
Abstract
Cancer cells acquire cell-autonomous capacities to undergo limitless proliferation and survival through the activation of oncogenes and inactivation of tumor suppressor genes. Nevertheless, the formation of a clinically relevant tumor requires support from the surrounding normal stroma, also referred to as the tumor microenvironment. Carcinoma-associated fibroblasts, leukocytes, bone marrow-derived cells, blood and lymphatic vascular endothelial cells present within the tumor microenvironment contribute to tumor progression. Recent evidence indicates that the microenvironment provides essential cues to the maintenance of cancer stem cells/cancer initiating cells and to promote the seeding of cancer cells at metastatic sites. Furthermore, inflammatory cells and immunomodulatory mediators present in the tumor microenvironment polarize host immune response toward specific phenotypes impacting tumor progression. A growing number of studies demonstrate a positive correlation between angiogenesis, carcinoma-associated fibroblasts, and inflammatory infiltrating cells and poor outcome, thereby emphasizing the clinical relevance of the tumor microenvironment to aggressive tumor progression. Thus, the dynamic and reciprocal interactions between tumor cells and cells of the tumor microenvironment orchestrate events critical to tumor evolution toward metastasis, and many cellular and molecular elements of the microenvironment are emerging as attractive targets for therapeutic strategies.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
From a cell autonomous to a microenvironmental view of cancer
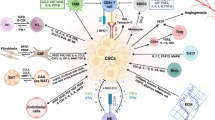
Cancer has been long viewed as a disease consisting of transformed cells acquiring cell autonomous hyperproliferative, invasive and limitless survival capacities. Accordingly, therapeutic anticancer strategies have been concentrated on and limited to targeting the tumor cell itself. Emerging evidence indicates that to effectively control cancer, we need to consider carcinogenesis and tumor progression not as a cell autonomous, cancer cell-centered condition, but rather as a disease involving complex heterotypic multicellular interactions within a newly formed tissue, the cancer tissue. Hence, the concept of tumor microenvironment as an integrated and essential part of the cancer tissue was coined (Bissell et al. 2002; Hanahan and Weinberg 2000). New data emerging from the study of the tumor microenvironment are forcing the cancer research community to revise basic concepts of cancer biology. The tumor microenvironment contains many distinct cell types, including endothelial cells and their precursors, pericytes, smooth muscle cells, fibroblasts, carcinoma-associated fibroblasts, myofibroblasts, neutrophils, eosinophils, basophils, mast cells, T and B lymphocytes, natural killer cells and antigen presenting cells (APC) such as macrophages and dendritic cells (Coussens and Werb 2002) (see Fig. 1). Experimental data have now demonstrated a role for these individual components, in particular endothelial cells, macrophages, and cancer-associated fibroblasts, in promoting tumor growth and progression. While most cellular components of the immune system are capable of rejecting tumors, in practice they are enslaved by cancer cells to promote their growth and invasion. Thus, knowledge and control of the tumor microenvironment is becoming as important as the knowledge and control of the transformed cancer cells to better understand cancer biology and to devise novel therapeutic approaches (Albini and Sporn 2007).

Heterotypic cellular interactions in the tumor microenvironment. a Tumor cells orchestrate directly (e.g. though the release of factors) or indirectly (though the induction of tissue hypoxia or appearance of necrosis) the modification of the microenvironment by attracting or activating many non-tumoral cells, including blood and lymphatic endothelial cells and pericytes, carcinoma associated fibroblast, bone marrow-derived cells, immune and inflammatory cells. Tumor cells can also deposit or modify the extracellular matrix. Most of these stromal modifications start early during tumor progression, often at the transition stage from premalignant to malignant lesions. In some cases they may even precede cancer formation, for example in situations of chronic inflammatory conditions. b In turn, tumor microenvironmental events promote tumor progression by stimulating tumor growth and survival, and facilitating invasion and metastasis. Collectively these events will contribute to determine the outcome of tumor progression: tumor growth, tumor dormancy, tumor invasion and metastasis and resistance to therapy. Abbreviations: B B lymphocyte; BMDC bone marrow-derived cells; BV blood vessel; CAF carcinoma associated fibroblast; EC endothelial cell; ECM extracellular matrix; EMT epithelial to mesenchymal transition; Gr granulocyte; LEC lymphatic endothelial cell; LV lymphatic vessel; Mo monocyte; MSC mesenchymal stem cell; PC pericyte; T T lymphocyte; TAM tumor associated monocyte/macrophage; TC tumor cells
The first evidence that non-cancerous tissue elements might affect tumor formation and growth came from the field of inflammation. A link between inflammation and cancer has been recognized already in 1863 by Rudolf Virchow, when he reported the presence of leucocytes in tumor tissues (Balkwill and Mantovani 2001). Based on this observation he proposed the idea that cancer originates at sites of chronic inflammation. The presence of leukocytes in tumors was subsequently interpreted as an aborted attempt of the immune system to reject the tumor. This observation remained largely neglected for over a century until it was demonstrated that innate immune cells, in particular phagocytes, play an active role in promoting the carcinogenesis process. In addition to leukocyte infiltration, angiogenesis is now being recognized as another stromal reaction promoting cancer progression. Thus, chronic inflammatory and neovascularization are critical, if not essential, for cancer progression (Rüegg 2006).
