
Abstract
The Cauliflower Mosaic Virus “35S promotor” (p35S) and the Agrobacterium “Nopaline Synthase” terminator (tNOS) are the most represented generic recombinant elements in commercial genetically modified crops to date. A set of four new SYBR®Green qPCR methods targeting the “p35S” and “tNOS” core elements have been developed. These qPCR methods generate short amplicons of 147 and 75 bp for the “p35S” element and 172 and 69 bp for the “tNOS” element. Single target plasmids containing these amplicons were constructed and allow determining the nominal melting temperature (T m value) of each amplicon. The four methods are specific for their respective targets, and moreover, three of them are highly sensitive (up to 1–2 copies detectable) at a PCR efficiency ranging between 95 and 100%. The latter methods can detect their respective targets at 0.1% (w/w) gDNA levels and are suitable for detecting low levels of genetically modified materials containing the “p35S” and/or “tNOS” elements.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
In the European Union, the development of genetically modified organisms (GMO) is subject to a complex legal framework. The most important GMO EC legislations are the environmental directive EC/2001/18 [1], the GM Food/Feed regulations EC/2003/1829 [2] and EC/2003/1830 [3], the EC Recommendation EC/787/2004 [4] and the Enforcement regulation EC/882/2004 [5]. Within these legislations, the detection of GMO represents an important element for compliance with the conditions set in the authorizations. Molecular characteristics (especially DNA sequence information) represent the most important identification criterion and legal basis for the presence of a particular GMO in a product [2–4].
Consequently, the EU enforcement framework is primarily based on molecular DNA methodology. Within the GM Food/Feed legislation, authorizations of new GM products require the availability of validated (quantitative) product-specific detection methods. Most elaborate in this respect are the so-called event-specific detection methods for GM crops validated by the Community Reference Laboratory for Genetically Modified Organisms (CRL-GMO) of the EC-JRC (Ispra, Italy) [6].
In 2007 on a global basis, about 114.3 million hectares GM crops were cultivated, especially soy, maize and oilseed rape [7]. The most common recombinant elements in these GM crops are the so-called “35S” promoter and “NOS” terminator sequences [8]. The 35S promoter (p35S) and NOS terminator (tNOS) are both transcription-regulating sequences [9, 10]. To date, many EU-authorized GMOs (17/24) contain either the “p35S” (15/24) or the “tNOS” (15/24) or both (9/24) [8, 11, 12] (for more details see Table 1). In order to assess the presence of GM material in a product, screening by “p35S” and/or “tNOS” PCR is very often performed [13]. Several detection methods have already been published for “p35S” and “tNOS” detection in a broad range of matrices. In most cases, either end-point detection on agarose gel or real-time qPCR with TaqMan® probe technology is applied [13–19].
In only a few cases, SYBR®Green qPCR methods were developed for detecting GM targets [e.g., 20, 21]. “SYBR®Green I”, is an asymmetrical cyanine dye [22] which has been reported to specifically detect the presence of double-stranded (ds) DNA [23]. Two criteria are routinely taken into account when assessing the outputs of PCR amplification by SYBR®Green qPCR analysis: the threshold cycle value (C t) and the melting temperature (T m). The C t value of qPCR amplification represents the time-point at which a PCR reaction reaches a prior-set threshold level for the reaction. This threshold level takes into account fluctuations in the background level during early reaction steps and the start of measurable exponential amplification [24, 25]. As such, the lack of a measurable C t value in a qPCR is to be interpreted as the absence of any (exponential) amplification above background level. The T m value represents the temperature at which 50% of the SYBR®Green fluorescence is lost due to denaturation and strand separation of the PCR end product. The T m is a physical parameter inherent to the sequence of the amplified product (esp. the GC content) and influenced by chemical factors that affect double-strand DNA stability (e.g., salt concentration, DMSO, formamide, etc.) [26].
In a GMO screening approach, SYBR®Green qPCR offers a number of advantages over other fluorescence-based PCR methods: (1) SYBR®Green qPCR monitors the increase in total fluorescence throughout the amplification, allowing to estimate the presence of non-specific amplification, (2) the melting temperature analysis allows post-PCR identification of the amplification not only of the expected target but also scoring the presence of closely related target(s), (3) the SYBR®Green technology is (rather) cost-effective as no dye-labeled oligonucleotide probes are required.
