
Abstract
Key message
Plant height of soybean is associated with a haplotype block on chromosome 19, which classified 211 soybean accessions into five distinct groups showing significant differences for the target trait.
Abstract
Genetic variation is pivotal for crop improvement. Natural populations are precious genetic resources. However, efficient strategies for the targeted utilization of these resources for quantitative traits, such as plant height (PH), are scarce. Being an important agronomic trait associated with soybean yield and quality, it is imperative to unravel the genetic mechanisms underlying PH in soybean. Here, a genome-wide association study (GWAS) was performed to identify single nucleotide polymorphisms (SNPs) significantly associated with PH in a natural population of 211 cultivated soybeans, which was genotyped with NJAU 355 K Soy SNP Array and evaluated across six environments. A total of 128 SNPs distributed across 17 chromosomes were found to be significantly associated with PH across six environments and a combined environment. Three significant SNPs were consistently identified in at least three environments on Chr.02 (AX-93958260), Chr.17 (AX-94154834), and Chr.19 (AX-93897200). Genomic regions of ~ 130 kb flanking these three consistent SNPs were considered as stable QTLs, which included 169 genes. Of these, 22 genes (including Dt1) were prioritized and defined as putative candidates controlling PH. The genomic region flanking 12 most significant SNPs was in strong linkage disequilibrium (LD). These SNPs formed a single haplotype block containing five haplotypes for PH, namely Hap-A, Hap-B, Hap-C, Hap-D, and Hap-E. Deployment of such superior haplotypes in breeding programs will enable development of improved soybean varieties with desirable plant height.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Cultivated soybean (Glycine max L.) is an important oilseed crop, grown globally as a major source of edible protein and oil (Kulkarni et al. 2016). However, in the past five decades, only a gradual increase in soybean yield has been observed in China. This can be witnessed by the fact that China imports > 80% of soybean and its products to meet its domestic requirements (Liu et al. 2018). Hence, development of soybean genotypes with desirable plant architecture and higher yield is a major objective of soybean breeders across the world, and especially from China. A significant positive correlation has been observed between PH and yield in soybean. However, taller soybean genotypes are highly prone to lodging, affecting the overall yield and seed quality (Liu et al. 2011; Lü et al. 2016). This makes PH a crucial trait in the breeding programs of soybean. Moreover, PH is a quantitative trait that is controlled by multiple loci/genes, besides being highly influenced by genotype × environment interactions (Wang et al. 2004; Cao et al. 2019). Over the years, breeding for desirable PH has been accomplished via both traditional and marker-assisted breeding (MAB) approaches (Bhat et al. 2020). In the case of environment-sensitive traits such as PH, the MAB approaches are much more effective and allow selection of plants at initial growth stages, such as germination or seedling, even for traits measured at maturity (Allen 1994; Chapman et al. 2003). Therefore, elucidating the genetic basis of PH is expected to greatly facilitate implementation of MAB in the development of high-yielding soybean varieties.
Some studies have documented the genetic basis of PH in soybean (Mansur et al. 1996; Orf et al. 1999; Liu et al. 2013; Yin et al. 2017). For instance, the past three decades have witnessed identification of > 200 quantitative trait loci (QTLs), as documented in the SoyBase (https://www.soybase.org/), and these QTLs are located across 20 soybean chromosomes. Most of the previously reported studies used linkage mapping with low-resolution markers to identify such QTLs, which often possess low genomic resolution. This has hampered successful deployment of PH QTLs in breeding programs to develop improved soybean varieties with desired PH (Cao et al. 2019). An alternative approach of trait mapping such as GWAS detects alleles/QTLs linked to the trait of interest at a higher resolution using diverse natural populations (Schmutz et al. 2010). Technological advances in next-generation sequencing-based genotyping technologies have enabled GWAS to be used routinely in crop genetics research (Song et al. 2016; Cao et al. 2019). Several studies have demonstrated the efficiency of GWAS to identify marker-trait associations (MTAs) regulating important agronomic traits and to detect the underlying putative genes in different plant species such as Arabidopsis thaliana (Ren et al. 2019), rice (Xu and Crouch 2008; Li et al. 2014), maize (Yang and Zhu 2005), wheat (Wang et al. 1999), cotton (Lai et al. 2016), soybean (Hyten et al. 2008; Cao et al. 2017; Fang et al. 2017; Li et al. 2017; Chang et al. 2018), chickpea (Varshney et al. 2019a, b; Thudi et al. 2021), pigeonpea (Varshney et al. 2017a, b), and pearl millet (Varshney et al. 2017a, b).
With the availability of whole genome sequencing data (WGS), sequencing-based trait mapping strategies have been successful in identifying QTLs/ MTAs in several crops (Varshney et al. 2019a, b). Notably, the use of WGS data has increased the mapping resolution of GWAS and enabled identification of haplotypes for the trait(s) of interest. In the post-sequencing era, ‘haplotype assembly’ has emerged as an efficient approach for the development of improved crop varieties (Bevan et al. 2017). In the past few years, haplo-pheno analysis has been successfully used for detecting superior haplotypes for agronomically important traits in some crop species. For example, Abbai et al. (2019) reported desirable haplotypes for 21 genes controlling quality and yield-related traits in rice, based on a panel of 3000 rice accessions. Similarly, superior haplotypes have been identified for drought and salinity stress adaptation in rice, soybean, and pigeonpea (Guan et al. 2014; Mishra et al. 2016; Kuroha et al. 2018; Chen et al. 2019; Sinha et al. 2020).
To understand the genetics of PH in soybean genotypes selected from major soybean growing areas of China, the present study used GWAS and haplotype analysis for identifying MTAs, candidate genes, and haplotypes. Superior haplotypes identified in this study hold potential for developing improved soybean varieties with desired plant architecture using haplotype-based breeding approach.
