
Abstract
Background
Miriplatin (formerly SM-11355), a novel lipophilic platinum complex developed to treat hepatocellular carcinoma, is administered into the hepatic artery using an oily lymphographic agent (Lipiodol Ultra-Fluide®) as a carrier. We clarified the usefulness of miriplatin as an agent for transarterial chemoembolization.
Methods
Platinum compounds released from miriplatin into serum, medium and Earle’s balanced salt solution were examined. Then, miriplatin and cisplatin were administered to rats bearing hepatoma AH109A tumors in livers. Platinum concentrations in tissues and DNA were assessed.
Results
Miriplatin showed a more sustained release than cisplatin. Dichloro[(1R, 2R)-1, 2-cyclohexane diamine-N, N′]platinum, the most abundant platinum compound released from miriplatin, was as effective as cisplatin in inhibiting the growth of cells. Miriplatin was selectively disposed of in tumors, maintained in tumors longer than cisplatin and caused apparent tumor regression inducing platinum-DNA adducts to form and massive apoptosis.
Conclusion
Miriplatin appears to be a suitable chemotherapeutic agent for transarterial chemoembolization.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Hepatocellular carcinoma (HCC) is one of the most common cancers, and its incidence is increasing worldwide due to the dissemination of hepatitis B and C virus infection [1–3]. It is clear that the most powerful risk factor is liver cirrhosis associated with a viral infection or high alcohol intake. Approximately 80% of Japanese HCC patients are diagnosed with hepatitis C virus (HCV)-associated cirrhosis or chronic hepatitis. Once cirrhosis is established, HCC develops at an annual rate of 1–7% [4, 5], although interferon therapy reduces the incidence of HCC among patients with HCV-related cirrhosis or chronic hepatitis [6–8]. Surveillance and advances in imaging have made early diagnosis possible. However, the prognosis is poor because surgical resection, liver transplantation, or percutaneous ablation is applicable to only a small proportion of patients, while most patients with HCC are diagnosed at the intermediate to advanced stage.
The liver receives a dual blood supply from the hepatic artery and the portal vein, whereas HCC is fed by only the hepatic artery [9–11]. On the basis of this peculiar feature of HCC, the efficacy and tumor selectivity of antitumor agents can be increased by injecting embolic material or the oily lymphographic agent (Lipiodol Ultra-Fluide®, LPD) through the hepatic artery. LPD is known to be selectively distributed and retained for a long period in hepatic tumor tissues after intra-hepatic arterial injection [12–15]. Several antitumor agents such as cisplatin, doxorubicin, epirubicin, mitomycin C, and zinostatin stimalamer, suspended or emulsified in LPD, have been clinically studied [16–23]. Zinostatin stimalamer has been used for primary unresectable HCC as an approved agent in Japan since 1994 [19, 20]. It is used as a suspension in LPD, given by arterial administration via the hepatic artery. Cisplatin suspended in LPD (cisplatin/LPD) has been reported to selectively accumulate in the HCC, and gradually release platinum after intra-hepatic arterial administration [17, 18]. However, there is still no standard transarterial chemoembolization (TACE) for patients with unresectable HCC. As most of the antitumor agents used for TACE are water-soluble, we have investigated a lipophilic platinum complex which utilizes LPD as the carrier solvent. Antitumor platinum complexes undergo leaving group exchange prior to the reaction with DNA in tumors, while, in physiological surroundings, any leaving groups are easily displaced by biological nucleophiles such as sulfur-containing amino acids, glutathione, and proteins, leading to inactivation [24, 25]. Therefore, platinum complexes containing lipophilic leaving groups are expected to be soluble or easily suspended in LPD, and be gradually released and active only at the tumor site, reducing systemic toxicity due to low circulation after intra-hepatic arterial administration.
Miriplatin (formerly SM-11355), (SP-4-2)-[(1R,2R)-cyclohexane-1,2-diamine-N,N′]bis (tetradecanoato-O)platinum, is one of the lipophilic platinum complexes synthesized by Maeda et al. [26], which can be easily suspended in LPD and release active platinum compounds into the aqueous phase gradually [27, 28]. Miriplatin suspended in LPD (miriplatin/LPD) had antitumor effects on hepatic tumors after intra-hepatic arterial administration in several animal models [27, 29, 30]. In vitro studies have shown that platinum compounds released from miriplatin/LPD were incorporated into rat hepatoma cells including cisplatin-resistant cells, and form platinum-DNA adducts [31, 32]. However, it has not been fully evaluated whether the sustained release of active platinum compounds from miriplatin/LPD could contribute the tumor-selective delivery, and whether the formation of platinum-DNA adducts by miriplatin could lead to tumor regression. In this study, confirming that miriplatin/LPD was active and retained in rat hepatic tumors after intra-hepatic arterial administration, we examined in vivo antitumor activities of miriplatin, cisplatin, and zinostatin stimalamer at the therapeutic dose of each agent. To further investigate which mechanism causes tumor regression after miriplatin treatment, we examined the formation of platinum-DNA adducts and induction of apoptosis in rat hepatic tumors.
Materials and methods
Chemicals
Miriplatin, dichloro[(1R,2R)-1,2-cyclohexanediamine-N,N′]platinum (DPC), chloro[(1R,2R)-1,2-cyclohexanediamine-N,N′]iodo platinum (DPCI) and [(1R,2R)-1,2-cyclohexanediamine-N,N′]diiodo platinum (DPI) were prepared by Dainippon Sumitomo Pharma Co., Ltd (Osaka, Japan). Cisplatin was obtained from Nihon Kayaku Co., Ltd (Tokyo, Japan) or SIGMA (St. Louis, MO). Zinostatin stimalamer and Lipiodol Ultra-Fluide® (Guerbet) were from Astellas Pharma Inc. (Tokyo, Japan) and TERUMO Corp. (Tokyo, Japan), respectively.