In this study, four SYBR®Green qPCR methods were developed allowing detecting core “p35S” and “tNOS” DNA sequences. Representative amplicons for each method were cloned in pENGL™-like vectors and characterized by DNA sequencing. The nominal T m value of the amplicons was determined by using these plasmids as template DNA with each of the SYBR®Green qPCR methods. The specificity of the methods was tested on a range of commodity crop species and on all EU-authorized GMO (date March 2009). Their respective sensitivity was estimated by applying different low-level detection criteria on various GM reference materials.
Materials and methods
Materials
Plant materials
To study the specificity of the different SYBR®Green qPCR methods, genomic DNA (gDNA) from either Certified Reference Materials (CRM) or from in-house grown plants is used. The CRM are obtained from the Institute of Reference Materials and Methods (IRMM) (Geel, Belgium), American Oil Chemists’ Society (AOCS) (Urbana, USA) or Bayer CropScience (Ghent, Belgium). In-house leaf material is produced from seeds obtained from the Biotech Companies or from the local commercial market. All plants are grown in a Snijders Scientific (Tilburg, The Netherlands) S1084 plant growth chamber under standard conditions (16/8 h day/night regime at 25 °C/80% humidity). A list of all applied materials is given in Table 1.
Chemicals, PCR reagents and PCR primers
All applied chemical products are analytical grade (NaCl, EDTA, Tris, boric acid, HCl, CTAB, chloroform, isopropanol, ethanol). The applied enzyme products are: Ribonuclase A (Sigma–Aldrich), Proteinase K (Sigma–Aldrich), EcoRI (Invitrogen) and T4 DNA polymerase (Invitrogen). DNase and RNase free water was purchased from ACROS organics.
All kit-products were used according to the manufacturer’s recommendation: Quant-iT™ PicoGreen® dsDNA Assay Kit, TOPO TA Cloning® Kit, pCR® 2.1-TOPO® vector, TOP10F′ competent cells (Invitrogen); Genome lab, Dye Terminator Cycle Sequencing (DTCS) kit (Beckman Coulter), QIAGEN Plasmid Midi kit (QIAGEN).
Pre-casted “Ready Agarose™ 96 Plus Gel (3%)” (BioRad) gels and “EZ Load HT molecular weight markers (100 bp–2 kb)” (Biorad) were used for agarose gel analysis.
In the PCR reactions, Amplitaq Gold DNA polymerase (Applied Biosystems), Oligold® oligonucleotides (Eurogentec), and SYBR®Green PCR Mastermix [Diagenode (ref: GMO-GS2X-A300)] were used.
Methods
The CTAB gDNA extraction, the qPCR analysis, the agarose gel analysis methods, the applied criteria and the analytical procedures were accredited under ISO-17025 by the official Belgian ISO accreditation organisation “Belac” (2006)
Bioinformatic development of primer pair
All bioinformatic analysis of DNA sequences are performed using the wEMBOSS software package [27–29]. Relevant DNA sequences were collected from public data bases (NCBI and EMBL), patents and scientific literature as well as from in-house DNA sequencing. A uniform primer design approach was applied in the development of primer pairs for the respective targets. A first step consists of identifying regions with high DNA sequence homology within the “p35S” and “tNOS” regions from the different GM events or retrieved DNA sequences. Next, several different primer pairs, comprised within the common target region(s), are designed using the “Primer Express” program from Applied Biosystems (version 3.0) using standard program configuration. An in silico specificity analysis for each primer is performed by probing it against several public and GMO DNA sequence dbases [30, 31] as well as the available in-house sequence information. Any primer showing homology with a non-relevant DNA sequence is discarded from further analysis. The remaining primers are organized in pairs, where as much as possible the primer pairs proposed by Primer Express are retained, and tested experimentally.
Extraction of genomic DNA
A CTAB-based extraction method was applied for the extraction of genomic DNA from all test matrices.
Prior to extraction, leaf tissue is homogenized to powder in a mortar and pestle after liquid nitrogen freezing. Small amounts of seeds (<30 g) are homogenized by crushing in a blender (Kika-Werke Corp.).