Material and methods
Plant material and field experiments
A natural population comprising 219 soybean germplasm accessions was evaluated for two consecutive years (2011 and 2012) at three different locations in China. This soybean germplasm represents the cultivated pool selected from different ecological regions of China, previously described by Wang et al. (2016) and Du et al. (2020) (Supplementary Table 1). The evaluated locations include the experimental fields of Nanjing Agricultural University (Nanjing); Jiangsu Yanjiang-Institute of Agricultural Sciences (Nantong); and Agricultural College of Yangzhou University (Yangzhou). Hence, the germplasm was evaluated at a total of six individual environments (location × year), namely E1–E6 (E1: Nanjing_2011, E2: Nantong_2011, E3: Yangzhou_2011, E4: Nanjing_2012, E5: Nantong_2012, and E6: Yangzhou_2012). These three locations differ substantially in temperature, precipitation, and soil quality. For example, Nanjing (32°12′ N, 118°37′ E) has a north subtropical humid climate with four distinct seasons. The average rainfall and relative humidity in Nanjing are 1106.5 mm and 76%, respectively. The annual average temperature in Nanjing is 15.4 °C, and the soil quality of Nanjing is mostly red soil with high iron content. Further, Nantong (31°58′ N, 120°53′ E) is in the alluvial plain at lower reaches of the Yangtze River with mild marine climate. The annual average temperature and precipitation in Nantong are 15.1 °C and ~ 1040 mm, respectively. There are four distinct seasons in Nantong, and the spring and autumn seasons are relatively short. The soil in Nantong is mainly yellow brown. Yangzhou (32°23′ N, 119°25′ E) is located at the southern end of the Yangtze Huaihe plain. It is affected by monsoon circulation, with four distinct seasons and a mild climate. The annual average temperature is 14.8 °C, and the average precipitation is 1020 mm. The soil texture of Yangzhou is mainly heavy soil, and a small part of it is sandy and light clay. The experiments were conducted in a complete randomized block design with three replications of each genotype. The field data for PH collected from six different environments are provided in Supplementary Table 2. Standard soybean agronomic cultural practices (Zhang et al. 2015) were followed at each location during the experiments.
Phenotypic data collection and statistical analysis
In each environment, five consecutive plants of each genotype were selected from the middle of each plot for PH phenotyping in each replication. PH was measured in centimeters (cm) by using a measuring tape stretched from the cotyledonary node to the top of the plant at the maturity stage. The data obtained from individual environments (location × year combination) and the combined environment were evaluated using “lme4” package in R environment. The analysis of variance (ANOVA) and predicted means [best linear unbiased predictions (BLUPs)] were calculated with genotype set as random. Predicted means (BLUPs) for individual environment were estimated using the model:
where Yijk is the trait (PH), µ is the overall mean effect, and Repi denotes the effect of the ith replicate/block. Genj represents the effect of the jth genotype; εij is the effect of the error associated with the ith replication/block, and jth genotype.
The ANOVA and predicted means (BLUPs) for the combined environment were estimated using the following model:
where Envi and Envi × Genk are the ith environment and the G × E interaction effects, respectively.
The broad-sense heritability for the combined and individual environment was estimated as σ2g/(σ2g + σ2ge/n + σ2ε/nr) and σ2g/(σ2g + σ2ε/r), respectively, where σg is the genetic variance, σ2ge is the interaction of genotype and environment, σ2ε is the residual error, n is the number of environments, and r is the number of replication/block (Knapp et al. 1985).
To identify the relationship between PH evaluated across different environments, we performed a Pearson’s correlation analysis. In brief, Pearson correlation analysis for soybean PH measured across different environment was conducted based on the predicted means calculated across the environment and visualized using MVApp (Julkowska et al. 2019).
Genome-wide association study
The NJAU 355 K SoySNP assay previously developed by our laboratory (Wang et al. 2016) was used in the present study. In brief, the sequencing data of 31 soybean genotypes (including 14 cultivated and 18 wild type) were used along with Williams 82 (Glyma.Wm82.a1.v1.1) as the reference genome (www.phytozome.net). The VIP variants include 60,800 SNPs from SoySNP50K15, 2103 SNPs from SoyBase, and 3671 SNPs from the sequencing data of 31 soybean genotypes. A total of 355,595 SNPs representing 609,883 probe sets were obtained following the Affymetrix filtering (Wang et al. 2016). The SNP dataset has been provided as Supplementary Table 3. In the current study, for the selection of high-quality SNPs, quality control analysis was conducted at minimum minor allele frequency (MAF) of 0.01 and missing genotype and taxa at 0.2 in TASSEL v5.2.73 (Bradbury et al. 2007).
Genome-wide association analysis was conducted with Genomic Association and Prediction Integrated Tool (GAPIT) package in R environment (Lipka et al. 2012) using a compressed mixed linear model (CMLM). This model was adopted to speed up computational time and optimize statistical performance (Lipka et al. 2012). UpSet plot was developed for significant SNPs by using the UpSetR package in R environment (Lax et al. 2014; Conway et al. 2017).
Haplotype analysis
The LD level of pairwise SNPs was calculated using Haploview 4.2 (Barnett et al. 2005). All markers associated with PH on chromosome 19 (Chr.19) within the LD ± 130 kb (Wang et al. 2016) were considered, and haplotype block was defined by “confidence intervals” algorithm (Gabriel et al. 2002). Genotypes were grouped into independent groups based on specific haplotype carried by them. To estimate the effect of haplotype on PH, groups were fitted into one-way ANOVA model in R environment as follows:
where phenotype denotes PH values in the individual and combined environments. A pairwise comparison of means was conducted using Turkey’s HSD test and visualized in R environment.
Identification of candidate genes
Within the LD decay distance of significant SNPs, all genes present within the ± 130 kb interval flanking the stable SNP positions were retrieved from SoyBase website (https://www.soybase.org/), using Glyma2.0 gene model. The functional annotations of genes were downloaded from SoyBase, and were screened manually. The RNA-seq data available at SoyBase (https://www.soybase.org/) were extracted for the genes underlying the QTL intervals from different soybean tissues (leaf, flower, pod shell, seed, root, and nodule) at different growth and developmental stages, viz., young leaf, flower, one cm pod, pod shell 10 days to flowering (DAF), pod shell 14DAF, seed 10DAF, seed 14DAF, seed 21DAF, seed 25DAF, seed 28DAF, seed 35DAF, seed 42DAF, root, and nodule) (Supplementary Table 4). These RNA-seq data were previously generated and deposited in the SoyBase by Severin et al. (2010). The RNA-seq dataset was used to analyze the expression of putative genes in different soybean tissues and across multiple development stages. For developing a heat map, the fragments per kilobase of transcript per million fragment mapped (FPKM) values of the candidate genes were used. Hierarchical clustering was performed using Euclidean Distance Method with complete linkage approach (Tao et al. 2013). A heatmap of these candidate genes was constructed using TBtools_JRE1.6 software (Chen et al. 2020). Based on functional annotations of genes, available literature, and gene expression patterns, some genes were defined as putative candidates regulating PH.
Results
Phenotypic characterization and correlation analysis of PH in soybean accessions
The descriptive statistics including mean, range (maximum and minimum values), standard deviation, skewness, kurtosis, broad-sense heritability, and coefficient of variation (CV) for PH in 219 soybean accessions evaluated across six different environments and the combined environment are presented in Table 1. In brief, the PH ranged from 8.24 cm in E6 to 311.67 cm in E4. The mean across individual environments ranged from 52.76 ± 0.83 cm in E3 to 105.30 ± 1.73 in E4. The CV in the combined environment was 49%, and ranged from 38% in E6 to 49.8% in E2. Furthermore, skewness and kurtosis were 1.34 and 2.84, respectively in the combined environment (Table 1). The broad-sense heritability ranged from 0.84 in E6 to 0.98 in E4, and 0.97 in the combined environment. The soybean accessions (G), environment (E), and the genotype × environment interaction (G × E) had significant effect (P < 0.0001) on PH (Table 2). Moreover, Pearson correlation analysis of PH showed a significantly positive correlation (P < 0.0001) across different environments evaluated (Supplementary Table 5).
