
Abstract
This paper reports the evaluation of the Quick, Easy, Cheap, Effective, Rugged and Safe (QuEChERS) method for the determination of polycyclic aromatic hydrocarbons (PAHs) in food of animal origin with GC–MS detection. Although in the available literature, there is a lot of information about sample preparation method for PAHs determination in food samples, but the QuEChERS method application for PAHs determination in food of animal origin has not been reported as yet. The results showed that the best recovery ratios 72.4–110.8 % with relative standard deviation lower than 10 % for all determined compounds were received for the method with ethyl acetate as an extraction solvent, primary–secondary amine and C18 sorbents and evaporation to dryness and dissolving the residues in the hexane. The limit of quantification ranged from 0.0003 to 0.0030 mg kg−1 for pyrene and benzo[a]anthracene, respectively. This method was also used for the determination of PAHs in 15 samples of pork ham. In 8 of 15 samples selected, PAHs were identified. It was observed that in 6 cooked ham and one smoked and cooked samples, any PAHs were found. In other samples, which were smoked and roasted, some low concentration of PAHs was detected. In one sample benzo[a]pyrene (0.0015 mg kg−1), in one sample benzo[b]fluoranthene (0.0015 mg kg−1) and in one sample chrysene (0.0024 mg kg−1) were detected. A number of other less harmful PAHs were also determined. There were no exceedances of maximum levels (according to Commission Regulation (EU) No 835/2011) for determined PAHs in any of the analysed samples.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Polycyclic aromatic hydrocarbons (PAHs) are a large group of organic compounds, containing two or more aromatic rings and belonging to the food and environmental contaminants [1]. The compounds containing five or more aromatic rings are known as ‘heavy’ PAHs, whereas those containing less than five rings are named ‘light’ PAHs. Both kinds of PAHs are non-polar compounds, showing high lipophilic nature, although heavy PAHs are more stable and toxic than the other group [2]. PAHs are ubiquitous environmental contaminants that are widespread in the air bonded to particulate matter. In spite of PAHs showing hydrophobic properties (especially heavy PAHs), they are also found in water.
PAHs originate from environmental sources (natural and anthropogenic), industrial food processing (e.g. heating, drying and smoking processes), packaging materials and certain cooking practices (e.g. grilling, roasting and frying processes). In fact, the main source of exposure to PAHs for non-smokers and non-occupationally exposed adults is food [3]. Apart from analytical discrepancy, this variation in PAHs levels in food is mainly due to the type and fat content of the food, cooking process (fried, grilled, roasted, boiled and smoked), temperature and duration of cooking, type of fuel used (electrical, gas, wood and charcoal) and proximity and direct contact with heat [4–7].
A number of PAHs are considered as genotoxic carcinogens, and other biological and mutagenic effects have also been reported. Other PAHs not defined as carcinogens may act as synergists. In general, PAHs are not present individually but in mixtures. PAHs that have been extensively monitored are the compounds included in the United States Environmental Protection Agency (USEPA) list of priority organic pollutants (the so-called 16 EPA PAHs) [2].
According to the Scientific Committee on Food [8], 15 PAHs showed clear evidence of mutagenicity/genotoxicity in somatic cells in experimental animals in vivo. They may be regarded as potentially genotoxic and carcinogenic to humans; their carcinogenicity is initiated by their metabolic conversion in mammalian cells to diol epoxides that bind covalently to cellular macromolecules, including DNA, causing errors in DNA replication and mutation [5].
According to the Scientific Committee on Food, benzo[a]pyrene can be used as a marker for the occurrence and effect of carcinogenic PAHs in food [9]. In view of the presence of PAHs in food and their significant toxicity, control of these compounds in food is necessary [1].
The methodology for PAHs analysis in environmental and plant samples is very well documented, and many examples are available in the literature. A number of recent reviews dealt with PAHs analysis in various foodstuffs [10–12].
It is well known that one of the main difficulties in the analysis of fatty matrices is due to their high fat content (e.g. lipids, triglycerides and fatty acids). Hence, the extraction of PAHs from these complex matrices is usually laborious and time-consuming. The removal of lipidic material is important not only to minimise the maintenance of the chromatographic system (especially when using GC) but also to reach low detection limits (LODs). The need for high sensitivity is justified by the low concentrations of PAHs fixed as maximum levels permitted in current legislation [2].