Genomic DNA (gDNA) is extracted using a CTAB-based method adapted from Dellaporta et al. 1983 [32]. To a particular powder mass, four volumes (w:v) of CTAB extraction buffer (NaCl 1.4 M, EDTA 0.02 M, Tris–Hcl 0.1 M, CTAB 2%), supplemented with Ribonuclase A (at a final concentration of 15 ng/μl) is added, mixed and incubated for 30 min at 65 °C. Next, Proteinase K (at a final concentration of 100 ng/μl) is added and incubated for 45 min at 65 °C. Upon centrifugation (20 min at 13,000g), 0.2 volume of chloroform is added to the supernatant. After mixing and centrifuging (20 min at 13,000g), the upper phase is collected and two volumes of CTAB precipitation buffer (NaCl 0.04 M, CTAB 0.5%) are added. After gently mixing, the gDNA is precipitated by incubation at room temperature for 1 h. Upon centrifugation (10 min at 13,000g), the gDNA pellet is resuspended in 700 μl NaCl (1.2 M) and 700 μl chloroform, mixed and centrifuged for 15 min at 13,000g. The aqueous phase is collected and 0.6 volume isopropanol is added, mixed and centrifuged (10 min at 13,000g). The pellet is washed with 500 μl of 70% ethanol and centrifuged after washing (10 min at 13,000g). Washing is repeated and the cleaned pellet is dried for 30 min at 28 °C in a dry bath (Fisher Bioblock). Finally, the pellet is resuspended in 200 μl of DNase and RNase free water and allowed to dissolve overnight at 4 °C under agitation. The extracted gDNA is quantified using a VersaFluorTM Fluorometer (Biorad) using the Quant-iT™ PicoGreen® dsDNA Assay Kit. Finally, the gDNA is stored at −20 °C.
Real-time PCR
All qPCR assays are performed on an ABI 7300 PCR System (Applied Biosystems) in 25 μl reaction volume containing 5 μl of template (10 ng/μl gDNA), 1× SYBR®Green PCR Mastermix, and 250 nM of each primer. The following thermal program is applied: a single cycle of DNA polymerase activation for 10 min at 95 °C followed by 40 amplification cycles of 15 s at 95 °C (denaturing step) and 1 min at 60 °C (annealing-extension step). Subsequently, melting temperature analysis of the obtained amplification products is performed by gradually increasing the temperature from 60 to 95 °C in 20 min (±0.6°/20 s). The fluorescent reporter signal is normalized against the internal reference dye (ROX) signal and the threshold limit setting is performed in automatic mode, according to the ABI Sequence Detection Software version 1.4, unless manual adjustment is considered necessary.
Amplicon cloning, sequencing and plasmid deposit
PCR fragments obtained by “classical” PCR amplification using Bt11 leaf gDNA as template are cloned in a pUC18 plasmid applying common “Good Laboratory Cloning Practices” [33]. The respective amplification products are subcloned in pCR®2.1 TOPO using the TOPO TA Cloning® Kit and characterized by restriction analysis. Plasmid DNA from a correct clone is then prepared (QIAGEN Plasmid Midi kit), and the corresponding gel-separated EcoRI fragment isolated and T4-ligated into pUC18 vector DNA (Invitrogen). These plasmids are designated as “Sybricons”, standing for “SYBR®Green amplicon”.
The respective amplicons are characterized by dideoxy-sequence analysis on a CEQ8000 Genetic Analysis System (Beckman Coulter) with the Genome lab, Dye Terminator Cycle Sequencing (DTCS) Quick start Kit. Each obtained sequence is verified by DNA sequence analysis using the alignment ClustalW2 program [34].
The Sybricon plasmids are registered under “Safe Deposit” or “Patent deposit” at the “Belgian Culture Collection for Micro-organisms” in the “Plasmid and DNA Library Collection” ([35] (BCCM/LMBP) (Ghent, Belgium) (see Table 1). Authenticity testing for each plasmid is performed by the BCCM/LMBP prior to acceptance and certification.
SYBR®Green qPCR assay specificity assessment
Primer pair specificity is assessed by testing amplification of reference materials for target-containing and target-lacking GM events (for an overview see Table 1). Four criteria were set to define what is considered as a “specific signal” generated in SYBR®Green qPCR analysis: (1) an (exponential) amplification above the threshold level is obtained with template DNA comprising the target sequence(s), while negative controls [the so-called “No Template Controls” (NTC) and the gDNA from wild-type crop plants] do not yield such amplification; with all target-containing template DNA, the obtained PCR product(s) represents (2) a single peak upon melting analysis with a unique T m value corresponding to the nominal T m value obtained with the respective Sybricon as template DNA (with an acceptable SD ± 1 °C), while no specific peaks are detectable in the negative controls, and (3) a single band on agarose gel analysis with (4) a molecular weight corresponding to the predicted size (SD ± 10 bp).
In each analysis, 50 ng of DNA template is applied. “No Template” controls (NTC) are included in each assay to assess primer dimers formation or specific background fluorescence.