Genome-wide distribution of SNPs in soybean natural population
In the present study, a total of 211 soybean accessions and 291,962 SNP markers were retained after quality control analysis (Table 3). These markers covered 949,992,372 bp (949.99 Mb) of the soybean genome, representing ~ 85% of the genome. The number of SNPs in each chromosome varied from 12,035 to 19,880, with the lowest number of SNPs present on Chr.11, whereas Chr.18 contained the highest number of SNPs. The lowest and highest SNP density of 264.27 SNPs/Mb and 361.97 SNPs/Mb was found on Chr.01 and Chr.13, respectively (Supplementary Fig. 1; Table 3).
GWAS revealed genetic architecture of PH in soybean
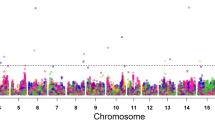
A total of 128 SNPs were found to be significantly associated with PH across six environments and the combined environment, based on GWAS results (Fig. 1; Supplementary Table 6). The distribution of markers across soybean chromosomes and their intersections based on the individual and combined environments are presented in Fig. 2 as UpSet plot (Lex et al. 2014). The UpSet plots provides a convenient alternative to visualize the data sets by frequency. In Fig. 2a, the horizontal bar graph on the left-hand side of the UpSet plot showed the size of each set, i.e., total number of significant SNPs identified in each individual environment and the combined environment. For example, the horizontal bar linked to E5 environment indicated the presence of 58 significant SNPs identified in this environment. Similarly, the horizontal bars corresponding to other five individual environments (E1, E2, E3, E4, and E6) and the combined environment showed the number of significant SNPs identified in each of these environments (Fig. 2a). Furthermore, the vertical bars on the top indicate the individual set participation sizes (Fig. 2a). For instance, the vertical bar on the left most side shows that out of the 58 significant SNPs identified in E5 environment, 49 SNPs were specific for E5 environment, two SNPs were detected in E4 and E5, one SNP in E2, E4, and E5; one SNP in E2, E3, and E5; four SNPs in E1, E3, E5, E6, and CE; and one SNP was detected in E1, E2, E3, E4, E5, E6, and CE (Fig. 2a). In Fig. 2b, each vertical bar reveals the number of significant SNPs associated with PH identified on each chromosome. Notably, three significant SNPs were consistently identified in three or more environments (Fig. 2; Table 4). These included a SNP (AX-93897200) on Chr.19, which was detected in all six environments and the combined environment, a SNP (AX-93958260) on Chr.02 and a SNP (AX-94154834) on Chr.17 that were detected in three different environments. The regions within ~ 130 kb interval [based on the LD decay previously reported for the same population by using NJAU 355 K SoySNP assay (Wang et al. 2016)] flanking the significant SNPs (AX-93958260, AX-94154834, and AX-93897200) present on Chr.02, Chr.17, and Chr.19, were referred as qPH2, qPH17 and qPH19, respectively. These QTLs/genomic regions represented stable genetic elements regulating PH in soybean.

GWAS signals for plant height evaluated across six environments. Manhattan plot and quantile–quantile (Q–Q) plot for the GWAS for PH evaluated at a Nanjing_2011 (E1), b Nantong_2011 (E2), c Yangzhou_2011 (E3), d Nanjing_2012 (E4), e Nantong_2012 (E5), and f Yangzhou_2012 (E6). The red lines on the Y-axis designate the significance threshold (− log10 P < 5.47). The numbers on the X-axis represent soybean chromosomes

Intersection of significant QTLs and their frequency across soybean chromosomes. a UpSet plot showing the intersection of significant QTLs across six different environment (E1, E2, E3, E4, E5, and E6) and combined environment (CE). b Frequency of QTLs across soybean chromosomes
Superior haplotypes for PH identified on soybean Chr. 19
The genomic region flanking three stable significant SNPs (AX-93958260, AX-94154834, and AX-93897200) within the LD decay of ± 130 kb was subjected to haplotype analysis. The results showed that only significant markers identified within the LD decay flanking the SNP AX-93897200 (detected in all six environments and the combined environment) on Chr.19 formed a haplotype block. No haplotype block was detected for other two significant SNPs within the referenced LD decay (± 130 kb) on Chr.02 and Chr.17. Hence, the SNP (AX-93897200) along with 11 other SNPs (AX-94195032, AX-93897192, AX-93659063, AX-94195035, AX-93897195, AX-93897196, AX-93955268, AX-93897197, AX-93897198, AX-93897199, and AX-93897201) were found to be in strong LD and formed a single haplotype block (Fig. 3). The length of this haplotype block was less than 0.15 Mb. These 12 SNPs within the haplotype block represented five haplotypes across 211 accessions of the soybean natural population. Notably, these haplotypes classified the genotypes of the studied soybean population into five groups. The PH of the genotypes varied significantly across the five haplotype groups. For instance, Haplotype-A (Hap-A) formed the largest group (n = 102), followed by Hap-B (n = 72), Hap-C (n = 16), Hap-D (n = 17), and Hap-E (n = 04) (Fig. 3c). The nucleotide variation detected for these haplotypes is represented in Supplementary Fig. 2.

GWAS signal, haplotype block analysis of qPH19, and haplotype effect for plant height. a Manhattan plot and Q-Q plot for the GWAS for PH analyzed in the combined environment. Red line represents the Bonferroni correction threshold. b GWAS signal for PH obtained on Chr.19 and pairwise LD analysis. The pairwise LD diagram for significant variants (P < 3.42 × 10–6) is displayed. c The boxplot for the haplotypes based on the predicted values of PH in the combined environment (CE). The genotypes were grouped and pairwise comparisons were conducted using Turkey’s HSD Test at P < 0.05. The boxes with a common alphabet indicate no significant difference in PH. Number of accessions (n) in each sub-class is represented on top of each box
The differences in PH based on different haplotype alleles identified within the haplotype block on Chr.19 in soybean GWAS population varied across environments. However, soybean accessions with Hap-D appeared to be the tallest across environments, those with Hap-E and Hap-B had a dwarf phenotype, and accessions with Hap-A and Hap-C were of intermediate height (Fig. 3c; Supplementary Fig. 3).
