
Abstract
Thirty-two genome sequences of various Vibrionaceae members are compared, with emphasis on what makes V. cholerae unique. As few as 1,000 gene families are conserved across all the Vibrionaceae genomes analysed; this fraction roughly doubles for gene families conserved within the species V. cholerae. Of these, approximately 200 gene families that cluster on various locations of the genome are not found in other sequenced Vibrionaceae; these are possibly unique to the V. cholerae species. By comparing gene family content of the analysed genomes, the relatedness to a particular species is identified for two unspeciated genomes. Conversely, two genomes presumably belonging to the same species have suspiciously dissimilar gene family content. We are able to identify a number of genes that are conserved in, and unique to, V. cholerae. Some of these genes may be crucial to the niche adaptation of this species.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The species concept for bacteria has long been under siege from several angles, and now with thousands of bacterial genomes being sequenced, the disputes have intensified [8]. One frequently used definition of a bacterial species is “a category that circumscribes a (preferably) genomically coherent group of individual isolates/strains sharing a high degree of similarity in (many) independent features, comparatively tested under highly standardized conditions” [12]. Such independent features are usually phenotypes that can easily be tested. For a new species to be defined, amongst other criteria, inter-species DNA–DNA hybridisation has to be below 70%, although this rule is not without its limitations [18]. In the late 1970s and 1980s, the 16S rRNA gene sequence was introduced as a molecular clock that could be used to infer phylogenetic relationships [50]. Ideally, isolates belonging to the same species have identical or nearly identical 16S rRNA genes, and these differ from isolates belonging to different species [32, 44]. In practice, this is not always the case. Examples exist of different species sharing identical rRNA genes (for instance, E. coli and Shigella [37] that are even placed in different genera); in addition, isolates of one species can have different rRNA genes beyond the 97% that is considered to demarcate species [4]. Lateral transfer of genetic material (to which ribosomal genes are believed to be resistant) destroys the phylogenetic relationship, so that phylogenies based on alternative housekeeping genes can differ from a 16S rRNA tree and frequently are not even in accordance to each other. Such observations question the validity of a phylogenetic tree as the most suitable model for bacterial ancestry, when multiple genetic transfers would produce a network-like evolutionary structure [6]. On the other hand, it is observed that lateral gene transfer is most frequent between genetically related members sharing a similar base content and occupying the same ecological niche [29]. Nevertheless, a core of genes can be recognised that produce coherent phylogenetic trees, though these may not represent the species’ complete evolutionary history as they comprise only a minor fraction of the genetic content of the organism [35].
Whether a tree or a network is more accurate to describe phylogeny, in either case bacterial species may be considered as a cloud of isolates having a higher level of genetic similarity to each other than to organisms belonging to a different species. When such clouds have fuzzy and overlapping borders, the species concept falls apart but that will only apply to certain cases [7]. Since 16S rRNA genes are not informative on the level of diversity within a species, the 'density' of a cloud of isolates making up a species cannot be determined by this gene. Those genes shared by all isolates belonging to one species comprise the core genome of that species [39], and the degree of diversity in the remaining non-core genes determines the density of the species cloud.
We hypothesised that certain genes can be recognised as specific to a particular species, to be conserved in that species but not present in related species. We tested our hypothesis with complete genome sequences of the bacterial family Vibrionaceae, which belong to the γ-Proteobacteria and comprises eight genera. Most available genome sequences belong to the genus Vibrio. This genus contains 51 recognised species [10, 46] which are mainly found in marine environments, frequently living in association with marine organisms such as corals, fish, squid or zooplankton. Most of them are symbionts and only a few are human pathogens, notably particular serotypes of V. cholerae producing cholera, Vibrio parahaemolyticus (causing gastroenteritis) and Vi vulnificus (causing wound infections) [46]. Other Vibrionaceae, including V. vulnificus, Aliivibrio salmonicida and V. harveyi, are fish or shellfish pathogens and have major economic impact. Photobacterium profundum, representing another genus within the Vibrionaceae, was also included.
The gene content of 32 available sequenced Vibrionaceae genomes was compared and the results were analysed in various ways. The data allowed us to identify possible V. cholerae-specific genes, since this species was represented by 18 genomes that was a sufficient number to test conservation both within the species and across species. We found that a two-component signal transduction pathway is uniquely conserved in V. cholerae but is not found outside this species. Our findings further indicated that possibly a relatively small set of genes could confer niche specialisation allowing V. cholerae to be adopted to a unique environment, so that over time V. cholerae have become a distinct species.