Isolation, identification and quantitative determination of PAHs in a complex food matrix suffer from three main problems. So far, most PAHs identified occur in food at microtrace levels, i.e., ppb or ppt levels, which makes their selective separation very difficult. Many other organic components are co-extracted from the matrix with the PAHs and make identification of the PAHs by chromatographic and spectral methods difficult; and PAHs are characterised by structural similarity and many occur as isomers, which again makes identification of individual compounds extremely difficult [13].
Extraction of PAHs from foodstuffs has traditionally relied on a three-stage methodology, including saponification, liquid–liquid extraction (LLE) and clean-up by column chromatography or, more recently, solid-phase extraction (SPE). Several methods have been described for the analysis of PAHs, with different techniques of extraction, purification and detection [1, 2, 13].
The need for a simple, rapid, cost-effective and multi-residue method able to provide high quality of analytical results led to develop a new sample treatment method. For multi-residue applications, Quick, Easy, Cheap, Effective, Rugged and Safe (QuEChERS) is a frequent and attractive alternative method for sample treatment. The QuEChERS method is particularly popular to determine moderately polar pesticide residues in various food matrices [11], although this methodology is also being used for the analysis of other family of compounds [12, 13]. The QuEChERS methodology has already been applied to the analysis of PAHs in fish and shrimp and acrylamide in various food matrices such as chocolate, peanut butter and coffee [14, 15].
It is based on the extraction with organic solvent/acetonitrile partitioned from the aqueous matrix using anhydrous MgSO4 and NaCl followed by a dispersive SPE (d-SPE) with MgSO4, primary–secondary amine (PSA) and other sorbents (GCB, C18 and SAX) and analysis by gas or liquid chromatography–mass spectrometry. Isotope dilution technique (deuterated standards used as an internal standard) is usually employed for the compensation of potential analyte losses and matrix-inducted chromatographic response enhancement.
Although in the available literature, there is a lot of information about sample preparation method for PAHs determination in food of animal origin, there is only few examples of QuEChERS method application. These studies are important because food of animal origin especially ham is widespread in the diet of Europeans.
The aim of this study was the application of the modified QuEChERS method for the analysis of twelve PAHs (acenaphthylene, fluorene, phenanthrene, anthracene, pyrene, benzo[a]anthracene, chrysene, benzo[b]fluoranthene, benzo[k]fluoranthene, benzo[a]pyrene, indeno[1,2,3-c,d]pyrene and dibenzo[a,h]anthracene) from selected samples of hams.
Materials and methods
Chemicals
Acetonitrile (MeCN), HPLC grade and ethyl acetate (EtAc) for liquid chromatography LiChrosolv® were purchased from Merck KGaA, Germany. Magnesium sulphate anhydrous p.a. and sodium chloride p.a. were purchased from Chempur SA, Poland. Bondesil PSA 40 μm, GCB and C18 sorbents were purchased from Agilent Technologies, USA. EPA 525 PAH Mix-B (for the study, the following PAHs were selected: acenaphthylene, fluorene, phenanthrene, anthracene, pyrene, benzo[a]anthracene, chrysene, benzo[b]fluoranthene, benzo[k]fluoranthene, benzo[a]pyrene, indeno[1,2,3-c,d]pyrene and dibenzo[a,h]anthracene) and anthracene d10 (IS) were obtained from Supelco, USA. Stock, intermediate and working standard solution PAHs at concentration 1 μg mL−1 and anthracene d10 at concentration 1 μg mL−1 were prepared in hexane. Deionised water (18 MΩ) was produced by a Milli-Q system (Millipore; USA).