Cell line and animals
The rat ascite hepatoma cell line AH109A was provided by Kumamoto University (Kumamoto, Japan). For in vitro use, AH109A cells taken from ascites of male Donryu rats (Charles Liver Laboratories, Japan) were grown in RPMI 1640 medium supplemented with antibiotics (penicillin and streptomycin) and 10–20% heat-inactivated fetal bovine serum (FBS). Cells were cultured at 37°C in 5% CO2. To establish the hepatic tumor model previously reported by Kishimoto et al. [29], AH109A cells originally taken from ascites of male Donryu rats were transplanted subcutaneously in rats. Subcutaneous tumors resected (approximately 1 × 1 × 1 mm cubes) were transplanted into the subcapsular parenchyma of the liver. The hepatic tumor model was maintained by repeated transplantation into the rat liver. The care and treatment of experimental animals were in accordance with institutional guidelines.
In vitro release of platinum compounds into rat serum
Miriplatin was easily suspended at 20 mg/mL by adding LPD and shaking. One milliliter of miriplatin/LPD and cisplatin/LPD (20 mg/mL) were layered over 12 mL of rat serum in test tubes. Test tubes were rotated vertically for 7 days at 5 rpm in an incubator at 37°C under protection from light [28]. The aqueous phase was obtained by centrifuging test tubes at 1,200 rpm for 30 min at room temperature. The platinum concentrations in serum after additional centrifuging at 12,000 rpm for 90 min at room temperature were quantitatively analyzed at 265.9 nm by flameless atomic absorption spectrometry (FAAS) Z-9000 (HITACHI, Tokyo, Japan).
In vitro release of platinum compounds into Earle’s balanced salt solution
Three milliliters of miriplatin/LPD (20 mg/mL) was layered over 6 mL of Earle’s balanced salt solution (Invitrogen Corp.) supplemented with magnesium sulfate at 97 mg/L in test tubes [33]. Test tubes were rotated vertically for 7 days at 5 rpm in an incubator at 37°C under protection from light [28]. The aqueous phase was obtained by centrifuging test tubes at 1,200 rpm for 30 min at room temperature. Platinum compounds in the aqueous phase after additional centrifuging at 12,000 rpm for 30 min at room temperature were analyzed by high performance liquid chromatography (HPLC). Samples (100 µL) were loaded on a 2.1 × 100 mm reverse phase column, XTerra MS C18 packed with 3.5-μm particles (Waters, Milford, MA, USA), and eluted at 40°C with 10% acetonitrile at a flow rate of 0.3 mL/min. Fractions from the reverse phase column were analyzed on-line with an inductively coupled plasma mass spectrometer (ICP-MS), Agilent 7500s (Agilent Technologies, Santa Clara, CA, USA), for the detection of platinum.
In vitro cell proliferation assay
Aliquots of AH109A cells were plated into 24-well microplates. Following cell adherence (1 day), LPD alone and agents suspended in LPD were added to Falcon cell culture inserts (Becton Dickinson and Company, Franklin Lakes, NJ, USA), equipped with a 0.4-μm pore membrane on their bottom. After 7 days of incubation at 37°C in 5% CO2, the numbers of viable cells were examined using AlamarBlue (Trek diagnostic systems, Westlake, OH, USA). The IC50 value was defined as the concentration inhibiting cell growth by 50% compared with treatment with LPD alone. To examine platinum concentrations in the medium, agents suspended in LPD were added to Falcon cell culture inserts in wells containing the culture medium alone. The platinum concentrations were quantitatively analyzed by FAAS. Alternatively, aliquots of AH109A cells were plated into 96-well microplates. Following cell adherence (1 day), agents in aqueous solution were added. After 3 days of incubation at 37°C in 5% CO2, the numbers of viable cells were examined using AlamarBlue.
Blood supply to implanted tumors
Tumor-bearing rats were anesthetized with an injection of sodium pentobarbital before laparotomy, and a 100% (w/v) barium sulfate suspension (Kaigen Inc, Osaka, Japan) was injected into the hepatic artery or the portal vein at the volume of 0.5 mL/head or 5.0 mL/head, respectively. For intra-hepatic arterial administration, a catheter with an inner and outer diameter of 0.28 and 0.61 mm, respectively, was inserted retrogressively into the gastroduodenal artery with the tip close to the origin of the proper hepatic artery [29]. Immediately after the administration, livers were resected, fixed in formalin, and radiographed with SRO-M50 soft X-ray machine (Sofron, Tokyo, Japan).
Distribution of LPD and measurement of platinum concentrations in rat livers
Miriplatin/LPD and cisplatin/LPD (400 µg/head) were injected into the hepatic artery of tumor-bearing rats at the volume of 0.02 mL/head. Immediately and 7 days after the administration, resected livers were radiographed with a soft X-ray machine and then divided into tumor tissues and normal liver tissues. Tissue homogenates incinerated using nitric acid and hydrogen peroxide were dissolved in 4% aqua regia and subjected to FAAS [34].