Materials and Methods
Genomes and Gene Annotations Used
Publicly available genome sequences of Vibrionaceae were selected that were provided in less than 300 contigs and in which full-length 16S rRNA sequence could be found using the rRNA gene finder RNAmmer [19]. The 32 genome sequences included are shown in Table 1.
The gene annotations as provided in GenBank were used, except for those genomes marked “Easygene” in Table 1 where protein annotation was not available in the RefSeq file at the time of analysis, and we used EasyGene [20] to identify the genes. As a control, an available GenBank annotation was compared to a generated Easygene annotation to confirm that the number of identified genes was comparable.
Ribosomal RNA Analysis
RNAmmer [19] was used to identify 16S rRNA sequences within the 32 genomes. Sequences were considered reliable if they were between 1,400 and 1,700 nucleotides long and had an RNAmmer score above 1,800. In cases where the program found multiple and variable 16S sequences within a genome, one of these (with satisfactory RNAmmer scores) was arbitrarily chosen. The sequences were aligned using PRANK [23, 24], and the program MEGA4 was used to elucidate a phylogenetic tree [45]. Within MEGA4, the tree was created using the Neighbor-Joining method with the uniform rate Jukes–Cantor distance measure and the complete-delete option. Five hundred resamplings were done to find the bootstrap values.
Pan-Genome Family Clustering
Clustering based on shared gene families from the Vibrio pan-genome was constructed, based on BLASTP similarity using default settings. A BLASTP hit was considered significant if the alignment produced at least 50% identity for at least 50% of the length of the longest gene (either query or subject). Using this criterion, each pair of genes producing a significant reciprocal best hit was scored as belonging to the same gene family. A genome matrix was constructed, containing one row for each genome and one column for each gene family. Cell (i, j) in this matrix is 1 if genome i has a member in gene family j, 0 otherwise. A hierarchical clustering, with average linkage based on the Manhattan distance between genomes was then performed. Two trees were made, one with more weight given to gene families present in most (90%, or between 27 and 30) Vibrio genomes (“stabilome”), and the other with more weight given to gene families present in only a few (two, three, or four) genomes (“mobilome”). Thus, the original Boolean matrix is now scaled differently, depending on the number of genomes in each gene family [44]. For both trees, singletons (families which are only found in one genome) have been excluded.
Pan- and Core Genome Analysis
The results of the BLAST analysis were also used to construct a pan- and core genome plot as follows. Based on clusterings from the pan-genome family tree, an ordered set of genomes was constructed with V. cholerae genomes at the start. For the first chosen genome, all BLAST hits found in the second genome were recorded and the accumulative number of gene families (as defined above) now recognised in total was plotted for the pan-genome. The number of gene families with at least one representative gene in both genomes was plotted for the core genome. A running total is plotted for the pan-genome which increases as more genomes are added, whilst the core genome representing conserved gene families slowly decreases with the addition of more genomes.
Whole-Genome BLAST Analysis and Construction of a BLAST Matrix
The predicted genes of every genome (annotated or found by Easygene) were translated and every gene was compared, by BLASTP against every other genome and its own genome. In the latter case, the hit to self was ignored. The 50/50 rule for BLAST hits as described above was used. If these requirements were met, genes were combined in a gene family. The BLAST results were visualised in a BLAST matrix [2], which summarises the results of genomic pairwise comparisons and reports, both as percentage and as absolute numbers, the number of reciprocal BLAST hits as a fraction of the total number of gene families found in the two genomes. For easier visual inspection, the cells in the matrix are coloured darker as the fraction of similarity increases. Hits identified within a genome are differently coloured.
BLAST Atlas
BLAST results were also visualised in a BLAST atlas, this time visualising, for all genes in the reference genome V cholerae N16961, their best hit in all other genomes, again with a threshold of 50% identity over at least 50% of the length of the query protein. The atlas displays the hits as they are located in the reference strain [14]. The BLAST scores obtained for each queried gene is plotted, so that conserved and variable regions are located with respect to the reference genome. Note that genes absent in the reference genome are not shown in the lanes of the query genomes.