Inflammatory lesions preceding early stages of neoplastic progression contribute to create an environment favorable to cancer development. For example, chronic inflammatory bowel diseases (IBD), such a Crohn’s disease or ulcerative colitis, or chronic reflux esophagitis in Barrett syndrome are associated with increased risk of developing colorectal or esophageal cancer, respectively (van der Woude et al. 2004). Cancers of infectious etiology have also been associated with chronic inflammation: for example, Hepatitis B and C viruses for hepatic cancer, Schistosomes for bladder cancer, and Helicobacter pylori for gastric cancer (Karin et al. 2006). In many epidemiological studies the role of chronic inflammation in the carcinogenesis process was examined through studies of pro-inflammatory and antiinflammatory cytokines along with other risk factors including virus-transmitted infections and genetic markers that take part in the inflammatory response. Experimentally, systemic administration of non-specific pro-inflammatory substances, like LPS, has been shown to favor tumor growth, angiogenesis and metastatic tumor spreading to secondary organs (Harmey et al. 2002).
Inflammation and tumor progression
Today some of the underlying cellular and molecular events connecting inflammation to cancer have been uncovered (Mantovani et al. 2008a). Chronic inflammatory events create a local microenvironment capable of promoting tumor progression though a series of dynamic and reciprocal interactions between tumor cells and inflammatory cells (Coussens and Werb 2002). Initially, tumor cells and cells of the tumor microenvironment, respond to tumor hypoxia and necrosis secondary to excessive tumor cell proliferation, by releasing a number of growth factors and cytokines that are chemoattractive for monocytes and macrophages, including colony stimulating factor (CSF)-1, granulocyte–monocyte (GM)-CSF, transforming growth factor (TGF)-β, and chemokines (e.g. CCL2, CCL7, CCL3, CCL4) (Robinson and Coussens 2005). In turn, recruited macrophages secrete growth factors that affect tumor cell behavior (e.g. induction of motility), activate tumor endothelium and perpetuate inflammation (Allavena et al. 2008b). Factors released by recruited monocytes/macrophages include vascular endothelial growth factor (VEGF)-A and -C, basic fibroblast growth factor (bFGF), tumor necrosis factor (TNF), hepatocyte growth factor (HGF), epidermal growth factor (EGF) family members, platelet-derived growth factor (PDGF), and chemokines such as CXCL12 and interleukin (IL)-8 (Benelli et al. 2006; Robinson and Coussens 2005). Monocytes and macrophages also bring in much of the cyclooxygenase-2 (COX-2) present in the tumor environment. COX-2 expression and prostaglandins production within the tumor environment stimulate tumor cell proliferation, survival and motility but also tumor angiogenesis (Prescott and Fitzpatrick 2000). Because of, COX-2 expression was long though to be restricted to inflammatory conditions, and potent COX-2 inhibitors (COXIBs) were available to treat inflammatory conditions, COX-2 appeared suddenly as a ‘magic’ therapeutic target for cancer prevention and treatment (Xu 2002). However, the appearance cardiovascular complications induced by potent COXIBs (Dogne et al. 2006) has damped enthusiasm and hampered the widespread use of COXIBs for cancer chemoprevention.