In vivo assay of the growth of tumors implanted in rat livers after the intra-arterial administration
Rats bearing a tumor approximately 100–250 mm2 in area were randomly allocated into different treatment groups and a control group, each of which consisted of seven rats. Tumor diameters were measured with calipers, and estimated tumor area was calculated by the formula: (smaller diameter) × (larger diameter). All agents suspended in LPD and LPD alone were injected into the hepatic artery of tumor-bearing rats at the volume of 0.02 mL/head. The therapeutic dose of each agent was defined in this study as follows: miriplatin (400 μg/head, 20 mg/mL in LPD) [35, 36], cisplatin (400 μg/head, 20 mg/mL) [17] and zinostatin stimalamer (20 μg/head, 1 mg/mL) [19, 20]. After the intra-hepatic arterial administration, the gastroduodenal artery and abdomen were closed with uninterrupted sutures. The tumor growth rate (%) was calculated with the following formula: A7/A0 × 100, where A7 is the estimated tumor area at day 7 and A0 is the estimated tumor area at the initiation of the treatment (day 0). The systemic toxicity of the treatments was assessed in terms of changes in body weight during the experiments. These were calculated as (W 7 − W 0)/W 0 × 100 where W 7 is body weight at day 7 and W 0 is body weight at day 0.
Platinum-DNA adducts within cultured cells and tumors implanted in rat livers
DNA was isolated from cells or homogenized tumors using DNAzol reagent (Invitrogen Corp., Carlsbad, CA, USA) in accordance with the manufacturer’s protocol. After purified DNA was solubilized in Tris-buffered EDTA, the amount of platinum in DNA was determined by FAAS. The DNA content was assessed fluorometrically using Hoechst 33258.
Induction of apoptosis within cultured cells and tumors implanted in rat livers
The apoptosis of cells was assessed as the fraction with sub-G1 DNA content. Cells were fixed in 80% ethanol for 20 min at room temperature. Following treatment with 1 mg/mL RNase A for 20 min at 37°C, cells were stained in propidium iodide solution (50 μg/mL in 0.1% sodium citrate and 0.1% Triton X-100) and then analyzed using FACScan flow cytometer (Becton Dickinson and Company). The apoptosis in resected liver in situ was visualized using the terminal deoxynucleotidyl transferase-mediated dUTP-biotin nick end labeling assay (Chemicon International Inc., Temecula, CA, USA) in accordance with the manufacturer’s protocol.
Statistical analysis
Differences in tumor growth rates or changes in body weight between the control group and the treated group were statistically analyzed by using the Dunnett test (P < 0.05, two-sided).
Results
In vitro release of platinum compounds from miriplatin/LPD
To confirm that miriplatin utilizes LPD as a carrier solvent leading to long-lasting release, the amounts of platinum released from miriplatin/LPD in rat serum were compared with those released from cisplatin/LPD (Fig. 1). While the amount in serum treated with miriplatin/LPD was 54 ± 3 μg after 7 days, cisplatin/LPD released 9,400 ± 100 μg of platinum in serum within a day. Thus, miriplatin/LPD showed a more sustained release over 7 days than cisplatin/LPD in this system using rat serum.

In vitro release of platinum from miriplatin/LPD and cisplatin/LPD. The amount of platinum released from miriplatin/LPD and cisplatin/LPD in rat serum. One milliliter of miriplatin/LPD (20 mg/mL, filled square) or cisplatin/LPD (20 mg/mL, open square) was layered over 12 mL of rat serum in test tubes. Test tubes were rotated vertically at 5 rpm in an incubator at 37°C under protection from light. The amounts of platinum in serum and LPD were measured at 265.9 nm using FAAS. All results are given as the mean ± SD (n = 3)
In vitro cell growth inhibition by platinum compounds released from miriplatin/LPD
The release characteristics for miriplatin/LPD and cisplatin/LPD using cell culture inserts are shown in Fig. 2a. While the platinum concentrations in the medium treated with miriplatin/LPD gradually increased, depending on incubation time, for 7 days, cisplatin/LPD caused a rapid increase in the platinum concentrations in the medium within a day, with the concentration almost the same until day 7. Thus, miriplatin/LPD showed a more sustained release over 7 days than cisplatin/LPD. First, we examined in vitro antitumor effects of miriplatin/LPD and cisplatin/LPD using cell culture inserts. In this system, the LPD suspension does not make contact with the cells directly, but via membranes with tiny pores. IC50 values for rat ascite hepatoma AH109A cells after 7 days of exposure to miriplatin/LPD and cisplatin/LPD were 0.89 ± 015 and 0.14 ± 0.09 μg/mL, respectively. Increasing the concentrations of miriplatin and cisplatin in LPD increased the platinum concentrations in medium at day 7 (Fig. 2b). In addition, both agents inhibited cell growth depending on their concentrations in the medium. Antitumor platinum complexes undergo leaving group exchange prior to biotransformation. To determine which platinum compounds were generated from miriplatin/LPD, we examined those released into Earle’s balanced salt solution. This solution consists of inorganic ingredients similar to a culture medium or serum, but lacks glutathione, amino acids, and proteins which inactivate platinum complexes [33]. Figure 2c shows a representative chromatogram. After 7 days, only three platinum complexes; DPC, DPCI, and DPI were detected in aqueous solution. DPC was the most abundant of the three. These results show that biological nucleophiles including chloride can displace leaving groups of miriplatin at concentrations which occur in the culture medium or serum. Next, in vitro antitumor effects of miriplatin, cisplatin, and zinostatin stimalamer were examined in addition to those of DPC or DPI, both of which were detected as compounds released from miriplatin/LPD. IC50 values for AH109A cells after 3 days of exposure to DPC, DPI, cisplatin, and zinostatin stimalamer were 0.14 ± 0.07, 0.83 ± 0.32, 0.30 ± 0.07, and 0.13 ± 0.00 μg/mL, respectively. Miriplatin did not inhibit cell growth at the highest concentrations tested (20 μg/mL) probably due to its low water solubility. DPC was about six times more active than DPI, and as potent as cisplatin and zinostatin stimalamer (Fig. 3). From these results, it was shown that platinum compounds released from miriplatin/LPD, but not miriplatin itself, inhibited the growth of AH109A cells.