Identification of candidate genes within three QTL intervals
A total of 169 genes were identified within the physical genomic interval of all three major QTLs (qPH2, qPH17 and qPH19), which consisted of 18, 35, and 116 genes, respectively (Supplementary Table 7). Of the 169 genes detected, the RNA-seq data for only 124 genes were available online at SoyBase (https://www.soybase.org/) for 14 soybean tissues across different stages of growth and development (Supplementary Table 4). Expression pattern of the genes obtained from the RNA-seq data is represented in the form of a heatmap in Fig. 4. Furthermore, based on in silico analysis of gene expression data and gene annotations, we defined a total of 22 candidate genes underlying three stable genomic regions. This includes two genes underlying qPH2, five genes underlying qPH17, and 15 genes underlying qPH19 (Table 5). The selection of these candidate genes was based on gene function annotation (i.e., genes regulating cell division, meristem growth, plant growth hormone biosynthesis and signaling, cell elongation, and transition from vegetative to reproductive phase), literature studies, and gene expression profiles (Table 5; Supplementary Table 4). These 22 genes can be considered as putative candidates controlling PH. However, further functional validation of these genes is needed to determine their exact role in regulating PH in soybean.

Heat map exhibiting the expression profiles of candidate genes underlying the target QTL intervals. The heat map represents the expression profiles of 124 candidate genes including 9, 27, and 88 genes present on Chr.02, Chr.17, and Chr.19 underlying three QTLs, a qPH2, b qPH17, and c qPH19. Heat map was generated using the RNA-seq data retrieved from online dataset at SoyBase. N, nodule; S10DAF, seed at 10 days after flowering; S14DAF, seed at 14 days after flowering; S21DAF, seed at 21 days after flowering; S25DAF, seed at 25 days after flowering; S28DAF, seed at 28 days after flowering; S35DAF, seed at 35 days after flowering; S42DAF, seed at 42 days after flowering; PS10DAF, pod shell at 10 days after flowering; PS14DAF, pod shell at 14 days after flowering; YL, young leaf; 1cmpd, 1 cm of pod; flower, flower; and R, root
Discussion
Plant height is an important determinant of soybean yield and quality (Lu et al. 2016; Yin et al. 2017). Taller soybean cultivars/varieties are often subjected to lodging, which leads to considerable reduction in soybean production as well as deterioration of its quality (Cao et al. 2019). Hence, tailoring PH in soybean to avoid qualitative and quantitative losses has been a long-term goal of soybean breeders. Determining the genetic basis of PH is a prerequisite to develop soybean varieties with desirable plant architecture. Studies have documented the complex inheritance of PH, besides the trait being strongly influenced by G × E interactions (Lee et al. 2015). Therefore, the present study was undertaken to identify the genetic loci/QTLs, and superior haplotypes regulating PH in soybean by using GWAS and haplotype analysis.
In the present study, ANOVA results revealed a significant difference among 211 accessions for PH (P < 0.01). In accordance with previous studies (Zhang et al. 2004; Cao et al. 2019), our study confirmed that PH is significantly affected by G, E, and G × E interactions. This suggests the complex genetic nature of PH in soybean. Furthermore, the estimate of broad-sense heritability for PH was high (98.95%) in the soybean natural population across all six environments, which is consistent with previous studies (Cao et al. 2019). Higher broad-sense heritability reported for PH in this study suggests a high probability of achieving the same phenotypic performance for PH when grown in the same environment. In agreement with our findings, many authors have earlier reported similar results for PH in the soybean germplasm collection (Zhang et al. 2015; Chang et al. 2018; Xue et al. 2019).
To date, the genetic basis of PH in soybean has been documented by multiple research groups (Orf et al. 1999; Chapman et al. 2003; Liu et al. 2013; Yin et al. 2017; Cao et al. 2019). However, the majority of previously detected QTLs have not been validated, which limit their deployment in MAB efforts. To this end, a more powerful and high-resolution mapping approach called the LD-based GWAS was used in the present study. Previous studies have demonstrated that GWAS is an effective strategy to determine the genetic basis of complex crop traits, such as PH, in a more precise and accurate manner (Kim et al. 2012; Cober and Morrison 2010). Hence, in the present study, we used GWAS for the identification of genomic regions associated with PH in soybean. We identified a total of 128 SNPs linked with PH across all the environments, which were found to be distributed on all chromosomes except Chr.06, Chr.18, and Chr.20. These results suggest the genome-wide distribution of loci/genes regulating PH in soybean and reveal the complex genetic architecture of this trait. In agreement with our results, Zhang et al. (2015) reported the distribution of significant MTAs for PH on 15 different chromosomes of soybean. Similar results have also been reported by other researchers (Fang et al. 2017, 2020; Chang et al. 2018). Further, the number of significant SNPs linked to PH was found to vary considerably across six different environments. For example, the highest number of SNPs were identified in E5, whereas the lowest number of SNPs were detected in E2. These findings suggest some effect of environment on the regulation of PH in soybean, which is in accordance with previous reports (Xue et al. 2019; Fang et al. 2020).
Importantly, three significant SNPs were consistently identified across multiple environments on Chr.02, Chr.17, and Chr.19. The genomic regions (~ 130 kb) flanking significant SNPs were referred as QTLs related to PH based on the LD decay. These QTLs/genomic regions represent stable genetic elements regulating PH in soybean. The QTL on Chr.02 (Plant height 26–9) associated with PH has been previously reported in the genomic region between 15,993,654 and 41,032,570 bp by Sun et al. (2006), and the genomic region underlying qPH2 was found to lie in the same physical interval. Therefore, qPH2 might be the same as Plant height 26–9, as earlier reported by Sun et al. (2006). However, when compared to Plant height 26–9, the physical interval of qPH2 was considerably narrowed down in the present study. Furthermore, qPH19 identified in the present study was found to co-locate with two previously identified QTLs viz., Plant height 6–1 (44,862,405–45,270,324 bp) and Plant height 10–4 (44,862,405- 45,270,324 bp) on Chr.19 (Lark et al. 1995; Orf et al. 1999). However, there is no QTL identified to date that falls within the physical interval of qPH17. Hence, qPH17 can be regarded as a novel QTL identified in the current study. The physical intervals of qPH2 and qPH19 were considerably reduced in the present study. This may be because the earlier reported QTLs (Plant height 26–9, Plant height 6–1, and Plant height 10–4) were identified via linkage mapping by using low-density markers such as simple sequence repeat and restriction fragment length polymorphism, possessing a relatively low genomic resolution. These marker systems have low selection accuracy, which has been the biggest obstacle for their deployment in the MAB programs. QTL mapping involves the use of bi-parental mapping populations, which are associated with poor resolution in the detection of target loci regulating traits of interest (Kraakman et al. 2004). To this end, GWAS has emerged as a powerful approach using the ancestral recombination events and has high-resolution in the detection of MTAs. Therefore, high resolution of GWAS in the identification of the stable QTLs across multiple environments will facilitate their effective utilization in MAB programs for breeding soybean varieties with desirable PH.