Results
Ribosomal RNA Analysis
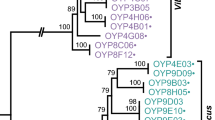
A phylogenetic tree based on the 16S rRNA gene extracted from the 32 analysed Vibrionaceae genomes is shown in Fig. 1. The 18 V. cholerae genomes build a tight subcluster, quite distanced from the other species. Above this in the figure, another subcluster comprising eight genomes representing at least six species is recognised, and within this cluster the two V. parahaemolyticus genes are not found on the same branch. A third cluster, a bit further removed, includes Aliivibrio fischeri and A. almonidica as well as V. splendidus and Vibrio species MED 222; the gene of Photobacterium profundum is the most distant.

Phylogenetic tree of the 16S rRNA gene extracted from 32 sequenced Vibrio genomes listed in Table 1. Environmental V. cholerae lacking the cholera enterotoxin genes are highlighted in bright green, whilst pathogenic V. cholerae genomes are in dark green. Further colouring was used for species for which two genomes are represented
Pan-Genome Family Trees
Starting with a database containing the total set of all Vibrio gene families, a profile of matching gene families was constructed for each individual genome. This was stored as a matrix, containing a column for each gene families, and a row for each genome. The rows contain a 0 or 1 representing the presence or absence of the gene family. This matrix was weighted to emphasise either the genes found in most genomes (the “stabilome”) or in only a few genomes (the “mobilome”); from these weighted matrices, clustering of gene families yielded the resulting trees shown in Fig. 2. Shorter distances represent genomes with many gene families in common, and larger distances reflect genomes with fewer gene families in common. As expected, in both trees, genomes from the same species cluster together, whereby the depth of resolution within a species is considerably better than can be seen in the 16S rRNA tree in Fig. 1. Similarity between the unspeciated Vibrio isolate MED222 and V. splendidus is suggested by their close clustering; this is a connection also suggested by others [21]. Note that the unspeciated Vibrio isolate Ex25 and V. parahaemolyticus 2210633 cluster together in the mobilome tree, but are more distant in the stabilome. This implies that the genes shared between these two genomes are less common genes within the Vibrio genomes examined here. As already indicated by the 16S rRNA tree, the two V. parahaemolyticus isolates are quite dissimilar, and appear on separate branches. The Aliivibrio cluster is placed within Vibrio genomes in both the stabilome and the mobilome, as was the case for their 16S rRNA gene. P. profundum is not such an outlier as in the 16S rRNA tree, and in the stabilome. It is even positioned close to the Aliivibrio genomes. Zooming in at the genomes of V. cholerae, a division into two subclusters can be seen; these clusters correspond to environmental vs. clinical isolates (with the exception of V52 in the stabilome).

Pan-genome family clustering of the 32 Vibrio genome sequences. The two plots represent weighted values for genes present in at least 90% of the genomes (stabilome) or genes found in only a few (two to four) genomes (mobilome). The colours highlighting the species are the same as in Fig. 1
Pan- and Core Genome Plot
BLAST results were analysed to construct a pan-genome, which is a hypothetical collection of all the gene families that are found in the investigated genomes [28]. The core genome was constructed from all gene families that were represented at least once in every genome. Thus, the gene families conserved in all genomes represent their core genome; adding the remaining gene families produces the pan-genome. The resulting pan- and core genome plot is shown in Fig. 3. The genomes start with the documented clinical isolates of V. cholerae and then follow the order suggested by the pan-genome family clustering (Fig. 2), although genomes from the same species were kept together (the two V. parahaemolyticus genomes were split in the trees). As more genomes are added in the plot, the number of gene families in the pan-genome (blue line) increases, and the number of conserved gene families (red line) in the core genome decreases, albeit at a lower rate. This is because every genome can add many novel (and frequently different) genes to the pan-genome but only decreases the core genome with a few genes that are absent in that particular strain but that were conserved in the previously analysed genomes. The pan-genome curve increases with a relative steep slope when a novel species is added, as is obvious when a V. parahaemolyticus genome is added after the last V. cholerae. A stable plateau can be seen for the pan-genome of V. cholerae around 6,500 genes. Nevertheless, a small increase occurs when adding V. cholerae 11587; this is caused by the difference between the two subclusters of V. cholerae seen in Fig. 2. V. cholerae strain 2740-80 behaves atypical in all the figures shown; although documented as an environmental isolate, it appears closer to the clinical isolates, in terms of overall genomic properties.