SYBR®Green qPCR assay sensitivity assessment
In this study the sensitivity of the assays was estimated according to the former AFNOR Norm XP V03-020-2 [36] and the IUPAC guidelines [37]. The so-called “LOD6” of a qPCR method for detection of a particular target represents the estimated haploid genome equivalent (HGE), at which level within a linear serial dilution analysis, each of the six repeats provides a positive signal (n = 6; 6/6 specific signals).
In this study, gDNA obtained from leaf tissue of Roundup Ready® soy GTS40-3-2 (RRS) is used as the model system. The calculation of the target copy numbers of “p35S” and “tNOS” in RRS genomic leaf tissue DNA took into consideration the following: (1) an estimated 1.25 pg Haploid Genome Weight for soy as described by Arumugunathan and Earle [38], (2) the homozygous status for the GTS40-3-2 locus in the applied reference material (gDNA from leaf tissue of homozygous seeds (Monsanto Company)), and (3) the available information on the inserted DNA present in RRS [8, 11, 12, 39, 40]. Based on these data, the “Roundup Ready GTS 40-3-2” locus comprises 1 copy of “p35S” and 1 copy of “tNOS” per haploid genome.
The SYBR®Green qPCR assay sensitivity is assessed by (1) serial dilution (in water) of leaf tissue DNA from homozygous Roundup Ready® soy GTS40-3-2 (RRS) (40.000–0.1 HGE), and (2) a dilution of the same leaf tissue DNA RRS in leaf tissue DNA Wt Soybean at 100, 1 and 0.1% RRS. All analyses are repeated sixfold and the LOD6 is determined. From these analyses, also the PCR efficiency (E) for each of the methods can be calculated according to: [41]
The PCR efficiency (E) could be expressed in percentage:
Agarose gel analysis
Agarose gel electrophoresis (3% precast gels, Biorad) is performed using 0.5× TBE (45 mM Tris–borate–1 mM EDTA) at 100 V for 15 min, including a 100 bp–2 kb Molecular Marker (BioRad).
Results and discussion
Identification of core target DNA regions in the “p35S and “tNOS” elements present in the “EU-authorized GMO” Universe (March 2009), primer design and selection
Most EU-authorized GMOs contain either the “p35S” or the “tNOS” element, or both of them (see Table 1) [8, 11, 12]. In order to develop primer sets that specifically amplify all the “p35S” or the “tNOS” elements as present in the EU-authorized GM plants, a Bioinformatics DBase was compiled containing all the available relevant DNA sequences. Within both elements, a highly conserved core region could be identified: a 366-bp sequence for the “p35S” (reference GenBank: V00141.1, position 7,072–7,437) and a 256 bp for “tNOS” (reference GenBank: V00087.1, position 1,844–2,099). A common strategy for the development and selection of primer sets for both core elements was then applied (see “Materials and methods”). Several primer pairs were developed and a limited assessment of their amplification efficiency, selectivity, and specificity on gDNA of several target-containing GMO was performed (data not shown). The primer pairs listed in Table 2 performed best in this assessment. The corresponding qPCR methods are further designated as “p35S-long”, “p35S-short”, “tNOS-long”, and “tNOS-short”, respectively. To guarantee that these qPCR methods amplified the correct target sequences, so-called “Sybricon” plasmids containing the respective amplification products are constructed using gDNA from Bt11 maize leaf tissue as template DNA. The DNA sequences of the cloned amplicons are shown in Fig. 1. The obtained sequences match perfectly with the sequence from which the primers were designed. “p35S-long” amplicon matches reference GenBank: V00141.1 (position 7,249–7,395), “p35S-short” amplicon matches reference GenBank: V00141.1 (position 7,323–7,397), and “tNOS-long” amplicon matches reference GenBank: V00087.1 (position 1,850–2,021). The “tNOS-short” amplicon matches reference GenBank: V00087.1 (position 1,996–2,064) with a single mismatch in position 2,055 (A → C) due to a degenerate nucleotide in the reverse primer (Table 2). The respective sequences match perfectly the expected ones as notified for Bt11 maize and recognize all to date “p35S” and “tNOS” containing EU-authorized GMO [as evaluated through blast analysis of the CCSIS Bioinformatics data analysis [40] (data not shown).