The stroma of a full-blown tumor becomes infiltrated with many different leukocyte populations, including neutrophils, eosinophils, basophils, monocytes/macrophages, dendritic cells, natural killer cells and lymphocytes (Lin and Pollard 2004). Tumor infiltrating macrophages, also referred to as tumor educated or tumor-associated macrophages (TAM), are known residents in most neoplastic tissues, where they play a critical role in orchestrating and promoting tumor growth (Allavena et al. 2008b; Pollard 2004). Deficient monocyte recruitment at tumor sites in mice lacking CSF-1 expression was shown to attenuate late-stage progression and metastasis formation, suggesting that monocytes contribute to tumor progression (Lin et al. 2001). A positive correlation between the number of TAMs and poor prognosis has been reported for many cancers. Importantly, monocytes/macrophages are often present already at the very early stage of tumor development, in close associations with areas of hyperplasia and atypia (Mantovani et al. 2006; Pollard 2004). This observation further reinforces the notion that macrophages themselves are driving force contributing to tumor initiation and/or initial tumor progression. Once recruited to tissue sites, monocytes can assume different phenotypes, epitomized by the M1 and M2 phenotypes, based on local environmental stimuli (Allavena et al. 2008a). The M1 phenotype is associated with inflammation and microbial killing activity, whereas the M2 phenotype is associated with tissue remodeling and pro-angiogenic activities. The latter are predominant and crucial events in cancer. Macrophage–tumor cell interaction leads to the release of macrophage-derived cytokines, chemokines and growth/motility factors (e.g. GM-CSF, IL-8, EGF) which in turn promote the recruitment of additional inflammatory cells, thereby amplifying the inflammatory reaction within the tumor microenvironment (Balkwill 2004). Chemokines play an important role in coordinating the stromal response to cancer, including the polarization of the immune responses to the tumor, the determination of the composition of the cellular infiltrate, and the induction of angiogenesis. Chemokine receptors have been detected on cancer cells and the relevant ligands were found expressed at the primary tumor site and at sites of tumor metastasis, suggesting a direct role for chemokines/chemokine receptors in tumor growth and metastasis (Balkwill 2004).
Inflammatory insults lead to upregulation of non-specific pro-inflammatory cytokines such as IL-1α/β, IL-6, interferon (IFN)-γ and TNF (Aggarwal et al. 2006; Robinson and Coussens 2005). Such unresolved chronic inflammation is associated with increased conversion of normal cells to preneoplastic foci. Accumulation of somatic mutations with gain of function can change preneoplastic foci into foci of fully transformed cells with tumor initiation capacities. Activated nuclear factor-kappa B (NF-κB) transcription factor, is one of the main link between inflammation and tumorigenesis and may be key in allowing both preneoplastic and malignant cells to escape apoptosis (Karin 2006; Naugler and Karin 2008). In addition to macrophages, mast cells and neutrophils can also support tumor progression by sustaining inflammation and secreting tumor-promoting cytokines, growth factors and proteases. For example, in skin carcinogenesis models, granulocytes were identified as source of MMP-9 expression required for tumor development (Coussens et al. 2000). Consistent with this observation, it has been recently reported that neutrophils also play a key role in ras oncogene-driven tumor progression (Ji et al. 2006).
Inflammatory cytokines may act as an initiator and promoter of carcinogenesis by directly increasing proliferation of epithelial cells. Inflammation may also promote tumorigenesis indirectly, by modulating the phenotype and function of cells mediating specific or innate immunity (Rollins 2006). Such effects can range from tissue remodeling, angiogenesis, immune suppression and growth promotion, all characteristic steps of chronic inflammation, to a tumor suppressive phenotype associated with intense tissue damaging events typical of acute inflammation. Thus, inflammatory cells within a tumor mass can be considered as a double-edged sword (Hagemann et al. 2007): under specific stimulation they can produce antiangiogenic cytokines, such as IL-12, resulting in suppressed tumor growth. However, in most tumors inflammatory cells promote tumor angiogenesis, tumor growth, tumor cell dissemination, tissue breakdown and remodeling. Most tumors are associated with the activation of tumor-promoting innate immune responses involving neutrophils, macrophages and NK cells, while on the other side, specific antitumor immune responses involving B or T lymphocytes are less efficient in suppressing tumor growth. Thus, in most cases, cancers exploit inflammatory and immune infiltrating cells for their own benefit.