In vitro cell growth inhibition by platinum compounds released from miriplatin/LPD. a Release characteristics of miriplatin/LPD and cisplatin/LPD into the culture medium. Aliquots (0.5 mL) of miriplatin/LPD (100 μg/mL, filled circle) and cisplatin/LPD (100 μg/mL, open circle) were added into Falcon cell culture inserts in wells containing 0.5 mL of RPMI 1640 medium supplemented with 10% FBS. The platinum concentrations in the medium were measured using FAAS. All results are given as the mean ± SD (n = 3 or 6). b The relation between platinum concentrations in the medium and cytotoxic activities following treatment with miriplatin/LPD and cisplatin/LPD. One day after the plating of cells into microplates, miriplatin/LPD and cisplatin/LPD were added into Falcon cell culture inserts. Cells were exposed to agents for 7 days at 37°C in 5% CO2. The platinum concentration was measured using FAAS. filled circle growth rates of cells treated with miriplatin, open circle growth rates of cells treated with cisplatin, filled square platinum concentrations in the medium after treatment with miriplatin, open square platinum concentrations in the medium after treatment with cisplatin. All results are given as the mean ± SD of triplicates. c HPLC/ICP-MS analysis of platinum compounds released from miriplatin/LPD. Three milliliters of miriplatin/LPD (20 mg/mL) was layered over 6 mL of Earle’s balanced salt solution supplemented with magnesium sulfate in test tubes. Test tubes were rotated vertically for 7 days at 5 rpm in an incubator at 37°C under protection from light. Platinum compounds in aqueous phase were analyzed by HPLC/ICP-MS

Effects of miriplatin, DPC, DPI, cisplatin, and zinostatin stimalamer on cell growth. One day after the plating of cells into microplates, miriplatin, DPC, DPI, cisplatin, and zinostatin stimalamer were added as aqueous solutions. Cells were exposed to agents at the indicated concentrations for 3 days at 37°C in 5% CO2. Open circle miriplatin, filled circle DPC, open triangle DPI, filled triangle cisplatin, open square zinostatin stimalamer. All results are given as the mean ± SD of triplicates
Formation of platinum-DNA adducts and induction of apoptosis in cells
Antitumor platinum complexes have growth inhibitory effects on cancer cells by causing platinum-DNA adducts to form, and apoptotic cell death [37, 38]. The amount of platinum in DNA was used as an overall measure of the formation of platinum-DNA adducts. After AH109A cells were treated with miriplatin/LPD (100 μg/mL) and cisplatin/LPD (15 μg/mL) for 3 days, 509 ± 100 and 34.1 ± 11.0 pg of platinum were incorporated into 1 μg of DNA, respectively. Miriplatin/LPD formed more platinum-DNA adducts than cisplatin/LPD at equitoxic concentrations. To compare the ability of miriplatin/LPD and cisplatin/LPD to induce apoptosis, we next evaluated the dose-dependent development of apoptosis in AH109A cells by measuring the population of sub-G1 apoptotic cells by flow cytometric analysis (Fig. 4a). As shown in Fig. 4b, there was a dose-dependent induction of apoptosis with both agents. Miriplatin/LPD, like cisplatin/LPD, showed the formation of platinum-DNA adducts and induction of apoptosis in AH109A cells. Both platinum-DNA adducts and apoptosis were also observed in cells treated with DPC or cisplatin as an aqueous solution for 3 days (data not shown).

Development of apoptosis in AH109A cells treated with miriplatin/LPD and cisplatin/LPD. a Representative DNA histograms after propidium iodide staining. Left LPD, middle miriplatin/LPD, right cisplatin/LPD. b The number of sub-G1 phase cells. AH109A cells were treated for 3 days with the indicated concentrations of miriplatin/LPD and cisplatin/LPD. The population of sub-G1 cells was determined by flow cytometry. Data are shown as the mean ± SD of triplicates
Blood supply to tumors, distribution of LPD, and platinum concentrations in rat livers
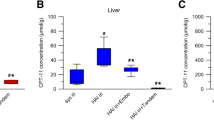
The liver receives a dual blood supply from the hepatic artery and the portal vein, whereas HCC is fed by only the hepatic artery [9–11]. To study the blood supply of AH109A tumors implanted in rat liver, a barium sulfate suspension was injected into the hepatic artery or the portal vein. The suspension injected into the portal vein distributed in non-tumor region, but not in the tumor, while that injected into the hepatic artery was selectively distributed in the tumor (Fig. 5a). LPD is known to be selectively distributed and retained for a long period in hepatic tumor tissues after intra-hepatic arterial injection [12–15]. When miriplatin/LPD was administered into the hepatic artery, LPD detected as white dots in the soft X-ray photograph was selectively retained in tumors, while it disappeared from non-tumor regions within 7 days (Fig. 5b). A similar distribution of LPD was observed when cisplatin/LPD and LPD alone were used (data not shown). Accordingly, the AH109A tumors implanted in rat livers were supplied exclusively by the hepatic artery, and LPD with or without agents administered into the hepatic artery could be selectively retained in tumors of rats. To examine whether miriplatin is retained in tumors by utilizing LPD as a tumor-selective carrier, the platinum concentrations in tissues were measured immediately and 7 days after the intra-hepatic arterial administration of miriplatin/LPD and cisplatin/LPD (Fig. 5c). The platinum concentrations in AH109A tumors were higher than those in non-tumor regions on both days, irrespective of the agents. On the other hand, the platinum concentrations in tumors were higher after the administration of miriplatin/LPD than cisplatin/LPD on both days. In addition, in the case of miriplatin/LPD, 18 ± 15 and 17 ± 13 μg of platinum were disposed of in tumors at day 0 and day 7, respectively, while 9.6 ± 6.0 μg of was disposed of in tumors at day 0 after the administration of cisplatin/LPD, and 2.8 ± 3.1 μg at day 7. These results indicate that platinum compounds were selectively disposed of in tumors after the intra-hepatic arterial administration of miriplatin/LPD, and maintained in tumors longer than after the administration of cisplatin/LPD.