DNA sequence of the “p35S-long”, “p35S-short”, “tNOS-long” and “tNOS-short” amplicons obtained by SYBR®Green qPCR using “Sybricon” reference plasmids as template DNA. a Sybricon010 (p35S-long qPCR). b Sybricon017 (p35S-short qPCR). c Sybricon001 (tNOS-long qPCR). d Sybricon006 (tNOS-short qPCR). The reverse and forward sequencing primers are indicated in bold
T m value determination for the “p35S” and “tNOS” SYBR®Green qPCR amplicons with “reference plasmids” as DNA template
To minimize bias due to the genetic background in determining the nominal value of the melting temperature for each target, the “Sybricon” plasmids containing the respective amplification products were used to generate each of the “p35S” and “tNOS” amplicons. The T m values for the different “p35S” and “tNOS” amplicons are distinct from each other with a T m value at 80 and 76.5 °C for the “p35S-long” and “p35S-short”, respectively, and at 73.3 and 72.5 °C for the “tNOS-long” and “tNOS-short”, respectively (Table 1A). It is generally accepted that the T m obtained with SYBR®Green qPCR could vary between 0.5 and 1 °C for the same amplicon [43, 44]. Therefore, to cover slight deviations in the T m value between reference materials (Sybricons) and samples due to analyte impurities, a standard deviation of ±1 °C on the nominal T m value will be applied, as the acceptance range, in further analysis.
Determination of “p35S” and “tNOS” SYBR®Green qPCR specificity
Using the 4 SYBR®Green qPCR methods, all target-containing GM-event samples give specific signal for “p35S” and/or “tNOS” (Table 1B). All NTC samples are negative and also all WT crop templates do not yield any specific signals. It can thus be concluded that all four methods are specific for their targets. In several CRM (8 out of 35 materials), however, weak-positive signals are detectable (indicated with “–a” in Table 1B.). These weak-positive signals are most probably due to the presence of low amounts of GMO impurities in the reference materials because the C t levels of the signals reside at or below the LOD of the methods (see below) and a ∆Ct > 6 between these aberrant signals and any target-positive element is observed. The CRM are certified for the presence of a specific target at a particular mass% but are not certified for the absence of any other GM targets that could be present at low level [45, 46]. Due to the very low quantities present, the nature of these impurities was not further investigated.
All specific signals in the target-containing GMO generate a unique peak in melting analysis and the T m values of the PCR products differ less than 1 °C from the nominal T m value of the corresponding Sybricon plasmids (see Table 1B and Fig. 2). No additional peaks were observed in these analyses. Thus, the 4 SYBR®Green qPCR reactions generate a single specific signal without major additional amplification products.

Linear amplification plots (panels a–d) and melting curves (panels a’–d’) obtained by SYBR®Green qPCR analysis of the target-containing GMO listed in Table 1. The different qPCR methods applied are the p35S-long qPCR in panel a and a’, the p35S-short qPCR in panel b and b’, the tNOS-long qPCR in panel c and c’ and the tNOS-short in panel d and d’. In the amplification curves (panels a–d), the cycle number is plotted on the X-axis versus the measured fluorescence increase (expressed as ∆Rn) on the Y-axis. In the melting curve analysis (panels a’–d’), the temperature (°C) is plotted on the X-axis versus the inverse of the first derivate of the best-fitted curve of the measured fluorescence decrease on the Y-axis
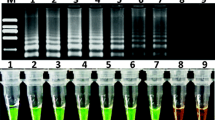
Agarose gel analysis of the respective PCR products yields a single band at the expected molecular weight in all target-containing GMO (147 bp for “p35S-long, 75 bp for “p35S-short”, 175 bp for “tNOS-long” and 69 bp for “tNOS-short”). Again, no major additional amplification products are observed (Fig. 3).