In the practical breeding programs, the identification of potential markers/candidate genes for the trait of interest underlying a particular genomic region is the ultimate objective (Ganie and Ahammed 2021; Ganie et al. 2021). To date, minimal efforts have been made to identify candidate genes regulating PH in soybean (Liu et al. 2013; Lee et al. 2015; Cao et al. 2017), and only few genes have been characterized (Liu et al. 2010; Ping et al. 2014; Zhang et al. 2018). Hence, based on literature research and gene annotations, we defined 22 candidate genes underlying qPH2, qPH17, and qPH19 that may possibly regulate PH. For example, Dt1 (Glyma.19G194300) gene lying within the physical interval of qPH19 was considered as the candidate gene. The same locus on Chr.19 underlying the Dt1 locus was also identified previously (Sonah et al. 2015; Kato et al. 2019). Liu et al. (2010) observed regulation of the growth habits and PH in soybean by Dt1. In addition to the growth habit genes, maturity and flowering time genes also have a major impact on PH (Cober and Morrison 2010; Xia et al. 2012; Zhang et al. 2015; Cao et al. 2017). Gene function related to growth hormones, mitosis, cell division, cell elongation or functions that are directly or indirectly related to the vegetative growth are involved in regulating PH. To this end, a total of 169 gene models were found underlying the physical intervals of three stable QTLs, and 22 genes (including Dt1) were defined as putative candidates controlling PH by taking into consideration their annotations and gene expression patterns. However, further verification and functional validation of these genes are required to precisely pinpoint their role in the regulation of PH in soybean.
Recently, haplotype-based breeding has emerged as a promising approach to develop custom-designed crop varieties (Varshney et al. 2021). However, this breeding approach needs detection of superior haplotypes for their ultimate use in breeding programs. Haplotype analysis allows plant breeders to utilize the genetic variation underlying key genes/loci to the greatest extent. For example, in soybean, a candidate gene regulating salinity tolerance (GmCHX1) was subjected to haplotype analysis. Among various haplotypes identified for GmCHX1, SV-2 was found to provide maximum salinity tolerance (Patil et al. 2016). Moreover, Wang et al. (2017) identified superior haplotypes for grain quality traits (cooking and eating quality traits) in rice. Sinha et al. (2020) performed haplotype analysis of five genes controlling drought tolerance in pigeonpea. In a given genomic region, many factors regulate haplotype variation, such as mutation, recombination rates, and selection (Zaitlen et al. 2005). Hence, it is a prerequisite to include heterozygous haplotypes to capture maximum haplotypic variation for the analysis of haplotype diversity (Sinha et al. 2020). In our study, the genomic region flanking the most stable significant SNP (AX-93897200) that was in strong LD with 11 significant SNPs formed a single haplotype block. Five haplotypes (Hap-A, Hap-B, Hap-C, Hap-D, and Hap-E) were detected in this haplotype block. The results showed that five haplotypes identified in the block on Chr.19 regulate PH in soybean from dwarf ➝ intermediate ➝ tall. However, our results also reported that these haplotype alleles showed significant environment interaction across six different environments in the regulation of PH. This suggests the presence of haplotype × environment interaction, which needs to be fixed before the utilization of superior haplotype(s) in crop improvement.
A previous study has documented only two haplotypes underlying the Dt1 locus viz., Gm19_Hap42b and Gm19_Hap42a regulating two different phenotypes of PH (Contreras-Soto et al. 2017). This haplotype block showed only two haplotypes (biallelic), and the significant SNP within this region was also biallelic. Therefore, this study showed no difference between the uses of the haplotype or SNP markers within the target region for MAB. The main objective behind the deployment of haplotype markers is that they are multi-allelic and thus provide better opportunity to modify the plant trait. For example, in the present study we identified five different haplotypes for PH underlying the Dt1 locus viz., Hap-A, Hap-B, Hap-C, Hap-D, and Hap-E which regulated five different phenotype classes of PH in soybean. This provides a greater possibility to modify PH in soybean. Therefore, the incorporation of these haplotypes via haplotype-based breeding in soybean breeding programs will be very useful to tailor plant architecture and improve yield and quality.
Conclusion
The present study identified 128 significant SNPs and three stable QTLs/genomic regions (qPH2, qPH17, and qPH19) associated with PH, which were consistently detected in at least three environments. Out of these three QTLs, the QTLs co-localizing with qPH2 and qPH19 have been previously detected by other researchers. However, no QTL was found to be associated with qPH17, implying this locus to be a novel genomic region regulating PH. A total of 22 candidate genes underlying these three QTL regions were also prioritized. The putative candidates defined within the intervals of qPH2, qPH17, and qPH19 need further functional validation before they can be used in soybean breeding programs. The haplotype analysis detected five haplotypes (Hap-A, Hap-B, Hap-C, Hap-D, and Hap-E) within the stable genomic region of Chr.19, which regulated PH ranging from dwarf ➝ intermediate ➝ tall. The superior haplotypes identified in the Chinese soybean germplasm will serve as a potential resource for haplotype-based breeding of PH in soybean.
Data availability
All relevant data can be found within the manuscript and in Supplementary Material.
Code availability
The scripts used for performing different analysis in the present study are provided in Supplementary File 1.