Blood supply to tumors, distribution of LPD, and platinum concentrations in rat livers. a Blood supply to AH109A tumors implanted in rat livers. A 100% (w/v) barium sulfate suspension was injected into the hepatic artery (right) or the portal vein (left) at the volume of 0.5 mL/head or 5.0 mL/head, respectively. Immediately after the administration, livers were resected, fixed in formalin, and radiographed with a soft X-ray machine. Yellow circles indicate tumor sites. b Distribution of LPD. Miriplatin/LPD (400 μg/head) was injected into the hepatic artery of tumor-bearing rats at the volume of 0.02 mL/head. Seven days after the administration, resected livers were radiographed with a soft X-ray machine. Yellow circle indicates tumor site. c Platinum concentrations and amounts in tumor and normal liver tissues. Miriplatin/LPD (400 μg/head) and cisplatin/LPD (400 μg/head) were injected into the hepatic artery of tumor-bearing rats at the volume of 0.02 mL/head. Immediately and 7 days after the administration, resected livers were divided into tumor tissues (filled square) and normal liver tissues (open square). Tissue homogenates incinerated using nitric acid and hydrogen peroxide were dissolved in 4% aqua regia and introduced to FAAS. All results are given as the mean ± SD (n = 7)
In vivo antitumor activities of miriplatin, cisplatin, and zinostatin stimalamer suspended in LPD after intra-hepatic arterial administration
The growth of AH109A tumors was dose-dependently inhibited by miriplatin/LPD, cisplatin/LPD, and zinostatin stimalamer suspended in LPD (zinostatin stimalamer/LPD) as shown in Table 1. Miriplatin/LPD, cisplatin/LPD, and zinostatin stimalamer/LPD treatment resulted in a significant reduction in the growth of AH109A tumors at a dose of 400, 200, and 100 μg/head, respectively. At these doses, both miriplatin/LPD and cisplatin/LPD induced apparent tumor regression with tumor growth rates of 79 ± 31 and 66 ± 29%, respectively. Although the decrease in body weight exceeded 10% in the group treated with the highest dose of cisplatin/LPD (400 μg/head), all agents inhibited tumor growth without markedly enhancing body weight loss, compared to treatment with LPD alone. Next, the in vivo antitumor activity of miriplatin/LPD at the therapeutic dose was compared with that of cisplatin/LPD and zinostatin stimalamer/LPD (Table 2). Miriplatin/LPD and cisplatin/LPD (400 μg/head) treatment resulted in a significant reduction in tumor growth compared to that with LPD alone, while the difference in the tumor growth rate between LPD alone and zinostatin stimalamer/LPD (20 μg/head) did not reach a level of statistical significance. In addition, no significant difference was found between the untreated group and the group treated with LPD alone or the sham-operated group. At the therapeutic dose, miriplatin/LPD, like cisplatin/LPD, was shown to be more active than zinostatin stimalamer/LPD against AH109A tumors.
Formation of platinum-DNA adducts and induction of apoptosis in tumors
In the treatment of AH109A tumors, both miriplatin/LPD and cisplatin/LPD caused tumor regression. To investigate the mechanism involved, we examined the formation of platinum-DNA adducts and induction of apoptosis in tumors. Three days after the intra-hepatic arterial administration of miriplatin/LPD and cisplatin/LPD (400 μg/head), 61 ± 52 and 6.6 ± 8.7 pg of platinum were incorporated into 1 μg of DNA in AH109A tumors, respectively. The in situ visualization of apoptosis at livers was evaluated using terminal deoxynucleotidyl transferase-mediated dUTP-biotin nick end labeling (TUNEL) by which fragmented DNA could be specifically stained [39]. TUNEL stained exclusively the nuclei in tumors after the treatment with miriplatin and cisplatin, but not with LPD alone (Fig. 6). On the other hand, few TUNEL-positive nuclei were observed in non-tumor regions in any groups. These results showed that miriplatin/LPD, as well as cisplatin/LPD, generated platinum-DNA adducts and induced massive apoptosis selectively in tumors after intra-hepatic arterial administration when these agents exerted their in vivo antitumor activities.