In spite of the important recruitment of inflammatory cells in the tumor microenvironment, these cells do not represent the major cell population of the tumor stroma (Tlsty and Coussens 2006). Carcinoma-associated fibroblasts (CAFs) are the most abundant cells of the tumor microenvironment (Orimo et al. 2005; Orimo and Weinberg 2006). CAFs are usually recognized by the expression of α-smooth muscle actin (a-SMA), similar to myofibroblasts present at the site of wound healing and chronic inflammation, which is absent in normal dermal fibroblasts. CAFs might differentiate locally from normal stromal fibroblasts of the surrounding tissue or from bone marrow-derived mesenchymal stem cells recruited to the tumor. The observation that CAFs lack genetic mutation or aneuploidy typically found in the neighboring tumor cells, tend to exclude a possible tumor origin of these cells following complete epithelial to mesenchymal transition (EMT) (Kalluri and Zeisberg 2006).
The immune system and its role in tumor progression
In the early 1900 Paul Ehrlich proposed the notion that the immune system could be exploited therapeutically to treat tumors by suggesting the use of antibodies to deliver toxins to tumor cells (immunotoxis as ‘magic bullets’) (Strom et al. 1990). Although in practice it has proven difficult to create safe and effective ‘magic bullets’, we have nevertheless learned that both innate and adaptive immune responses can both promote tumor progression or mediate tumor rejection, as a balance between pro-tumor and antitumor immunity (Ostrand-Rosenberg 2008). In the tumor microenvironment the cellular components of the inflammatory infiltrate and immunomodulatory mediators (cytokines and chemokines), polarize host immune response toward a specific phenotype, in favoring or suppressing tumor progression (DeNardo and Coussens 2007; Johansson et al. 2008). It is nowadays considered that the main mechanism of tumor immunity is due to an antitumoral T cell response (Mantovani et al. 2008b). This antitumor response can be due to the direct killing of tumor cells by CD8 cytotoxic T lymphocytes (CTL) recognizing major histocompatibility complex (MHC) class I restricted antigens expressed on the surface of tumor cells. Oligoclonal tumor-reactive CTLs can be detected in the blood or among tumor infiltrating lymphocytes (TILs) in cancer patients. In some cases the presence of TILs in primary tumors is associated with a better prognosis, suggesting that TILs might control tumor growth (Yu and Fu 2006). However, in most cases the adaptive immune response against tumor cells is very weak and largely inefficient, since the tumor itself and the surrounding microenvironment down-regulate CTL responses (tumor induced immunosuppression). The fact that CTLs against human and animal tumors can be easily generated in vitro using freshly isolated peripheral blood lymphocytes indicate that the tumor microenvironment has immunosuppressive capacities (Mantovani et al. 2008b; Verdegaal et al. 1999).
Evidence for the existence of a specific antibody response against tumor cells in cancer patients stems from the observation that sera from some tumor patients specifically recognize antigens selectively expressed on autologous tumor cells but not on autologous normal cells. Recently, using the serological analysis of recombinantly expressed tumor antigens, a large number of tumor antigens have been identified from the antibody repertoire of cancer patients. Antitumor antibodies have been detected against many intracellular antigens, including p53 in patients with melanoma, hepatocellular carcinoma, colorectal and breast cancers, and against membrane-associated antigens, such as Her-2 in patients with breast and prostate cancer (Mittelman et al. 2002). Strikingly, the cellular localization of the recognized antigen has prognostic relevance. Antibodies detected against intracellular proteins appear to be correlated with poor prognosis, while antibodies reactive against cell surface components appear to be correlated with a better prognosis (Taylor and Gercel-Taylor 1998).