Agarose gel electrophoresis of the “p35S” and “tNOS” PCR products amplified by SYBR®Green qPCR from gDNA extracted from reference material containing these elements. The respective qPCR methods applied were: panel a the p35S-long qPCR (expected amplicon length: 147 bp), panel b the p35S-short qPCR (expected amplicon length: 75 bp), panel c the tNOS-long qPCR (expected amplicon length: 172 bp), and panel d the tNOS-short qPCR (expected amplicon length: 69 bp). Tested GMO events containing these elements are: 1 “MON1445”, 2 “MON531”, 3 “MON15985”, 4 “LL25”, 5 “BT11”, 6 “BT176”, 7 “DAS 59122”, 8 “GA21”, 9 “MIR604”, 10 “MON810”, 11 “MON863”, 12 “NK603”, 13 “T25”, 14 “TC1507”, 15 “RF3”, 16 “T45”, 17 “MS8”, 18 “EH92-527-1”, 19 “LL62” (Bayer material), 20 “LL62” (AOCS material), 21 “A2704-12”, 22 “GTS 40-3-2”. M EZ load HT molecular marker, 100 bp–2 kb (5 bands: 100, 200, 500, 1,000, 2,000 bp)
Sensitivity of the 4 SYBR®Green qPCR methods for “p35S” and “tNOS” analytes on “model” reference materials
The results for the LOD6 determination for the 4 SYBR®Green qPCR methods by serial dilution of leaf DNA from RRS is shown in Table 3. For the “p35S-long”, “p35S-short” and “tNOS-short” qPCR methods, the LOD6 can be set at 1–2 estimated HGE of the respective targets (Table 3). In the dilution series of the “p35S-long” analysis, the one copy dilution showed an initial deviation from the 6/6 positives, what would make the 2-copy level the LOD6. However, at the consecutive estimated 0.5-copy dilution in this particular series, again 6/6 positives were found and a single positive was found at 0.1 copy. To clarify this statistically highly improbable observation, the latter dilution series was repeated and this time, the 1-copy dilution yielded 6/6, the 0.5 copy 3/3 and the 0.1 copy a 0/6 positives, respectively (data not shown). This allows to conclude that the LOD6 for the “p35S-long” method is indeed to be set at 2 copies. These three qPCR methods meet as such the criteria set by Kay and Van den Eede [47] (LOD < 20 copies) and by the ENGL method performance guidelines (2008) [41]. The PCR efficiencies of these 3 SYBR®Green qPCR methods (92.4, 94.2 and 96.1% for the “p35S-long”, “p35S-short” and “tNOS-short” qPCR methods, respectively), also meet the ENGL acceptance criteria (accepted PCR efficiency between 89.6 and 110.2%) [41]. The performance of the “tNOS-long” method is however not acceptable with respect to both its sensitivity (LOD6 > 400 estimated copies) and its PCR efficiency (75.4%). With the “p35S-short” SYBR®Green qPCR method one false positive is observed in a NTC; this weak signal (C t = 36.75) is probably the result of aerosol contamination (e.g., from the co-analyzed RRS samples).
Finally, the performance of the 4 SYBR®Green qPCR methods on admixed leaf tissue gDNA preparation at 0.1, 1 and 100% RRS (w/w) was evaluated (Table 4). The “p35S-long”, “p35S-short” and “tNOS-short” methods reliably detect 0.1% RRS, whereas the “tNOS-long” method fails at the 0.1% level (only 2/6 detected). Again, one weak-false positive signal was observed with the “p35S-short” SYBR®Green qPCR method in a NTC sample (C t = 36.95). The lesser PCR sensitivity of the “tNOS-long” method is also reflected in a much larger ΔC t with the “tNOS-short” method (ΔC t = 10), compared to the ΔC t between both “p35S” methods (ΔC t = 4.5), Together, these results confirm that only three of the developed SYBR®Green qPCR methods are suitable in detecting low levels of GM material comprising “p35S” or “tNOS” elements.
Conclusion
Four different SYBR®Green qPCR methods for detecting “p35S” and “tNOS” elements, currently the two major targets in GMO screening analysis, have been developed. All four methods perform reliably with respect to target specificity, as (1) only target-positive DNA templates generate an exponential amplification, (2) the melting temperature analysis of the generated amplicons represents a single peak at the expected temperature, (3) a single band is visualized by agarose gel analysis with target-containing GM-event samples, and (4) the MW and DNA sequence of the respective amplification products matches the expected size and predicted DNA sequence. Three SYBR®Green qPCR methods (“p35S-long”, “p35S-short” and “tNOS-short”) have a high PCR efficiency (between 91 and 96%,) and are highly efficient at detecting low target concentrations [LOD < 20 HGE; 0.1% RRS (w/w)]. These three SYBR®Green qPCR methods offer a new valuable tool in screening for GMO presence in products. Combining these methods for generic targets with appropriate methods for GMO discriminating targets such as trait and/or endogenous markers, may enable the development of a cost-efficient GMO screening platform.