TUNEL staining of rat livers bearing tumors. Miriplatin/LPD (400 μg/head), cisplatin/LPD (400 μg/head), and LPD alone were injected into the hepatic artery of tumor-bearing rats at the volume of 0.02 mL/head. Three days after the administration, livers were resected and fixed in formalin. Paraffin sections of livers were stained by the TUNEL method and counter-stained with hematoxylin. Left upper untreated, right upper LPD alone, left lower miriplatin/LPD, right lower cisplatin/LPD. Yellow circles indicate tumor sites
Discussion
Most HCC patients with intermediate to advanced stage tumors are not eligible for curative treatments due to poor liver function or the presence of advanced liver disease. For these patients, TACE is treatment option with a survival benefit. Actually, recent randomized controlled trials indicated that TACE induced marked objective responses and improved the survival of selected patients with HCC [23, 40]. Antitumor agents used for TACE should be soluble or easily suspended in LPD, be stable in LPD, and be gradually released in the tumor. However, few antitumor agents currently available have such properties. The strategy to improve the impact of TACE on survival and to reduce systemic toxicity should be based on the development of new agents which utilize LPD as a solvent and carrier. Miriplatin containing myristates as leaving groups to improve lipophilicity was chemically designed to be a more suitable antitumor agent for TACE [18]. Some platinum compounds released from miriplatin/LPD, but not miriplatin itself, showed antitumor activities in vitro by generating platinum-DNA adducts and inducing apoptosis. Tetraplatin, oxaliplatin, NDDP, and malonatoplatin have diaminocyclohexane as a carrier ligand. All of these platinum complexes undergo a transient conversion to DPC in physiological surroundings because their leaving groups are easily displaced by chlorides which are abundant biological nucleophiles [24, 25, 41, 42]. DPC was also detected as a major platinum compound released into saline and Earle’s balanced salt solution from miriplatin/LPD [28], indicating that chlorides could displace leaving groups of miriplatin at the concentrations which occurred in the culture medium or serum. DPIC and DPI were also observed as released platinum compounds, suggesting that leaving groups were also displaced by iodides from LPD, which consists of ethyl esters of iodized poppy seed oil. In addition, DPC was as potent as cisplatin in inhibiting the growth of hepatic cancer cell lines. From these findings, it was suggested that miriplatin/LPD exhibited in vitro antitumor activity by releasing platinum compounds, and DPC is one of the most likely candidates as described by Kishimoto et al. [28]. This was supported by the findings that miriplatin/LPD as well as cisplatin/LPD formed platinum-DNA adducts and induced apoptosis in AH109A cells.
Cisplatin has been used widely for systemic chemotherapy for various malignancies. Although no systemic chemotherapy using a single antitumor agent produced a response rate of more than 10%, cisplatin was relatively effective against HCC [43]. Shibata et al. has reported that the survival was significantly longer in patients treated with cisplatin/LPD as compared with those treated with LPD containing neocarzinostatin, an active component of zinostatin stimalamer [17]. While cisplatin/LPD are not easy to prepare under sterilized conditions and physically stable [44], miriplatin is easily suspended in LPD with just mild shaking by hand, and releases active platinum compounds gradually [27, 28]. Based on these characteristics, it is expected that the pharmacokinetics in livers containing tumors after the intra-hepatic administration of miriplatin/LPD could be improved compared with that after the administration of cisplatin/LPD. Actually, in a rat hepatic tumor model administered miriplatin/LPD, platinum compounds were selectively disposed of at higher concentrations in tumors than in non-tumor regions and maintained in tumors longer than those released from cisplatin/LPD. After intra-hepatic arterial administration of 14C-miripaltin/LPD in dogs, unchanged miriplatin was the major component retained in the liver for a long period [34]. Accordingly, it was suggested that by utilizing LPD as a carrier solvent, unchanged miriplatin was selectively disposed of and retained for a long time in tumors. To confirm whether the selective and long-lasting disposition in tumors contributes to the in vivo efficacy, antitumor activities of miriplatin/LPD were compared with those of cisplatin/LPD. Like cisplatin/LPD, miriplatin/LPD was quite effective against AH109A tumors at the doses where severe body weight loss was not observed. In addition, the formation of platinum-DNA adducts and massive apoptosis were induced selectively in tumors treated with miriplatin/LPD as well as cisplatin/LPD. Although we could not determine which platinum compounds were active at tumor sites after miriplatin treatment, it appears that some of the platinum compounds released, in which leaving groups of miriplatin were displaced by biological nucleophiles, most probably chlorides, contribute to the in vivo activities of miriplatin because miriplatin showed a mode of action common to most antitumor platinum complexes including cisplatin in vivo.
The volume of LPD suspension administered in TACE is decided according to the size of the tumor, the condition of the tumor vessels, and the degree of liver dysfunction, and the injection is discontinued when full accumulation of the suspension in the tumor vessels is obtained under fluoroscopic monitoring. As it is thought that the efficacy of an antitumor agent depends on its concentration in LPD, we compared the in vivo antitumor activity between three agents at the therapeutic dose. Under such conditions, miriplatin/LPD and cisplatin/LPD (400 μg/head, 20 mg/mL in LPD), but not zinostatin stimalamer/LPD (20 μg/head, 1 mg/mL), caused apparent tumor regression. At a dose fivefold higher than the therapeutic one, zinostatin stimalamer/LPD had a significant growth inhibitory effect on AH109A tumors. Based on the growth and histological examination of VX2 tumors implanted in rabbit livers, remarkable antitumor activities have been observed with zinostatin stimalamer/LPD at the therapeutic dose [45, 46]. The differences in the type of tumor implanted and dosing volume might influence the antitumor effect of zinostatin stimalamer/LPD. LPD by itself induced neither growth inhibition nor apoptosis in AH109A tumors. These results suggested that the cytotoxicity at the tumor site after the administration of miriplatin/LPD was resulted from the effect of miriplatin, and was not caused by either antiproliferative effects or vascular damage due to LPD.
In this study, it was suggested that after intra-hepatic arterial administration, miriplatin/LPD was selectively removed and retained for a long time in the tumor, and then released active platinum compound(s) gradually at the tumor site where massive apoptosis was induced after platinum-DNA adducts had formed. At the therapeutic dose, miriplatin/LPD as well as cisplatin/LPD was shown to be more active than zinostatin stimalamer/LPD against AH109A tumors after a single intra-hepatic arterial administration without enhancing body weight loss. Our results suggest miriplatin to be a suitable candidate for use in TACE. To confirm whether miriplatin was active against human HCC, additional studies are needed in which miriplatin/LPD is administered into the hepatic artery of animals bearing human hepatic tumors.