Equipment
Varian 4000 GC/MS (Varian, Inc., USA) system consisted of 3800 GC and 4000 Ion Trap MS detector was used to perform the GC–MS analyses. The autosampling injector was CP-1177 Split/Splitless Capillary Injector, with a temperature of 270 °C, and a volume of 1.0 μL, the splitless time being 1.0 min for all standards and samples. Each injection was repeated three times. Chromatographic separations were conducted using a Phenomenex, Inc. Zebron Multiresidue-1 (30 mL × 0.25 mm i.d. x 0.25 μm df; Phenomenex, Inc., USA). The GC oven was operated with the following temperature programme: initial temperature 50 °C (1.0 min)–15 °C min−1–320 °C (6.0 min). The total analysis time was 25 min. Helium (Linde Gas, Poland) was used as the GC carrier gas at a flow rate of 1.0 mL min−1.
The ion trap mass spectrometer was operated on the internal ionisation mode in the range 45–500 m/z. Analysis was conducted in the selected ion monitoring mode (SIM) based on the use of one quantitative ion. Analysed compounds were identified according to their qualitative ions and retention times (Table 1). The trap and the transfer line temperatures were set at 180 and 220 °C, respectively. The analyses were carried out with the solvent delay of 8.0 min. The emission current of the ionisation filament was set at 15 μA. Acquisition and processing data were performed using Varian Start Workstation software and NIST 2.0 library.
Accu™ Thermoblock (Labnet, USA) with nitrogen (Linde Gas, Poland) was used to evaporate the solvent and concentrate the extracts. MPW-350R centrifuge (MPW Med. Instruments, Poland) was used for sample preparation.
QuEChERS sample preparation method for PAHs determination
The study involved two experiments. In the first step, we focused on adaptation of QuEChERS method for PAHs determination in samples of animal origin. Then, in the second part, the optimised procedure was applied for real samples of ham, available on retailed market.
A series of experiments were performed for the optimisation the sample preparation techniques, including selecting the appropriate, additional sorbent for clean-up the samples (C18 or GCB), apart from PSA sorbent, which is an essential material in QuEChERS method. Additionally, different types of solvents (MeCN, EtAc) and the final method of sample preparation by solvent evaporation to dryness (and dissolving the residues in the hexane) or the solvent exchange from acetonitrile to hexane were also tested (microscale LLE). The methods are named as follows: A (EtAc, PSA + C18 and solvent evaporation), B (EtAc, PSA + C18 + GCB and solvent evaporation), C (MeCN, PSA + C18 and solvent evaporation), D (MeCN, PSA + C18 and LLE), E (MeCN, PSA + C18 + GCB and solvent evaporation) and F (MeCN, PSA + C18 + GCB and LLE). For the better illustration, the breakdown of the methods is shown in Table 2).
The usefulness of the method was verified on the basis of the recovery ratio of analysed compounds (analysis of spiked samples). Homogenised samples of ham with no PAHs detected previously were used for recovery studies. Recovery study involved two samples of food of animal origin being spiked with the standard solution of PAHs to the fortification level of 0.005 mg kg−1. This level has been adapted to the MRL’s limit set in EU (5 μg kg−1) [16]. Chromatogram of a spiked sample is shown in Fig. 1.

Chromatogram of spiked sample of ham (1—acenaphthylene, 2—fluorene, 3—phenanthrene, 4—anthracene d10, 5—anthracene, 6—pyrene, 7—benzo[a]anthracene, 8—chrysene, 9—benzo[b]fluoranthene, 10—benzo[k]fluoranthene, 11—benzo[a]pyrene, 12—indeno[1,2,3-c,d]pyrene and 12—dibenzo[a,h]anthracene)
A representative portion of ham was cut, macerated and homogenised in a blender. Eight grams of each homogenised sample was weighed into a 50-mL centrifuge tube, and the samples were spiked with mixture of PAHs, mixed and left to stand for 15 min at room temperature prior to the extraction. Then, 10 mL of a suitable solvent (MeCN or EtAc) was added to each tube, and the mixture was shaken vigorously for 1 min. After that, 1 g NaCl and 4 g MgSO4 were added, and the tubes were shaken immediately after addition of the salt. Then, each sample was shaken vigorously for 1 min and centrifuged for 15 min at 8700 RCF. Six millilitres of the supernatant was transferred into a PP 15-mL tube containing 0.150 g PSA, 0.300 g C18 (and/or 0.045 g GCB) and 0.900 g MgSO4. The tubes were shaken for 30 s and centrifuged for 5 min at 5000 RCF. Four millilitres from each extracts was transferred into a screw cup vial, and 100 μL of the anthracene d10 solution was added. The extracts were kept in a freezer over 24 h to freeze out the fat. In the last step of the procedure, after removing the fat from the samples, part of the samples (according to the scheme presented in Table 2) were evaporated under a stream of N2 at a temperature of 40 °C to dryness and the residues were dissolved in 1 mL of hexane (evaporation, E). To the other samples, 4 mL of hexane was added, the samples were shaken vigorously, and 3.5 mL of the supernatant was taken (LLE). The extracts were also evaporated to dryness and dissolved in 1 mL of hexane. Finally, all extracts were analysed by GC–MS.