Pan- and core genome plot of the 32 Vibrionaceae genomes. The colours highlighting species are the same as in Fig. 1
When the first genome of A. fischeri is added, which is not a member of the Vibrio genus, it does not add significantly more novel genes to the pan-genome than Vibrio genomes did. This contrasts with P. profundum which produces a sharp increase in the pan-genome, as does, interestingly, V. shilonii. Note that there are approximately 20,200 total gene families within the 32 sequenced Vibrionaceae genomes, whereas the core genome decreases to approximately 1,000 gene families.
BLAST Comparison Visualised in a BLAST Matrix
A BLAST matrix provides a visual overview of reciprocal pairwise whole-genome comparisons, as shown in Fig. 4. The stronger a matrix cell is coloured, the more similarity was detected between the gene content of two genomes. As can be seen in the lower right triangle, all V. cholerae genomes are highly similar, with similarity ranging between 64% and 93% for any given pair of genomes. No statistical difference was observed when comparing clinical isolates to environmental isolates. The two A. fischeri and the two V. vulnificus genomes also share a high degree of identity within their species (75% and 67%, respectively), visible at the bottom of the matrix. In contrast, the two V. parahaemolyticus genomes only share 35% identity, which is not higher than the similarity detected between genomes of different species. With 72% similarity, isolate MED222 most closely matches V. splendidus and with 65% isolate EX25 again shares most similarity with V. parahaemolyticus 2210633.

BLAST matrix of the 32 Vibrionaceae genomes. The colours highlighting the species are the same as in Fig. 1. Since the reciprocal similarity (reported as percent) is not readable at this resolution, every matrix cell is coloured using the scales as indicated. The bottom row identifies hits (other than hits-to-self) found within a genome. Four matrix cells reporting high pairwise similarities are outlined; their numbers are specified in the text
BLAST Atlas
A BLAST atlas was constructed using V. cholerae N16961 (O1, El Tor) as the reference genome, shown in Fig. 5. The best blast hits identified in the query genomes are plotted in the lanes around the reference genome, with different colours for different species. In general, chromosome 1 is more strongly conserved than chromosome 2. A large part of chromosome 2 of N16961 displays very little conservation in the other genomes; this area represents a super integron [40] that contains the V. cholerae-specific repeat (VCR) sequences, as well as a high number of gene cassettes. The repeat sequences are visible as black boxes in the repeat lane of the reference genome (second inner lane). Although all V. cholerae genomes contain a superintegron, its genes are very diverse between isolates [34] which explains the lack of blast hits in this region.

BLAST atlas with V. cholerae strain N16961 as a reference strain, showing chromosomes 1 (top) and 2 (bottom). The best BLAST hits identified with genes from N16961 in the other V. cholerae genomes are represented in dark red, for the location as it appears in N16961. Blast hits in the other genomes are shown in various colours as indicated to the right. Major areas conserved in V. cholerae but not in other Vibrionaceae are identified as gap B, gap C, gap D and gap F in green; areas that are found in toxigenic V. cholerae only are marked black as gap A, gap E and gap G. The superintegron on chromosome 2 of V. cholerae is also indicated
Several regions of the atlas have been highlighted. Gaps B, C, D and F on chromosome 1 (indicated in green) contain genes that are conserved in the represented genomes of V. cholerae but not in the other Vibrionaceae. The gaps marked A, E and G indicate regions that are specific to the toxigenic, clinical isolates only. Annotated, V. cholerae-specific genes present in all these regions are listed in Table 2 (hypothetical genes are excluded). Genes specific for toxinogenic V. cholerae identified in gap A include, amongst others, biosynthesis genes for the toxin co-regulated pilus (which is required for transmission of the prophage CTXΦ carrying the enterotoxin genes), as well as genes encoding citrate lyase. Note that the genes in gap A are also found in the environmental isolate V. cholerae 2740-80.
Gap B contains a number of outer membrane protein genes involved in sugar modification that are found in all V. cholerae genomes. Genes from gap C encoding a histidine kinase two-component signal transduction regulatory system are also conserved within the species, as genes in gaps D and F, involved in chemotaxis and possible multidrug resistance.