References
Llovet JM, Burroughs A, Bruix J (2003) Hepatocellular carcinoma. Lancet 362:1907–1917
Bruix J, Sherman M, Llovet JM, Beaugrand M, Lencinoni R, Burroughs AK et al (2001) Clinical management of hepatocellular carcinoma. Conclusions of the Barcelona-2000 EASL conference. J Hepatol 35:421–430
Llovet JM, Beaugrand M (2003) Hepatocellular carcinoma. J Hepatol 38:S136–S149
El-Serag HB (2002) Hepatocellular carcinoma and hepatitis C in the United States. Hepatol 36:S74–S83
Fattovich G, Stroffolini T, Zagni I, Donato F (2004) Hepatocellular carcinoma in cirrhosis: incidence and risk factors. Gastroentarol 127:S35–S50
Nishiguchi S, Kuroki T, Nakatani S et al (1995) Randomized trial of effects of interferon-α on incidence of hepatocellular carcinoma in chronic active hepatitis C with cirrhosis. Lancet 346:1051–1055
Kasahara A, Hayashi N, Mochizuki K et al (1998) Risk of hepatocellular carcinoma and its incidence after interferon treatment inpatients with chronic hepatitis C. Hepatol 27:1394–1402
Ikeda K, Saitoh S, Arase Y et al (1999) Effect of interferon therapy on hepatocellular carcinogenesis in patients with chronic hepatitis type C: a long-term observation study of 1, 643 patients using statistical bias correction with proportional hazard analysis. Hepatol 29:1124–1130
Mann JD (1953) Alterations in the vasculature of the diseased liver. Gastroenterol 25:540–546
Breedis C, Young G (1954) The blood supply of neoplasms in the liver. Am J Pathol 30:969–977
Nilsson LAV, Zettergren L (1967) Blood supply and vascular pattern of induced primary hepatic carcinoma in rats. Acta Pathol Microbiol Scand 71:179–186
Nakakuma K, Tashiro S, Hiraoka T et al (1983) Studies on anticancer treatment with an oily anticancer drug injected into the ligated feeding hepatic artery for liver cancer. Cancer 52:2193–2200
Nakakuma K, Tashiro S, Hiraoka T, Ogata K, Ootsuka K (1985) Hepatocellular carcinoma and metastatic cancer detected by iodized oil. Radiol 154:15–17
Yumoto Y, Jinno K, Tokuyama K et al (1985) Hepatocellular carcinoma detected by iodized oil. Radiology 154:19–24
Kan Z (1996) Dynamic study of iodized oil in the liver and blood supply to hepatic tumors: an experimental investigation in several animal species. Acta Radiol 408:1–25
Konno T, Maeda H, Iwai K et al (1984) Selective targeting of anti-cancer drug and simultaneous image enhancement in solid tumors by arterially administered lipid contrast medium. Cancer 54:2367–2374
Shibata J, Fujiyama S, Sato T, Kishimoto S, Fukushima S, Nakano M (1989) Hepatic arterial injection chemotherapy with cisplatin suspended in an oily lymphographic agent for hepatocellular carcinoma. Cancer 64:1586–1594
Maeda S, Shibata J, Fijiyama S et al (2003) Long-term follow-up of hepatic arterial chemoembolization with cisplatin suspended in iodized oil for hepatocellular carcinoma. Hepatogastroenterology 50:809–813
Taguchi T, Saito T, Ota J et al (1991) Phase I study of YM 881 (zinostatin stimalamer) suspension by hepatic arterial infusion. Jpn J Cancer Chemother 18:1657–1663
Taguchi T, Saito T, Ota J et al (1991) Phase II study of YM 881 (zinostatin stimalamer) suspension injected into the hepatic artery. Jpn J Cancer Chemother 18:1665–1675
Watanabe S, Nishioka M, Ohta Y, Ogawa N, Ito S, Yamamoto Y (1994) Cooperative study group for liver cancer treatment in shikoku area Prospective and randomized controlled study of chemoembolization therapy in patients with advanced hepatocellular carcinoma. Cancer Chemother Pharmacol 33:S93–S96
Buttacharya S, Novell JR, Dusheiko GM, Hilson AJ, Dick R, Hobbs KE (1995) Epirubicin-lipiodol chemotherapy versus 131iodine-lipiodol radiotherapy in the treatment of unresectable hepatocellular carcinoma. Cancer 76:2202–2210
Lo CM, Nygan H, Tso WK et al (2002) Randomized controlled trial of transarterial lipiodol chemoembolization for unresectable hepatocellular carcinoma. Hepatology 35:1164–1171
Mauldin SK, Plescia M, Richard FA, Wyrick SD, Voyksner RD, Chaney SG (1988) Displacement of the bidentate malonate ligand from (d,l-trans-1,2-diaminocyclohexane)malonatoplatinum (II) by physiologically important compounds in vitro. Biochem Pharmacol 37:3321–3333
Luo FR, Yen T-Y, Wyrick SD, Chaney SD (1999) High-performance liquid chromatographic separation of the biotransformation products of oxaliplatin. J Chromatograph B 724:345–356
Maeda H, Uchida NA, Sasaki T (1986) Liposoluble platinum (II) complexes with antitumor activity. Jpn J Cancer Res 77:523–525
Kishimoto S, Fukui M, Fukushima S, Nakano M (1992) Application of a lipophilic platinum derivative contained in an oily lymphographic agent to intra-hepatic arterial chemotherapy. Reg Cancer Treat 1–2:25–29