A series of standard solutions in pure solvent were prepared by the dilution of the standard mixture solution in acetonitrile at the same ranges from 2 to 100 ng mL−1. An example of chromatogram of standard solution at the concentration 40 ng mL−1 is presented in Fig. 2.

Chromatogram of PAHs standard solution at the concentration 40 ng mL−1 (1—acenaphthylene, 2—fluorene, 3—phenanthrene, 4—anthracene d10, 5—anthracene, 6—pyrene, 7—benzo[a]anthracene, 8—chrysene, 9—benzo[b]fluoranthene, 10—benzo[k]fluoranthene, 11—benzo[a]pyrene, 12—indeno[1,2,3-c,d]pyrene and 12—dibenzo[a,h]anthracene)
Based on the results from the first part of the experiment, the most appropriate variant of the method was chosen and applied for the determination of PAHs in real samples. The usefulness of the method was verified on the basis of the recovery ratio of analysed compounds.
In the second part of the study, 15 types of ham were taken from retailed market to determine PAHs in food of animal origin. All products were purchased from local shops and were manufactured by leading meat companies in Poland. The modified and optimised QuEChERS sample preparation method, described above, was employed for extraction and clean-up of the samples.
Results and discussion
Gas chromatographic determination
The PAHs involved in this study were identified by comparing the retention time and three ions (one target and two qualifiers) with the NIST library. Calibration curves were constructed by plotting the ratio of the peak area, divided by the peak area of the internal standard, against concentration of the analyte. The retention times and characteristic ions for all analysed PAHs are summarised in Table 1.
Analytical performance of the method
The analytical performance of the selected variant of the QuEChERS method outlined above (method A) was examined by looking at its linearity, selectivity, recovery, repeatability, the limit of detection and limit of quantification.
A sequence of least squares regression models was fitted, a form of goodness of fit being given by the coefficient of determination (R 2). No evidence for nonlinearity was observed for all PAHs in the range of concentrations from 2 to 100 ng mL−1. All values of r were higher than 0.99 (Table 3).
All correlation coefficients were statistically significant and slope at a significance level (α = 0.05). For the same hypothesis, the intercept from the regression models was not significantly different to zero, there being no evidence that the absence of compound in any observed sample would imply that the analytical signal would be anything other than zero.
Recovery studies were conducted after fortification to the level of 0.005 mg kg−1. The recovery values ranged from 72.4 to 110.8 %, being in agreement with the most recent EU guidelines (50–120 %) [17].
The repeatability, expressed as the relative standard deviation (RSD) of the spiked sample concentrations, was lower than 10 % for all target analytes.
The limit of detection (LOD) and limit of quantification (LOQ) for the selected method (A) calculated as signal to noise ratio were ranged from 0.0001 to 0.0010 and from 0.0003 to 0.0030 mg kg−1 for pyrene and benzo[a]anthracene, respectively (Table 3). The level of noise was measured from the chromatograms of the lowest standard at a concentration of 2 ng mL−1. The limit of detection was calculated as three times higher than the level of noise, and the limit of quantification was equal to ten times of the noise level. LOQs for all determined compounds were lower than value in EU guidelines [17]. The sensitivity calculated as calibration slope coefficient was much higher for benzo[b]fluoranthene isomers. The highest sensitivity was obtained for benzo[k]fluoranthene, the lowest for indeno[1,2,3-c,d]pyrene.