References
Abbai R, Singh VK, Nachimuthu VV, Sinha P, Selvaraj R, Vipparla AK, Singh AK, Singh MS, Varshney RK, Kumar A (2019) Haplotype analysis of key genes governing grain yield and quality traits across 3K RG panel reveals scope for the development of tailor-made rice with enhanced genetic gains. Plant Biotechnol J 17:1612–1622
Allen FL (1994) Usefulness of plant genome mapping to plant breeding. In: Gresshoff PM (ed) Plant genome analysis. CRC Press, Boca Raton, pp 11–18
Bevan MW, Uauy C, Wulff BB, Zhou J, Krasileva K, Clark MD (2017) Genomic innovation for crop improvement. Nature 543:346
Bhat JA, Deshmukh R, Zhao T, Patil G, Deokar A, Shinde S, Chaudhary J (2020) Harnessing high-throughput phenotyping and genotyping for enhanced drought tolerance in crop plants. J Biotechnol 324:248–260
Bradbury PJ, Zhang Z, Kroon DE, Casstevens TM, Ramdoss Y, Buckler ES (2007) TASSEL: software for association mapping of complex traits in diverse samples. Theor Appl Genet 23:2633–2635
Cao Y, Li S, He X, Chang F, Kong J, Gai J, Zhao T (2017) Mapping QTLs for plant height and flowering time in a Chinese summer planting soybean RIL population. Euphytica 213:39
Cao Y, Li S, Chen G, Wang Y, Bhat JA, Karikari B, Kong J, Gai J, Zhao T (2019) Deciphering the genetic architecture of plant height in soybean using two RIL populations sharing a common M8206 parent. Plants 8:373
Chang F, Guo C, Sun F, Zhang J, Wang Z, Kong J, He Q, Sharmin RA, Zhao T (2018) Genome-wide association studies for dynamic plant height and number of nodes on the main stem in summer sowing soybeans. Front Plant Sci 9:1184
Chapman A, Pantalone V, Ustun A, Allen F, Landau-Ellis D, Trigiano R, Gresshoff P (2003) Quantitative trait loci for agronomic and seed quality traits in an F2 and F4: 6 soybean population. Euphytica 129:387–393
Chen K, Zhang Q, Wang CC, Liu ZX, Jiang YJ, Zhai LY, Zheng TQ, Xu JL, Li ZK (2019) Genetic dissection of seedling vigour in a diverse panel from the 3000 Rice (Oryza sativa L.) genome project. Sci Rep 9:4804
Chen C, Chen H, Zhang Y, Thomas HR, Frank MH, He Y, Xia R (2020) TBtools: An integrative toolkit developed for interactive analyses of big biological data. Mol Plant 13:1194–1202
Cober ER, Morrison MJ (2010) Regulation of seed yield and agronomic characters by photoperiod sensitivity and growth habit genes in soybean. Theor Appl Genet 120:1005–1012
Contreras-Soto RI, Mora F, de Oliveira MAR, Higashi W, Scapim CA, Schuster I (2017) A genome-wide association study for agronomic traits in soybean using SNP markers and SNP-based haplotype analysis. PLoS ONE 12:e0171105
Du W, Ning L, Liu Y, Zhang S, Yang Y, Wang Q, Chao S, Yang H, Huang F, Cheng H, Yu D (2020) Identification of loci and candidate gene GmSPX-RING1 responsible for phosphorus efficiency in soybean via genome-wide association analysis. BMC Genom 21:1–16
Fang C, Ma Y, Wu S, Liu Z, Wang Z, Yang R, Hu G, Zhou Z, Yu H, Zhang M, Pan Y, Zhou G, Ren H, Du W, Yan H, Wang Y, Han D, Shen Y, Liu S, Liu T, Zhang J, Qin H, Yuan J, Yuan X, Kong F, Liu B, Li J, Zhang Z, Wang G, Zhu B, Tian Z (2017) Genome-wide association studies dissect the genetic networks underlying agronomical traits in soybean. Genome Biol 18:161
Fang Y, Liu S, Dong Q, Zhang K, Tian Z, Li X, Li W, Qi Z, Wang Y, Tian X, Song J, Wang J, Yang C, Jiang S, Li W-X, Ning H (2020) Linkage analysis and multi-locus genome-wide association studies identify QTNs controlling soybean plant height. Front Plant Sci 11:9
Gabriel SB, Schaffner SF, Nguyen H, Moore JM, Roy J, Blumenstiel B, Higgins J, DeFelice M, Lochner A, Faggart M, Liu-Cordero SN, Rotimi C, Adeyemo A, Cooper R, Ward R, Lander ES, Daly MJ, Altshuler D (2002) The structure of haplotype blocks in the human genome. Science 296:2225–2229
Ganie SA, Ahammed GJ (2021) Dynamics of cell wall structure and related genomic resources for drought tolerance in rice. Plant Cell Rep 40:437–459
Ganie SA, Wani SH, Henry R, Hensel G (2021) Improving rice salt tolerance by precision breeding in a new era. Curr Opin Plant Biol 60:101996
Guan R, Qu Y, Guo Y, Yu L, Liu Y, Jiang J, Chen J, Ren Y, Liu G, Tian L, Jin L, Liu Z, Hong H, Chang R, Gilliham M, Qiu L (2014) Salinity tolerance in soybean is modulated by natural variation in GmSALT3. Plant J 80:937–950
Hyten DL, Song Q, Choi IY, Yoon MS, Specht JE, Matukumalli LK, Nelson RL, Shoemaker RC, Young ND, Cregan PB (2008) High-throughput genotyping with the golden gate assay in the complex genome of soybean. Theor Appl Genet 116:945–952
Kato S, Sayama T, Taguchi-Shiobara F, Kikuchi A, Ishimoto M, Cober E (2019) Effect of change from a determinate to a semi-determinate growth habit on the yield and lodging resistance of soybeans in the northeast region of Japan. Breed Sci 69:151–159
Kim KS, Diers BW, Hyten DL, Mian MAR, Shannon JG, Nelson RL (2012) Identification of positive yield QTL alleles from exotic soybean germplasm in two backcross populations. Theor Appl Genet 125:1353–1369
Knapp SJ, Stroup WW, Ross WM (1985) Exact confidence intervals for heritability on a progeny mean basis1. Crop Sci 25:192–194
Kraakman ATW, Niks RE, Van den Berg PMMM, Stam P, Van Eeuwijk FA (2004) Linkage disequilibrium mapping of yield and yield stability in modern spring barley cultivars. Genetics 168:435–446
Kulkarni KP, Kim M, Shannon JG, Lee JD (2016) Identification of quantitative trait loci controlling soybean seed weight in recombinant inbred lines derived from PI 483463 (Glycine soja) × Hutcheson (G. max). Plant Breed 135:614–620
Kuroha T, Nagai K, Gamuyao R, Wang DR, Furuta T, Nakamori M, Kitaoka T, Adachi K, Minami A, Mori Y, Mashiguchi K, Seto Y, Yamaguchi S, Kojima M, Skakibara H, Wu J, Ebana K, Mitsuda N, Ohme-Takagi M, Yanagisawa S, Yamasaki M, Yokoyama R, Nishitani K, Mochizuki T, Tamiya G, McCouch SR, Ashikari M (2018) Ethylene-gibberellin signaling underlies adaptation of rice to periodic flooding. Science 361:181–186