References
EC/2001/18 (2001) Directive 2001/18/EC of the European Parliament and of the Council of 12 March 2001 on the deliberate release into the environment of genetically modified organisms and repealing Council Directive 90/220/EEC
EC/1829/2003 (2003) Regulation (EC) No 1829/2003 of the European Parliament and of the Council of 22 September 2003 on genetically modified food and feed
EC/1830/2003 (2003) Regulation (EC) No 1830/2003 of the European Parliament and of the Council of 22 September 2003 concerning the traceability and labelling of genetically modified organisms and the traceability of food and feed products produced from genetically modified organisms and amending Directive 2001/18/EC
EC/787/2004 (2004) Commission recommendation of 4 October 2004 on technical guidance for sampling and detection of genetically modified organisms and material produced from genetically modified organisms as or in products in the context of Regulation (EC) No 1830/2003
EC/882/2004 (2004) Regulation (EC) No 882/2004 of the European Parliament and of the Council of 29 April 2004 on official controls performed to ensure the verification of compliance with feed and food law, animal health and animal welfare rules
Community Reference Laboratory (CRL) Status of dossier web-page: http://gmo-crl.jrc.ec.europa.eu/statusofdoss.htm
James C (2007) ISAAA brief 37-2007: executive summary global status of commercialized biotech/GM Crops 2007. Available at http://www.isaaa.org/resources/publications/briefs/37/executivesummary/default.html
Bruderer S, Leitner KE, Lindenmeyer J (2003) Genetically modified (GM) crops: molecular and regulatory details, BATS, centre for biosafety and sustainability, version 2. Available at http://www.bats.ch/gmo-watch/index.php
Odell JT, Nagy F, Chua NH (1985) Identification of DNA sequences required for the activity of the cauliflower mosaic virus 35S promoter. Nature 313:810–812
Depicker A, Stachel S, Dhaese P, Zanbryski P, Goodman HM (1982) Nopaline synthase: transcript mapping and DNA sequence. J Mol Appl Genet 1(6):561–573
GMO compass web site: http://www.gmo-compass.org
Agbios website: http://www.agbios.com/dbase.php
Waiblinger HU, Ernst B, Anderson A, Pietsch K (2008) Validation and collaborative study of p35S and T-nos duplex real-time PCR screening method to detect genetically modified organisms in food products. Eur Food Res Technol 226:1221–1228
Fernandez S, Charles-Delobel C, Geldreich A, Berthier G, Boyer F, Collonier C, Coué-Philippe, Diolez A, Duplan MN, Kebdani N, Romanick M, Feinberg M, Bertheau Y (2005) Quantification of the 35 S promoter in DNA extracts from genetically modified organisms using real-time polymerase chain reaction and specificity assessment on various genetically modified organisms, part I: operating procedure. J AOAC Int 88(2):547–557
Höhne M, Santisi CR, Meyer R (2002) Real-time multiplex PCR: an accurate method for detection and quantification of 35S-CaMV promoter in genetically modified maize-containing food. Eur Food Res Technol 215:59–64
Corbisier P, Trapmann S, Gancberg D, Hannes L, Van Iwaarden P, Berben G, Schimmel H, Emons H (2005) Quantitative determination of Roundup Ready soybean (Glycine max) extracted from highly processed flour. Anal Bioanal Chem 383(2):282–290
Yang LT, Shen HF, Pan AH, Chen JX, Huang C, Zhang DB (2005) Screening and construct-specific detection methods of transgenic Huafan No 1 tomato by conventional and real-time PCR. J Sci Food Agric 85(13):2159–2166
Pardigol A, Guillet S, Pöpping B (2003) A simple procedure for quantification of genetically modified organisms using hybrid amplicon standards. Eur Food Res Technol 216:412–420
Reiting R, Broll H, Waiblinger HU, Grohman L (2007) Collaborative study of T-nos real-time PCR method for screening of genetically modified organisms in food products. J Verbr Lebensm 2:116–121
Hernandez M, Rodriguez-Lazaro D, Esteve T, Prat S, Pla M (2003) Development of melting temperature-based SYBR Green I polymerase chain reaction methods for multiplex genetically modified organism detection. Anal Biochem 323:164–170
Li Y, Xing D, Zhang C (2009) Rapid detection of genetically modified organisms on a continuous-flow polymerase chain reaction. Anal Biochem 385:42–49
Zipper H, Brunner H, Bernhagen J, Vitzthum F (2004) Investigations on DNA intercalation and surface binding by SYBR Green I, its structure determination and methodological implications. Nucleic Acids Res 32(12):e103
