE). Plant Biotechnol J 4:289–301
Zale JM, Clancy JA, Ullrich SE, Jones BL, Hayes PM, Project NABGM (2000) Summary of barley malting-quality QTLs mapped in various populations. Barley Genet Newsl 30:44–54
Acknowledgments
We acknowledge Mehdi Fazel, Leon van Eck, and Hong Wang for technical support. This project was partially funded by the USDA North American Barley Project, Anheuser-Busch, and Hatch Funds 644 to NL.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by A. Graner.
Electronic supplementary material
Below is the link to the electronic supplementary material.
122_2008_951_MOESM1_ESM.xls
Table S1. Expression data for genes showing differential expression between different malting stages relative to dry seed* (XLS 152 kb)
122_2008_951_MOESM2_ESM.doc
Table S2. List of genes falling under functional categories (GO category) listed in Table 1 based on PLAN software program (XLS 227 kb)
122_2008_951_MOESM3_ESM.xls
Table S3. Expression data for genes showing differential expression between pairs of cultivars during day 1 germination stage (XLS 806 kb)
122_2008_951_MOESM4_ESM.xls
Table S4. Expression data for genes showing differential expression between pairs of cultivars during day 4 germination stage* (XLS 555 kb)
122_2008_951_MOESM8_ESM.tif
Hierarchial clustering of 70 probe sets representing genes that were previously shown to be associated with malting quality traits. Red indicates up-regulated, green indicates down-regulated, and black indicates no change (TIFF 316 kb)
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Lapitan, N.L.V., Hess, A., Cooper, B. et al. Differentially expressed genes during malting and correlation with malting quality phenotypes in barley (Hordeum vulgare L.). Theor Appl Genet 118, 937–952 (2009). https://doi.org/10.1007/s00122-008-0951-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00122-008-0951-8