Lai Y, Cheng J, He Y, Yang B, Wang Z, Zhan H (2016) Identification of QTLs with additive, epistatic, and QTL× seed maturity interaction effects for seed vigor in rice. Plant Mol Biol Rep 34:160–171
Lark KG, Chase K, Adler F, Mansur LM, Orf JH (1995) Interactions between quantitative trait loci in soybean in which trait variation at one locus is conditional upon a specific allele at another. Proc Natl Acad Sci 92:4656–4660
Lee S, Jun TH, Michel AP, Mian MAR (2015) SNP markers linked to QTL conditioning plant height, lodging, and maturity in soybean. Euphytica 203:521–532
Li JY, Wang J, Zeigler RS (2014) The 3000 rice genomes project: new opportunities and challenges for future rice research. Gigascience 3:8
Li B, Fan S, Yu F, Chen Y, Zhang S, Han F, Yan S, Wang L, Sun J (2017) High-resolution mapping of QTL for fatty acid composition in soybean using specific-locus amplified fragment sequencing. Theor Appl Genet 130:1467–1479
Lipka AE, Tian F, Wang Q, Peiffer J, Li M, Bradbury PJ, Gore MA, Buckler ES, Zhang Z (2012) GAPIT: genome association and prediction integrated tool. Bioinformatics 28:2397–2399
Liu BH, Watanabe S, Uchiyama T, Kong FJ, Kanazawa A, Xia ZJ, Nagamatsu A, Arai M, Yamada T, Kitamura K, Masuta C, Harada K, Abe J (2010) The soybean stem growth habit gene Dt1 is an ortholog of Arabidopsis TERMINAL FLOWER1. Plant Physiol 153:198–210
Liu W, Kim MY, Van K, Lee YH, Li H, Liu X, Lee SK (2011) QTL identification of yield-related traits and their association with flowering and maturity in soybean. J Crop Sci Biotechnol 14:65–70
Liu YL, Li YH, Reif JC, Mette MF, Liu ZX, Liu B, Zhang SS, Yan L, Chang RZ, Qiu LJ (2013) Identification of quantitative trait loci underlying plant height and seed weight in soybean. Plant Genome 6:841–856
Liu J, Dou Y, Batistella M, Challies E, Connor T, Friis C, Millington JDA, Parish E, Romulo CL, Silva RFB, Triezenberg H, Yang H, Zhao Z, Zimmerer KS, Huettmann F, Treglia ML, Basher Z, Chung MG, Herzberger A, Lenschow A, Mechiche-Alami A, Newig J, Roche J, Sun J (2018) Spillover systems in a tele-coupled anthropocene: typology, methods, and governance for global sustainability. Curr Opin Environ Sustain 33:58–69
Lü HY, Li H, Fan R, Li H, Yin J, Zhang J, Zhang D (2016) Genome-wide association study of dynamic developmental plant height in soybean. Canad J Plant Sci 97:308–315
Mansur LM, Orf JH, Chase K, Jarvik T, Cregan PB, Lark KG (1996) Genetic mapping of agronomic traits using recombinant inbred lines of soybean. Crop Sci 36:1327–1336
Mishra S, Singh B, Panda K, Singh BP, Singh N, Misra P, Rai V (2016) Association of SNP haplotypes of HKT family genes with salt tolerance in Indian wild rice germplasm. Rice 9:15
Orf JH, Chase K, Jarvik T, Mansur LM, Cregan PB, Adler FR, Lark KG (1999) Genetics of soybean agronomic traits: I. comparison of three related recombinant inbred populations. Crop Sci 39:1642–1651
Patil G, Do T, Vuong TD, Valliyodan B, Lee J-D, Chaudhary J, Shannon JG, Nguyen HT (2016) Genomic-assisted haplotype analysis and the development of high-throughput SNP markers for salinity tolerance in soybean. Sci Rep 6:19199
Ping J, Liu Y, Sun L, Zhao M, Li Y, She M, Sui Y, Lin F, Liu X, Tang Z, Nguyen H, Tian Z, Qiu L, Nelson RL, Clemente TE, Specht JE, Ma J (2014) Dt2 is a gain-of-function MADS-domain factor gene that specifies semi-determinacy in soybean. Plant Cell 26:2831–2842
Ren D, Wang X, Yang M, Yang L, He G, Deng XW (2019) A new regulator of seed size control in Arabidopsis identified by a genome-wide association study. New Phytol 222:895–906
Schmutz J, Cannon SB, Schlueter J, Ma JX, Mitros T, Nelson W, Hyten DL, Song QJ, Thelen JJ, Cheng J, Xu D, Hellsten U, May GD, Yu Y, Sakurai T, Umezawa T, Bhattacharyya MK, Sandhu D, Valliyodan B, Lindquist E, Peto M, Grant D, Shu S, Goodstein D, Barry K, Futrell-Griggs M, Abernathy B, Du J, Tian Z, Zhu L, Gill N, Joshi T, Libault M, Sethuraman A, Zhang XC, Shinozaki K, Nguyen HT, Wing RA, Cregan P, Specht J, Grimwood J, Rokhsar D, Stacey G, Shoemaker RC, Jackson SA (2010) Genome sequence of the palaeopolyploid soybean. Nature 465:120
Severin AJ, Woody JL, Bolon YT, Joseph B, Diers BW, Farmer AD, Muehlbauer GJ, Nelson RT, Grant D, Specht JE, Graham MA (2010) RNA-seq atlas of Glycine max: a guide to the soybean transcriptome. BMC Plant Biol 10:1–16
Sinha P, Singh VK, Saxena RK, Khan AW, Abbai R, Chitikineni A, Desai A, Molla J, Upadhyaya HD, Kumar A, Varshney RK (2020) Superior haplotypes for haplotype-based breeding for drought tolerance in pigeonpea (Cajanus cajan L.). Plant Biotechnol J 18:2482–2490
Sonah H, O’Donoughue L, Cober E, Rajcan I, Belzile F (2015) Identification of loci governing eight agronomic traits using a GBS-GWAS approach and validation by QTL mapping in soya bean. Plant Biotechnol J 13:211–221
Song Q, Jenkins J, Jia G, Hyten DL, Pantalone V, Jackson SA, Schmutz J, Cregan PB (2016) Construction of high-resolution genetic linkage maps to improve the soybean genome sequence assembly Glyma1. 01. BMC Genomics 17:1–11
Sun D, Li W, Zhang Z, Chen Q, Ning H, Qiu L, Sun G (2006) Quantitative trait loci analysis for the developmental behavior of soybean (Glycine max L. Merr.). Theor Appl Genet 112:665–673
Tao T, Zhao L, Lv Y, Chen J, Hu Y, Zhang T, Zhou B (2013) Transcriptome sequencing and differential gene expression analysis of delayed gland morphogenesis in Gossypium australe during seed germination. PLoS ONE 8:e75323
Thudi M, Chen Y, Pang J, Kalavikatte D, Bajaj P, Roorkiwal M, Chitikineni A, Ryan MH, Lambers H, Siddique KHM, Varshney RK (2021) Novel genes and genetic loci associated with root morphological traits, phosphorus-acquisition efficiency and phosphorus-use efficiency in chickpea. Front Plant Sci 12:636973