Haugland RP (2002) Handbook of fluorescence probes and research products, Ninth edn. Molecular Probes, Eugene, OR
Bustin SA (2000) Absolute quantification of mRNA using real-time reverse transcription polymerase chain reaction assays. J Mol Endocrinol 25:169–193
Tse C, Capeau J (2003) Quantification des acides nucléiques par PCR quantitative en temps réel. Ann Biol Clin 61(3):279–293
Ye J, Parra EJ, Sosnoski DM, Hiester K, Underhill PA, Shriver MD (2002) Melting curve SNP (McSNP) genotyping: a useful approach for diallelic genotyping in forensic science. J Forensic Sci 47:593–600
wEMBOSS is an Open Source software: http://wemboss.sourceforge.net/
Rice P, Longden I, Bleasby A (2000) EMBOSS the European molecular biology open software suite. Trends Genet 16(6):276–277
Sarachu M, Colet M (2005) wEMBOSS: a web site interface for EMBOSS. Bioinformatics 21(4):540–541
EMBL database: http://www.ebi.ac.uk/embl/
RefSeq NCBI database: http://www.ncbi.nlm.nih.gov/RefSeq/
Dellaporta SL, Wood J, Hicks JB (1983) A Plant DNA Minipreparation Version II. Plant Mol Biol Report 1(4):19–21
Sambrook J, Russell DW (2001) Molecular cloning, a laboratory manual, vol 1, 2, 3, 3rd edn. Cold Spring Harbor Laboratory Press, Cold Spring Harbor
ClustalW2 program on EMBL-EBI website:http://www.ebi.ac.uk/Tools/clustalw2/index.html
BCCM/LMBP plasmids catalogue website: http://bccm.belspo.be/db/lmbp_search_form.php
Norme AFNOR XP V03-020-2 (04/2005) Produits alimentaires–Détection et quantification des organismes végétaux génétiquement modifiés et produits dérivés–Partie 2: méthodes basées sur la réaction de polymérisation en chaîne
Thompson M, Ellison SLR, Wood R (2002) Harmonized guidelines for single-laboratory validation of methods of analysis, (IUPAC Technical Report). Pure Appl Chem 74(5):835–855
Arumugunathan K, Earle ED (1991) Nuclear DNA content of some important plant species. Plant Mol Biol Report 9:208–218
Windels P, Taverniers I, Depicker A, Van Bockstaele E, De Loose M (2001) Characterisation of the Roundup Ready soybean insert. Eur Food Res Technol 213:107–112
Central Core DNA Sequence Information System (CCSIS): http://bgmo.jrc.ec.europa.eu/home/bioinformatics/bioinformatics.html
Definition of minimum performance requirements for analytical methods of GMO testing, European Network of GMO Laboratories (ENGL), 13 October 2008. Available at http://gmo-crl.jrc.ec.europa.eu/
Berben G, Dardenne P (2001) Traçage et authentification des produits à base d’organismes génétiquement modifiés, Recherches financées par les SSTC dans le cadre du programme de “Recherche pré-nominative dans le secteur alimentaire”, CRA-W, rapport finale contract NP/42/026
Donohoe GG, Laaksonen M, Pulkki K, Rönnemaa T, Kairisto V (2000) Rapid single-tube screening of the C282Y hemochromatosis mutation by real-time multiplex allele-specific PCR without fluorescent probes. Clin Chem 46(10):1540–1547
Herrmann MG, Durtschi JD, Wittwer CT, Voelkerding KV (2007) Expanded instrument comparison of amplicon dna melting analysis for mutation scanning and genotyping. Clin Chem 53(8):1544–1548
AOCS certified material: http://members.aocs.org/source/orders/index.cfm?section=orders&task=1&CATEGORY=H-CRMS&DESCRIPTION=Certified%20Reference%20Materials&CFTOKEN=38450281&continue=1&SEARCH_TYPE=find
IRMM certified material catalogue: http://irmm.jrc.ec.europa.eu/html/reference_materials_catalogue/catalogue/index.htm
Kay S, Van den Eede G (2001) The limit of GMO detection. Nat Biotechnol 19:405 405
Acknowledgments
The authors would like to greatly thank Els Vandermassen and Dirk van Geel for their technical assistance. Gilbert Berben (CRA-W, Belgium) and his team are acknowledged for providing the “p35S-long” primers sequences prior to publication. This study was financially supported by the European Commission through the Integrated Project Co-Extra, Contract No. 007158, under the 6th Framework Program, and by the GMODETEC project (RT-06/6) of the Belgian federal ministry of “Health, Food Chain safety and Environment”.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Barbau-Piednoir, E., Lievens, A., Mbongolo-Mbella, G. et al. SYBR®Green qPCR screening methods for the presence of “35S promoter” and “NOS terminator” elements in food and feed products. Eur Food Res Technol 230, 383–393 (2010). https://doi.org/10.1007/s00217-009-1170-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00217-009-1170-5