Gap E, containing genes conserved in toxigenic strains only, holds the prophage CTXΦ that contains the genes encoding cholera enterotoxin subunits A and B; this enterotoxin is responsible for the excessive, watery diarrhoea typical for cholera. Upon binding to target cell GM1 gangliosides, enterotoxin enters the cell and stimulates adenylate cyclase by ADP ribosylation. The resultant increased cyclic AMP levels induce excessive electrolyte movement and sodium plus water secretion [43]. Strain M66-2 is believed to be a precursor of the seventh pandemic V. cholerae that lacks the prophage CTXΦ and the enterotoxin genes [11]. Gap E bears the RTX toxin operon, which encodes a pore-forming cytotoxin [22]. An RTX toxin is also present in environmental isolate 2740-80 and in V. vulnificus.
Gap G on chromosome 2 consists of a set of five genes, all in the same orientation, in a putative operon, flanked by genes on the complimentary strand. This appears to be a remnant of a mobile element, as these genes are flanked by a transposase gene on the 3′ end, and there is a small global repeat on the 5′ end. Only the first two of the five genes have an assigned function, with the first gene being a GMP reductase, and the second a putative DNA methyltransferase. The remaining three genes are hypothetical, but their strikingly strong conservation in all pathogenic strains and complete absence of homologues in the other Vibrio genomes strongly point towards a potential biological significance.
Discussion
The recent availability of many Vibrionaceae genomes, including a substantial number of V. cholerae genomes, allows the possibility to take a closer look at the similarities and differences of species within the genus Vibrio. This can examine, on a genome scale, what distinguishes V. cholerae from the other Vibrio species. Since not all V. cholerae isolates are pathogenic, the presence of the prophage-bearing cholera enterotoxin, the main virulence factor for cholera, is not a suitable marker for this species. We attempted to identify a set of V. cholerae-specific genes, and also explored the internal diversity within the V. cholerae genomes that have been sequenced to date.
On a phylogenetic tree based on the 16S ribosomal RNA gene, those isolates that do not belong to the genus Vibrio were positioned as outliers, as expected. This tree further indicated the closest resembling 16S rRNA sequence for the two sequenced Vibrio strains that are currently not assigned to a species. It was observed that the two sequenced V. parahaemolyticus strains were not placed together. The complete gene content of each genome was next compared by BLAST and the results were pooled into gene families which were subjected to cluster analysis. This provided evidence that the 18 V. cholerae genomes fall into two subclusters, one mainly containing clinical isolates and the other environmental isolates.
The gene family clustering, subsequent pan-genome analysis and the pairwise BLAST results, as summarised in the BLAST matrix, all supported the relatedness of Vibrio species Ex25 to V. parahaemolyticus 2210633 but not to V. parahaemolyticus 16. This latter genome was quite different from V. parahaemolyticus 2210633 in all analyses. Although it is possible that the species V. parahaemolyticus is far more genetically diverse than V. cholerae, A. fischeri or V. vulnificus, an alternative explanation is that one of the sequenced isolates is perhaps incorrectly named as V. parahaemolyticus. The similarity between Vibrio species MED222 and V. splendidus based on gene families is in agreement with their related 16S rRNA genes and published data [21]. However, in contrast to what the ribosomal gene suggests, our whole-genome comparison indicates that the three Aliivibrio genomes (A. salmonicida and two A. fischeri) are not so different from Vibrio after all. Their recent placement in the genus Aliivibrio, a decision based on five genes (the 16S rRNA gene and four housekeeping genes) and phenotypical characteristics [47], appears not to be reflective of the whole genome picture presented here.
The BLAST results were graphically summarised in a BLAST atlas, which visualised V. cholerae-specific gene clusters. These coded for polysaccharide biosynthesis enzymes, response regulators and chemotaxis proteins, amongst others. In addition, a V. cholerae-specific, histidine kinase two-component signal transduction regulatory system was identified. The two-component signal transduction pathway is a powerful regulating system for bacteria to adapt to a particular ecological niche. There is a precedent for this claim, as the introduction of a single regulatory protein in Vibrio fischeri strain MJ11 has been shown to specifically enable colonization of the squid Euprymna scolopes [26].