Different variants of QuEChERS methods comparison—recovery study
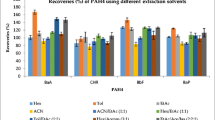
The recovery values for all six variants of methods are presented in Figs. 3, 4, 5, 6, 7 and 8. The received results showed that the best recovery ratios (72.4–110.8 %) with RSD lower than 10 % for all determined compounds were received for the method A (EtAc as an extraction solvent, sorbents: PSA + C18 and solvent evaporation to dryness), and this variant was applied to real sample analysis. In the other variants of the QuEChERS method, the recovery values were diversified. In method B (EtAc as an extraction solvent, sorbents: PSA + C18 + GCB and solvent evaporation to dryness), the recovery values ranged from 11.2 to 69.0 % (with RSD from 4.6 to 17.0 %). For method C (MeCN as an extraction solvent, sorbents: PSA + C18 and solvent evaporation to dryness), the recovery values were in the range from 25.0 to 116.6 % and RSD from 7.3 to 12.1 %. In method D (MeCN as an extraction solvent, sorbents: PSA + C18 and LLE), the recovery values ranged from 14.9 to 64.4 % (with RSD from 7.3 to 17.3 %). For method E (MeCN as an extraction solvent, sorbents: PSA + C18 + GCB and solvent evaporation to dryness), the recovery values were in the range from 3.2 to 44.6 % and RSD from 6.2 to 13.2 %. Finally, in method F (MeCN as an extraction solvent, sorbents: PSA + C18 + GCB and LLE), the recovery values from 1.1 to 35.7 % were obtained with RSD from 8.8 to 14.1 %.

PAHs recovery for method A (EtAc as an extraction solvent, sorbents: PSA + C18 and E)

PAHs recovery for method B (EtAc as an extraction solvent, sorbents: PSA + C18 + GCB and E)

PAHs recovery for method C (MeCN as an extraction solvent, sorbents: PSA + C18 and E)

PAHs recovery for method D (MeCN as an extraction solvent, sorbents: PSA + C18 and LLE)

PAHs recovery for method E (MeCN as an extraction solvent, sorbents: PSA + C18 + GCB and E)

PAHs recovery for method F (MeCN as an extraction solvent, sorbents: PSA + C18 + GCB and LLE)
Overall, it was observed that GCB sorbent used during sample preparation decreased the value of recoveries. For the method using MeCN as an extraction solvent and PSA and C18 sorbents (C and D methods), the LLE in the final step of sample preparation decreased the value of recoveries, and finally, evaporation to dryness increased the value of recoveries. For the method using additionally GCB sorbent (E and F methods), received results were diversified and no correlation was found. It was also noted that the use of each of six modifications of the QuEChERS method (except of method A) resulted in losses of PAHs such as benzo[b]fluoranthene and benzo[k]fluoranthene. Similarly, there was a compound that remained quite stable recovery regardless of the modification of the method. Benzo[a]anthracene (except methods E and F) has always had quite similar recovery value (48.2–88.0 %).
Analysis of real samples
To verify the effectiveness of the method, it was decided to examine 15 samples of meat of animal origin (pork ham). The results of real sample analysis are presented in Table 4.
In 8 of 15 samples selected, PAHs were identified. It was observed that in 6 cooked ham (S1, S3, S4, S8, S9 and S13) and one smoked and cooked (S12) samples, any PAHs were found. The absence of PAHs in smoked and boiled ham is probably caused by the fact that PAHs formed during the smoking processing passed into infusion during the cooking process.
In other samples, which were smoked and roasted, some low concentration of PAHs was detected. In one sample (S6) benzo[a]pyrene (0.0015 mg kg−1), in one sample (S11) benzo[b]fluoranthene (0.0015 mg kg−1) and in one sample (S2) chrysene (0.0024 mg kg−1) were detected. A number of other less harmful PAHs were also determined: acenaphthylene (S6 and S7) in the range from 0.0031 to 0.0047 mg kg−1, fluorene (S6, S10, S14 and S15) in the range from 0.0029 to 0.0077 mg kg−1, anthracene (S2, S5, S11, S14 and S15) in the range from 0.0033 to 0.0068 mg kg−1, phenanthrene (S2, S5, S6 and S15) in the range from 0.0015 to 0.003 mg kg−1, pyrene (S5 and S10) in the range from 0.0006 to 0.0012 mg kg−1, benzo[k]fluoranthene (S11, S14) from 0.0009 to 0.0015 mg kg−1, dibenzo[a,h]anthracene (S2, S5, S10, S14 and S15) from 0.0027 to 0.0069 mg kg−1. Benzo[a]anthracene and indeno[1,2,3-c,d]pyrene were not identified in any of samples. There was no correlation observed between the content of individual PAHs and manufacturer-investigated ham samples.