It is generally assumed that growing tumors develop strategies to evade or limit the effects of the host’s immune responses (Drake et al. 2006). These assumptions are based on the evidence that an efficient tumor-specific immune response is rarely detectable in patients and that tumor progression is often associated with the secretion of immunosuppressive factors and down-regulation of MHC class I antigen presentation. Furthermore, as suggested by Gilboa, increasing immune activity against a tumor in any active or passive immunotherapy will exacerbate the rate of immune escape and select for a tumor sub-population, which will be resistant to immunotherapy. Immune escape can be broken down into several mechanisms, including down-regulation or loss of tumor-associated antigens (TAA), or the release of immunosuppressive cytokines (Gilboa 1999). Loss of TAA expression is one of the main mechanisms by which tumors escape CTL recognition in vivo. Since intratumoral TAA expression frequently appears to be heterogeneous, the emergence of antigen-loss variants due to selection pressure mediated by a relevant antitumor response induced by a vaccination strategy is to be expected. The secretion of immunosuppressive cytokines and chemokines into the tumor microenvironment, such as TGF-β, IL-6 and IL-10, which can interfere with multiple steps and pathways in the generation of an effective immune response, has been described in many types of tumors (Mantovani et al. 2008b). In particular, TGF-β is an extremely potent immunosuppressive factor that affects proliferation, activation and differentiation of the immune effector cells and has been associated with malignant progression (Teicher 2001). The presence of this factor also inhibits the production of immunoregulatory cytokines and may also modify the expression of surface receptors essential for T cell activation and growth. Concomitantly, TGF-β is a potent inducer of EMT and contributes to promote the invasive and metastatic behavior of tumor cells (Bierie and Moses 2006b; Lee et al. 2006). IL-6 is expressed in the tumor microenvironment in 83% of patients with colorectal carcinoma correlates with unfavorable disease progression (Piancatelli et al. 1999). IL-6 is also known to have multiple effects on T cell function, and it may play a significant role in modulating T cell reactivity at the tumor site. Recently, has been reported the IL-6/IL-8 interleukin-dependent inflammatory network contributes to connecting oncogene-induced cellular senescence, with an inflammatory phenotype and tumor progression (Kuilman et al. 2008). In particular IL-6 acts as a central regulator of this inflammatory network, also mediates cellular senescence, reminding a striking parallel with TGF-β (Bierie and Moses 2006a). Both cytokines are able to restrain proliferation of non-malignant cells present in the tumor microenvironment, but at the same time they promote proliferation, invasion and metastasis, thereby reinforcing the close connection between inflammatory mediators and neoplastic progression.
IL-10 has pleiotropic effects on T cell functions, including the suppression of GM-CSF, IFN-γ and IL-2 production by T helper cells, inhibition of T cell proliferation, down-regulation of expression of adhesion molecules and MHC class I and class II antigens (Mocellin et al. 2005). Patients with ovarian carcinoma frequently present abundant levels of this cytokine in serum, in the peritoneal exudate and in the tumor tissue. These cytokines inhibit the expression of immune activating cytokines, such as IL-2 and IL-4 (Benelli et al. 2006; Punnonen et al. 1998). This can inhibit the natural homeostatic mechanisms that control the specific cellular immunity and could be responsible for the signaling defects in T lymphocytes, routinely observed in late stage cancer patients, rendering them ineffective in mounting a TAA specific effector activity.
Tumor angiogenesis
Angiogenesis occurs in physiological conditions during embryonic development, wound healing and tissue regeneration (Carmeliet 2005). In these circumstances, angiogenesis is a highly regulated process leading to structured, hierarchically organized and well functioning vascular networks. In contrast, in pathological conditions such as cancer or chronic inflammation, angiogenesis leads to the formation of disorganized, chaotic and poorly functioning vascular networks (McDonald and Choyke 2003). The formation of a tumor-associated (angiogenic) vasculature is a stromal reaction essential for tumor progression (Carmeliet and Jain 2000; Kerbel 2008). Tumor-associated vessels promote tumor growth by providing oxygen and nutrients and favor tumor metastasis by facilitating tumor cell entry into the circulation. In the absence of sufficient vascularization most tumors cannot exceed a few mm3 in volume, and remain clinically silent (Folkman 1995). Tumor angiogenesis is initiated at discrete time points during tumor progression (‘angiogenic switch’), which are determined by the balance between the genetic status of the tumor itself, signals from stromal and recruited inflammatory cells and by the appearance of hypoxia (Bergers and Benjamin 2003; Coussens and Werb 2002). The angiogenic switch often occurs at the time of the conversion from premalignant to malignant lesions (Hanahan and Folkman 1996).