As expected, the main differences observed between V. cholerae clinical isolates and the environmental strains are due to genes related to virulence. Two exceptions are the presence of a number of virulence genes in the environmental strain V. cholerae 2740-80 and the absence of enterotoxin genes in clinical isolate M66-2. It has already been suggested that M66-2 might be a predecessor of pandemic, enterotoxic V. cholerae [11]. From sequence comparison of four housekeeping genes, it was concluded that V. cholerae 2740-80 is intermediary between toxigenic and non-toxigenic isolates [30]. This view is confirmed by the data presented here, although we propose to consider the possibility that the isolate arose from a pandemic clone that has lost the CTXΦ prophage, rather than being a precursor of a pathogen.
In conclusion, several different methods of genome comparisons have yielded a picture of V. cholerae genomes as forming a distinct cluster, compared to related species, and a relatively small number of genes might be responsible for environmental niche adaptation and hence for generation of this distinct species. Likely candidates include multiple two-component signal transduction regulatory proteins as well as chemotaxis proteins.
References
Bassler B et al. (2007) CP000789.1: Direct submission to GenBank
Binnewies TT, Hallin PF, Staerfeldt HH, Ussery DW (2005) Genome update: proteome comparisons. Microbiol 151:1–4
Chen CY, Wu KM, Chang YC, Chang CH, Tsai HC, Liao TL, Liu YM, Chen HJ, Shen AB, Li JC, Su TL, Shao CP, Lee CT, Hor LI, Tsai SF (2003) Comparative genome analysis of Vibrio vulnificus, a marine pathogen. Genome Res 13:2577–2587
Clayton RA, Sutton G, Hinkle PS, Bult C, Fields C (1995) Intraspecific variation in small-subunit rRNA sequences in GenBank: why single sequences may not adequately represent prokaryotic taxa. Int J Syst Bacteriol 45:595–599
Colwell R, Grim CJ, Young S, Jaffe D, Gnerre S, Berlin A, Heiman D, Hepburn T, Shea T, Sykes S, Alvarado L, Kodira C, Heidelberg J, Lander E, Galagan J, Nusbaum C, Birren B (2008) NZ_AAKF00000000: Direct submission to GenBank
Doolittle WF (1995) Phylogenetic classification and the universal tree. Science 284:2124–2129
Doolittle WF, Papke RT (2006) Genomics and the bacterial species problem. Genome Biol 7:116
Doolittle WF, Zhaxybayeva O (2009) On the origin of prokaryotic species. Genome Res 19:744–756
Edwards R, Ferriera S, Johnson J, Kravitz S, Beeson K, Sutton G, Rogers Y-H, Friedman R, Frazier M, Venter JC (2008) NZ_ACCV00000000: Direct submission to GenBank
Farmer JJ, Janda JM (2005) Vibrionaceae. In: Bergey’s manual of systematic bacteriology, 2nd edn, vol 2 part B. Springer, New York, pp 491–546
Feng L, Reeves PR, Lan R, Ren Y, Gao C, Zhou Z, Ren Y, Cheng J, Wang W, Wang J, Qian W, Li D, Wang L (2008) A recalibrated molecular clock and independent origins for the cholera pandemic clones. PLoS ONE 3:e4053
Gevers D, Cohan FM, Lawrence JG, Sprat BG, Coeyne T, Feil EJ, Stackebrandt E, Van de Peer Y, Vandamme P, Thompson FL, Swings J (2005) Re-evaluating prokaryotic species. Nat Rev Microbiol 3:733–739
Hagstrom A, Ferriera S, Johnson J, Kravitz S, Beeson K, Sutton G, Rogers Y-H, Friedman R, Frazier M, Venter JC (2007) NZ_ABGR00000000: Direct submission to GenBank
Hallin PF, Binnewies TT, Ussery DW (2008) The genome BLASTatlas—a GeneWiz extension for visualization of whole-genome homology. Mol Biosyst 4:363–371