There were no exceedances of maximum levels (according to Commission Regulation (EU) No 835/2011) for determined PAHs in any of the analysed samples [16]. The results show that the QuEChERS method can be successfully applied for the determination of PAHs in food of animal origin.
Overall, it was observed that in 7 cooked ham samples (S1, S3, S4, S8, S9, S12 and S13), any PAHs were not identified.
In this study, the results show that the QuEChERS method can be successfully applied for the determination of PAHs in food of animal origin. The experiment revealed that the use of EtAc as an extraction solvent, PSA and C18 sorbents in combination with the evaporation of the extract to dryness is the optimal variant of QuEChERS method for the PAHs analysis in food of animal origin.
References
Veyrand B, Brosseaud A, Sarcher L, Varlet V, Monteau F, Marchand P, Andre F, Le Bizec B (2007) J Chromatogr A 1149:333–344
Plaza-Bolanos P, Frenich AG, Martinez Vidal JL (2010) J Chromatogr A 1217:6303–6326
Alomirach H, Al-Zenki S, Al-Hooti S, Zaghloul S, Sawaya W, Ahmed N, Kannan K (2011) Food Control 22:2028–2035
Akpambanga VOE, Purcrob G, Lajidea L, Amooa IA, Conteb LS, Moretb S (2009) Food Control 22:2028–2035
Farhadian A, Jinap S, Abas F, Sakar ZI (2010) Food Control 21:606–610
Knize MG, Salmon CP, Paris P, Felton JS (1999) Food heating and the formation of heterocyclic aromatic hydrocarbons and PAH mutagens/carcinogens. Kluwer, New York
Perello G, Marti-Cid R, Castell V, Llobet JM, Domingo JL (2009) Food Chem Toxicol 47(4):709–715
Scientific Committee on Ford (SCF) (2002) Opinion on the Scientific Committee on Food on the Risks to Human Health of Polycyclic Aromatic Hydrocarbons in Food (expressed on 4th December 2002), SCF/CS/CNTM/PAH/29 final 2002
Janoszka B, Warzecha L, Błaszczyk U, Bodzek D (2004) Acta Chromatogr. 14:115–128
Serpe FP, Esposito M, Gallo P, Serpe L (2010) Food Chem 122:920–925
Cieślik E Sadowska-Rociek A, Molina Ruiz JM, Surma-Zadora (2011) Food Chem. 125(2):773-778
Sadowska-Rociek A, Surma M, Cieślik E (2013) Bull Environ Contam Toxicol 90:508–513
Sadowska-Rociek A, Surma M, Cieślik E (2014) Environ Sci Pollut Res 21(2):1326–1338
Ramos L (2012) J Chromatogr A 1221:84–98
Nunez O, Gallart-Ayala H, Martins CPB, Lucci P (2012) J Chromatogr A 1228:298–323
Commission Regulation (EU) No 835/2011. Official Journal of the European Union L 215/4
Commission Regulation (EU) No 836/2011. Official Journal of the European Union L 215/9
Acknowledgments
The research was financed by the University of Agriculture from its own funds.
Conflict of interest
None.
Compliance with Ethics Requirements
This article does not contain any studies with human or animal subjects.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article
Cite this article
Surma, M., Sadowska-Rociek, A. & Cieślik, E. The application of d-SPE in the QuEChERS method for the determination of PAHs in food of animal origin with GC–MS detection. Eur Food Res Technol 238, 1029–1036 (2014). https://doi.org/10.1007/s00217-014-2181-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00217-014-2181-4