Varshney RK, Saxena RK, Upadhyaya HD, Khan AW, Yu Y, Kim C, Rathore A, Kim D, Kim J, An S, Kumar V, Anuradha G, Yamini KN, Zhang W, Muniswamy S, Kim J-S, Penmetsa RV, Wettberg E, Datta SK (2017a) Whole-genome resequencing of 292 pigeonpea genotypes identifies genomic regions associated with domestication and agronomic traits. Nat Genet 49:1082
Varshney RK, Shi C, Thudi M, Mariac C, Wallace J, Qi P, Zhang H, Zhao Y, Wang X, Rathore A, Srivastava RK, Chitikineni A, Fan G, Bajaj P, Punnuri S, Gupta SK, Wang H, Jiang Y, Couderc M, Katta MAVSK, Paudel DR, Mungra KD, Chen W, Harris-Shultz KR, Garg V, Desai N, Doddamani D, Kane NA, Conner JA, Ghatak A, Chaturvedi P, Subramaniam S, Yadav OP, Berthouly-Salazar C, Hamidou F, Wang J, Liang X, Clotault J, Upadhyaya HD, Cubry P, Rhoné B, Gueye MC, Sunkar R, Dupuy C, Sparvoli F, Cheng S, Mahala RS, Singh B, Yadav RS, Lyons E, Datta SK, Hash CT, Devos KM, Buckler E, Bennetzen JL, Paterson AH, Ozias-Akins P, Grando S, Wang J, Mohapatra T, Weckwerth W, Reif JC, Liu X, Vigouroux Y, Xu X (2017b) Pearl millet genome sequence provides a resource to improve agronomic traits in arid environments. Nat Biotechnol 35:969–976
Varshney RK, Thudi M, Roorkiwal M, He W, Upadhyaya HD, Yang W, Bajaj P, Cubry P, Rathore A, Jian J, Doddamani D, Khan AW, Garg V, Chitikineni A, Xu D, Gaur PM, Singh NP, Chaturvedi SK, Nadigatla GVPR, Krishnamurthy L, Dixit GP, Fikre A, Kimurto PK, Sreeman SM, Bharadwaj C, Tripathi S, Wang J, Lee SH, Edwards D, Polavarapu KKB, Penmetsa RV, Crossa J, Nguyen HT, Siddique KHM, Colmer TD, Sutton T, von Wettberg E, Vigouroux Y, Xu X, Liu X (2019a) Resequencing of 429 chickpea accessions from 45 countries provides insights into genome diversity, domestication and agronomic traits. Nat Genet 51:857–864
Varshney RK, Pandey MK, Bohra A, Singh VK, Thudi M, Saxena RK (2019b) Toward the sequence-based breeding in legumes in the post-genome sequencing era. Theor Appl Genet 132:797–816
Varshney RK, Bohra A, Yu J, Graner A, Zhang Q, Sorrells ME (2021) Designing future crops: genomics-assisted breeding comes of age. Trends Plant Sci 26:631–649
Wang D, Zhu J, Li Z, Paterson AH (1999) Mapping QTLs with epistatic effects and QTL× environment interactions by mixed linear model approaches. Theor Appl Genet 99:1255–1264
Wang D, Graef GL, Procopiuk AM, Diers BW (2004) Identification of putative QTL that underlie yield in interspecific soybean backcross populations. Theor Appl Genet 108:458–467
Wang J, Chu S, Zhang H, Zhu Y, Cheng H, Yu D (2016) Development and application of a novel genome-wide SNP array reveals domestication history in soybean. Sci Rep 6:1–10
Wang X, Pang Y, Zhang J, Wu Z, Chen K, Ali J, Ye G (2017) Genome-wide and gene-based association mapping for rice eating and cooking characteristics and protein content. Sci Rep 7:17203
Xia Z, Watanabe S, Yamada T, Tsubokura Y, Nakashima H, Zhai H, Anai T, Sato S, Yamazaki T, Lu S, Wu H, Tabata S, Harada K (2012) Positional cloning and characterization reveal the molecular basis for soybean maturity locus E1 that regulates photoperiodic flowering. Proc Natl Acad Sci USA 109:E2155–E2164
Xu Y, Crouch JH (2008) Marker-assisted selection in plant breeding: from publications to practice. Crop Sci 48:391–407
Xue H, Tian X, Zhang K, Li W, Qi Z, Fang Y, Li X, Wang Y, Song J, Li WX, Ning H (2019) Mapping developmental QTL for plant height in soybean [Glycine max (L.) Merr.] using a four-way recombinant inbred line population. PLoS ONE 14:e0224897
Yang J, Zhu J (2005) Methods for predicting superior genotypes under multiple environments based on QTL effects. Theor Appl Genet 110:1268–1274
Yin Z, Qi H, Chen Q, Zhang Z, Jiang H, Zhu R, Hu Z, Wu X, Li C, Zhang Y, Liu C, Hu G, Xin D, Qi Z (2017) Soybean plant height QTL mapping and meta-analysis for mining candidate genes. Plant Breed 136:688–698
Zaitlen NA, Kang HM, Feolo ML, Sherry ST, Halperin E, Eskin E (2005) Inference and analysis of haplotypes from combined genotyping studies deposited in dbSNP. Genome Res 15:1594–1600
Zhang WK, Wang YJ, Luo GZ, Zhang JS, He CY, Wu XL, Gai JY, Chen SY (2004) QTL mapping of ten agronomic traits on the soybean (Glycine max L. Merr.) genetic map and their association with EST markers. Theor Appl Genet 108:1131–1139
Zhang JP, Song QJ, Cregan PB, Nelson RL, Wang XZ, Wu JX, Jiang GL (2015) Genome-wide association study for flowering time, maturity dates and plant height in early maturing soybean (Glycine max) germplasm. BMC Genomics 16:1
Zhang X, Wang W, Guo N, Zhang Y, Bu Y, Zhao J, Xing H (2018) Combining QTL-seq and linkage mapping to fine map a wild soybean allele characteristic of greater plant height. BMC Genomics 19:226
Funding
Open Access funding enabled and organized by CAUL and its Member Institutions. This work was supported in part by National Natural Science Foundation of China (No. 32090065), Ministry of Science and Technology (No. 2017YFE0111000), and Horizon 2020 of European Union (EUCLEG/ No. 727312).
Author information
Authors and Affiliations
Contributions
JAB, DY, and RKV conceived and planned the experiments. JAB and DH performed the experiments. KAA, JAB, BK, SAG, RB, and DH analyzed the data. KAA, JAB, BK, and SAG wrote the initial draft of the manuscript. JAB, DY, and RKV supervised the project. All the authors read and revised the final manuscript.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Ethics approval
The authors declare that no ethical standards have been violated during the course of the study.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Additional information
Communicated by Volker Hahn.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Bhat, J.A., Karikari, B., Adeboye, K.A. et al. Identification of superior haplotypes in a diverse natural population for breeding desirable plant height in soybean. Theor Appl Genet 135, 2407–2422 (2022). https://doi.org/10.1007/s00122-022-04120-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00122-022-04120-0