Heidelberg JF, Eisen JA, Nelson WC, Clayton RA, Gwinn ML, Dodson RJ, Haft DH, Hickey EK, Peterson JD, Umayam L, Gill SR, Nelson KE, Read TD, Tettelin H, Richardson D, Ermolaeva MD, Vamathevan J, Bass S, Qin H, Dragoi I, Sellers P, McDonald L, Utterback T, Fleishmann RD, Nierman WC, White O, Salzberg SL, Smith HO, Colwell RR, Mekalanos JJ, Venter JC, Fraser CM (2000) DNA sequence of both chromosomes of the cholera pathogen Vibrio cholerae. Nature 406:477–483
Heidelberg J, Sebastian Y. NZ_AAKJ00000000, NZ_AAUT00000000, NZ_AAKK00000000, NZ_AAUR00000000, NZ_AAWF00000000: Direct submission to GenBank
Hjerde E, Lorentzen MS, Holden MT, Seeger K, Paulsen S, Bason N, Churcher C, Harris D, Norbertczak H, Quail MA, Sanders S, Thurston S, Parkhill J, Willassen NP, Thomson NR (2008) The genome sequence of the fish pathogen Aliivibrio salmonicida strain LFI1238 shows extensive evidence of gene decay. BMC Genomics 9:616
Konstantinidis T, Ramette A, Tiedje JA (2006) The bacterial species definition in the genomic era. Phil Trans R Soc B 361:1929–1940
Lagesen K, Hallin P, Rødland EA, Staerfeldt HH, Rognes T, Ussery DW (2007) RNAmmer: consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res 35:3100–3108
Larsen TS, Krogh A (2003) EasyGene—a prokaryotic gene finder that ranks ORFs by statistical significance. BMC Bioinformatics 4:29
Le Roux F, Zouine M, Chakroun N, Binesse J, Saulnier D, Bouchier C, Zidane N, Ma L, Rusniok C, Lajus A, Buchrieser C, Médigue C, Polz MF, Mazel D (2009) Genome sequence of Vibrio splendidus: an abundant planctonic marine species with a large genotypic diversity. Environ Microbiol 11:1959–1970
Lin W, Fullner KJ, Clayton R, Sexton JA, Rogers MB, Calia KE, Calderwood SB, Fraser C, Mekalanos JJ (1999) Identification of a Vibrio cholerae RTX toxin gene cluster that is tightly linked to the cholera toxin prophage. Proc Natl Acad Sci U S A 96:1071–1076
Loytynoja A, Goldman N (2005) An algorithm for progressive multiple alignment of sequences with insertions. Proc Natl Acad Sci U S A 102:10557–10562
Loytynoja A, Goldman N (2008) Phylogeny-aware gap placement prevents errors in sequence alignment and evolutionary analysis. Science 320:1632–1635
Makino K, Oshima K, Kurokawa K, Yokoyama K, Uda T, Tagomori K, Iijima Y, Najima M, Nakano M, Yamashita A, Kubota Y, Kimura S, Yasunaga T, Honda T, Shinagawa H, Hattori M, Iida T (2003) Genome sequence of Vibrio parahaemolyticus: a pathogenic mechanism distinct from that of V. cholerae. Lancet 361:743–749
Mandel MJ, Wollenberg MS, Stabb EV, Visick KL, Ruby EG (2009) A single regulatory gene is sufficient to alter bacterial host range. Nature 458:215–218
Mazel D, Le Roux F (2008) FM954973.1: Direct submission to GenBank
Medini D, Donati C, Tettelin H, Masignani V, Rappuoli R (2005) The microbial pan-genome. Curr Opin Genet Dev 15:589–594
Medrano-Soto A, Moreno-Hagelsieb G, Vinuesa P, Christen JA, Collado-Vides J (2001) Succesful lateral transfer requires codon usage compatibility between foreign genes and recipient genomes. Mol Biol Evol 21:1884–1894
Mohapatra SS, Ramachandran D, Mantri CK, Colwell RR, Singh DV (2009) Determination of relationships among non-toxigenic Vibrio cholerae O1 biotype El Tor strains from housekeeping gene sequences and ribotype patterns. Res Microbiol 160:57–62
Munk A, Tapia R, Green L, Rogers Y, Detter JC, Bruce D, Brettin TS, Colwell R, Grim C, Vonstein V, Bartels D. CP001485.1, NZ_ACHV00000000, NZ_ACHY00000000, NZ_ACHW00000000, NZ_ACHX00000000, NZ_ACHZ00000000, NZ_ACIA00000000, NZ_ACFQ00000000: Direct submission to GenBank
Murray RG, Stackebrandt E (1995) Taxonomic note: implementation of the provisional status Candidatus for incompletely described procaryotes. Int J Syst Bacteriol 45:186–187