Kishimoto S, Noguchi T, Yamaoka T, Fukushima S, Takeuchi Y (2000) In vitro release of SM-11355, cis[((1R,2R)-1,2-cyclohexanediamine-N,N′)bis(myristato)] platinum (II) suspended in lipiodol. Biol Pharm Bull 23:637–640
Kishimoto S, Noguchi T, Yamaoka T, Fukushima S, Takeuchi Y (2000) Antitumor effects of a novel lipophilic platinum complex (SM-11355) against a slowly-growing rat hepatic tumor after intra-hepatic arterial administration. Biol Pharm Bull 23:344–348
Ono Y, Yoshioka H, Ohkusa A et al (1992) The effect of liposoluble cis-platinum (II) complex administered via hepatic artery on rat hepatic carcinoma. J Jpn Soc Cancer Ther 27:49–58
Kishimoto S, Miyazawa K, Fukushima S, Takeuchi Y (2000) In vitro antitumor activity, intracellular accumulation, and DNA adduct formation of cis-[((1R,2R)-1,2-cyclohexanediamine-N,N′)bis(myristato)] platinum (II) suspended in lipiodol. Jpn J Cancer Res 91:99–104
Kishimoto S, Miyazawa K, Terakawa Y et al (2000) Cytotoxicity of cis-[((1R,2R)-1,2-cyclohexanediamine-N,N′)bis(myristato)] platinum (II) suspended in lipiodol in a newly established cisplatin-resistant rat hepatoma cell line. Jpn J Cancer Res 91:1326–1332
Earle WR, Schilling EL, Stark TH, Straus NP, Brown MF, Shelton E (1943) Production of malignancy in vitro. IV. The mouse fibroblast cultures and changes seen in the living cells. J Natl Cancer Inst 4:165–212
Shimakura J, Fujimoto K, Komuro S, Nakano M, Kanamaru H (2002) Long-term disposition of a novel lipophilic platinum complex SM-11355 in dog after intrahepatic arterial administration: highly sensitive detection of platinum and radioactivity. Xenobiotica 32:399–409
Fujiyama S, Shibata J, Maeda S et al (2003) Phase I clinical study of a novel lipophilic platinum complex (SM-11355) in patients with hepatocellular carcinoma refractory to cisplatin/lipiodol. Br J Cancer 89:1614–1619
Okusaka T, Okada S, Nakanishi T, Fujiyama S, Kubo Y (2004) Phase II trial of intra-arterial chemotherapy using a novel lipophilic platinum derivative (SM-11355) in patients with hepatocellular carcinoma. Invest New Drugs 22:169–176
Gonzalez VM, Fuertes MA, Alonso C, Perez JM (2001) Is cisplatin-induced cell death always produced by apoptosis? Mol Pharmacol 59:657–663
Kartalou M, Essigmann JM (2001) Recognition of cisplatin adducts by cellular proteins. Mutat Res 478:1–21
Gavrieli Y, Sherman Y, Ben-Sasson SA (1992) Identification of programmed cell death in situ via specific labeling of nuclear DNA fragmentation. J Cell Biol 119:493–501
Llovet JM, Real MI, Montaña X et al (2002) Arterial embolisation or chemoembolisation versus symptomatic treatment in patients with unresectable hepatocellular carcinoma: a randomized controlled trial. Lancet 359:1734–1739
Chaney SG, Wyrick S, Till GK (1990) In vitro biotransformations of tetrachloro(d,l-trans)-1,2-diaminocyclohexanepaltinum(IV)(tetraplatin) in rat plasma. Cancer Res 50:4539–4545
Han I, Khokhar AR, Perez-Soler R (1996) Intraliposomal conversion of lipophilic cis-bis-carboxylato-trans-R,R-1,2-diaminocyclohexane-paltinum (II) complexes into cis-bis-dichloro-trans-R,R-1,2-diaminocyclohexane-paltinum (II). Cancer Chemother Pharmacol 39:17–24
Nagahama H, Okada S, Okusaka T et al (1997) Predictive factors for tumor response to systemic chemotherapy in patients with hepatocellular carcinoma. Jpn J Clin Oncol 27:321–324
Fukushima S, Kishimoto S, Hayashi Y, Kaneko M, Nakano M (1988) Intra-hepatic artery infusional chemotherapy with cisplatin suspension in lipiodol (LPS) for hepatocellular carcinoma (I): preparation of LPS and its anticancer effect on rabbit liver cancer model after injection into the hepatic artery (in Japanese). J Jpn Soc Cancer Ther 23:2743–2749
Kimura M, Konno T, Ohtsuka N, Mizumachi R, Oda T (1989) Antitumor activities of oily suspended YM881 (SMANCS) against VX2 carcinoma. Jpn J Cancer Chemother 16:2183–2188
Oda T, Konno T, Ohtsuka N, Kimura M, Mizumachi R (1991) Antitumor activities of oily suspended zinostatin stimalamer (YM 881) against VX2 carcinoma implanted in the liver of rabbits—comparison with another anticancer agent. Jpn J Cancer Chemother 18:2423–2428
Acknowledgments
The authors are grateful to Yoko Tsunashima, Miki Takatsuka, and Junko Yasui for their expert technical assistance in the experiments in vitro and in vivo.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Hanada, M., Baba, A., Tsutsumishita, Y. et al. Intra-hepatic arterial administration with miriplatin suspended in an oily lymphographic agent inhibits the growth of tumors implanted in rat livers by inducing platinum-DNA adducts to form and massive apoptosis. Cancer Chemother Pharmacol 64, 473–483 (2009). https://doi.org/10.1007/s00280-008-0895-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00280-008-0895-3