Nierman WC (2006) NZ_AATY00000000: Direct submission to GenBank
Pang B, Yan M, Cui Z, Ye X, Diao B, Ren Y, Gao S, Zhang L, Kan B (2007) Genetic diversity of toxigenic and nontoxigenic Vibrio cholerae serogroups O1 and O139 revealed by array-based comparative genomic hybridization. J Bacteriol 189:4837–4879
Philippe H, Douady CJ (2003) Horizontal gene transfer and phylogenetics. Curr Opin Microbiol 6:498–505
Pinhassi J, Pedros-Alio C, Ferriera S, Johnson J, Kravitz S, Halpern A, Remington K, Beeson K, Tran B, Rogers Y-H, Friedman R, Venter JC (2006) NZ_AAND00000000: Direct submission to GenBank
Pupo GM, Lan R, Reeves PR (2000) Multiple independent origins of Shigella clones of Escherichia coli and convergent evolution of many of their characteristics. Proc Natl Acad Sci U S A 97:10567–10572
Rhee JH, Kim SY, Chung SS, Lee SE, Choy HE (2002) AE016795.2: Direct submission to GenBank
Riley MA, Lizotte-Waniewski M (2009) Population genomics and the bacterial species concept. Methods Mol Biol 532:367–377
Rowe-Magnus DA, Guérout AM, Mazel D (1999) Super-integrons. Res Microbiol 150:641–651
Rosenberg E, Ferriera S, Johnson J, Kravitz S, Beeson K, Sutton G, Rogers Y-H, Friedman R, Frazier M. Venter JC (2006) NZ_ABCH00000000: Direct submission to GenBank
3Ruby EG, Urbanowski M, Campbell J, Dunn A, Faini M, Gunsalus R, Lostroh P, Lupp C, McCann J, Millikan D, Schaefer A, Stabb E, Stevens A, Visick K, Whistler C, Greenberg EP (2005) Complete genome sequence of Vibrio fischeri: a symbiotic bacterium with pathogenic congeners. Proc Natl Acad Sci U S A 102:3004–3009
Sánchez J, Holmgren J (2005) Virulence factors, pathogenesis and vaccine protection in cholera and ETEC diarrhoea. Curr Opin Immunol 17:388–398
Stackebrandt E, Frederiksen W, Garrity GM, Grimont PA, Kämpfer P, Maiden MC, Nesme X, Rosselló-Mora R, Swings J, Trüper HG, Vauterin L, Ward AC, Whitman WB (2002) Report of the ad hoc committee for the re-evaluation of the species definition in bacteriology. Int J Syst Evol Microbiol 52:1043–1047
Tamura K, Dudley J, Nei M, Kumar S (2007) MEGA4: Molecular Evolutionary Genetics Analysis (MEGA) software version 4.0. Mol Biol Evol 24:1596–1599
Thompson FL, Iida T, Swings J (2004) Biodiversity of vibrios. Microbiol Mol Biol Rev 68:403–431
Urbanczyk H, Ast JC, Higgins MJ, Carson J, Dunlap PV (2007) Reclassification of Vibrio fischeri, Vibrio logei, Vibrio salmonicida and Vibrio wodanis as Aliivibrio fischeri gen. nov., comb. nov., Aliivibrio logei comb. nov., Aliivibrio salmonicida comb. nov. and Aliivibrio wodanis comb. nov. Int J Syst Evol Microbiol 57:2823–2829
Vezzi A, Campanaro S, D'Angelo M, Simonato F, Vitulo N, Lauro FM, Cestaro A, Malacrida G, Simionati B, Cannata N, Romualdi C, Bartlett DH, Valle G (2005) Life at depth: Photobacterium profundum genome sequence and expression analysis. Science 30:1459–1461
Wang L, Feng L, Reeves P, Lan R, Ren Y, Gao C, Zhou Z, Ren Y, Wang W (2008) CP001233.1. CP001235.1: Direct submission to GenBank
Woese CR (1987) Bacterial evolution. Microbial Rev 51:221–271
Acknowledgements
We would like to thank Tim Binnewies for early work on this project, and also to the Danish Research Councils and the DTU Globalization funds for financial support.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Vesth, T., Wassenaar, T.M., Hallin, P.F. et al. On the Origins of a Vibrio Species. Microb Ecol 59, 1–13 (2010). https://doi.org/10.1007/s00248-009-9596-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00248-009-9596-7