Angiogenesis is a multi step process involving different cell types, in particular endothelial, perivascular cells (i.e. pericytes, smooth muscle cells), inflammatory and stromal cells. The cellular and molecular mechanisms of angiogenesis have been largely uncovered in recent years (Carmeliet 2000). In order to form new vessels, endothelial cells have to exit dormancy, proliferate, migrate, differentiate, polarize and organize themselves into new tubular structures within the remodeled stroma. These events are induced and regulated by diffusible growth and chemotactic factors, cell adhesion receptors, and instructive molecules. Many molecular mediators and regulators of angiogenesis are now known. VEGFs have emerged as critical mediators of angiogenesis (Ferrara 2005). The principal member of the VEGF family of factors, VEGF-A binds to and activates two tyrosine kinase receptors, VEGFR-1 and VEGFR-2. Of the two, VEGFR-2 is the major mediator of the mitogenic, angiogenic and permeability-enhancing effects of VEGF. The significance of VEGFR-1 in the regulation of angiogensis is more complex (Shibuya 2001). Under some circumstances, the higher affinity VEGFR-1 may function as a ‘decoy’ receptor that sequesters VEGF and prevents its interaction with VEGFR-2. Alternatively, VEGFR-1 may regulate function of VEGFR-2 (cross talk). There is growing evidence that VEGFR-1 has significant roles in haematopoiesis and in the recruitment of monocytes and other bone-marrow-derived cells that may home in on the tumor vasculature and promote angiogenesis. VEGFR-1 is involved in the induction of matrix metalloproteinases (MMPs) and in the release of matrix-bound growth factors. Furthermore, in some cases VEGFR-1 is expressed by tumor cells and may mediate a chemotactic signal, thus potentially extending the role of this receptor in cancer growth (Gerber and Ferrara 2003).
VEGF is strongly induced by hypoxia forming within the tumor microenvironment. The transcription factor hypoxia inducible factor (HIF) 1α plays a central role in the hypoxic regulation of VEGF (Semenza 2002). HIF1α is constitutively produced in excess but rapidly degraded under normoxic conditions. The von Hippel–Lindau (VHL) tumor suppressor protein targets it for ubiquitination and subsequent proteasomal degradation (Semenza 2001). In situ hybridization studies demonstrate that VEGF mRNA is expressed in many tumors. Renal cell carcinomas have a particularly high level of VEGF expression, consistent with the notion that inactivating VHL mutations occur in about 50% of such tumors, thus providing a further explanation for the responsiveness of this tumor type to a VEGF blockade (Haase 2006). Strikingly, HIFs have been shown to activate specific signaling pathways such as Notch and Oct4 that control stem cell self renewal and multipotency, suggesting a possible link between hypoxia and stemness (Keith and Simon 2007). Hypoxia, however, is not the only mechanism regulating VEGF expression in tumors. A broad and diverse spectrum of oncogenes can induce VEGF expression, including mutant ras, erbB-2/Her2, activated EGF receptor and bcr-abl. Besides VEGF, other vascular growth factors, including b-FGF, IL-8, TGF-β, TNF and molecules promoting, mediating or regulating angiogenesis, have been identified and characterized in recent years. They include adhesion molecules of the integrin and cadherin families, such as αVβ3, α5β1, and VE-cadherin; extracellular matrix proteins, such as fibronectin, collagens, and laminins; remodeling and morphogenic molecules and their receptors, in particular Angiopoietins/Ties, Notch and Delta-like ligands, Eph and Ephrins, semaphorins, neuropilins and the plexins; proteinases such as MMP-2 and -9, plasminogen activators (u-PA and t-PA) and their inhibitors (TIMPs and PAIs); intracellular signaling molecules, most notably protein kinases (e.g. raf and mitogen activated protein kinase (MAPK), protein kinase A (PKA), protein kinase B (PKB/Akt) and GTPases (e.g. Ras and Rho families), and many transcription factors and transcriptional regulators (besides HIF-1α), such as inhibitors of differentiation (Id1/3), NF-κB and homeobox gene products (e.g. Hox D3 and B3). (For review on mechanisms see the following publications and references herein: Avraamides et al. 2008; Bergers and Benjamin 2003; Jain 2003; Karamysheva 2008; Kerbel 2008; Neufeld and Kessler 2008; Pugh and Ratcliffe 2003; Rüegg and Mariotti 2003; Semenza 2007).
The recent discovery that VEGFR-3 and its ligands VEGF-C and -D promote lymphatic vessel formation (lymphangiogenesis) has boosted research on tumor lymphangiogenesis (Alitalo and Carmeliet 2002). Expression of VEGF-C or -D by tumor cells induces lymphatic vessel formation around the tumors and promotes metastatic tumor spreading at draining lymph nodes and distant organs. While it is clear that lymphatic vessels facilitate metastatic tumor spreading, the exact mechanism by which this occurs has not been fully elucidated (Alitalo et al. 2005).
Inflammatory cells promote tumor angiogenesis
It is becoming increasingly evident that inflammatory cells recruited in the tumor stroma play a pivotal role in triggering tumor angiogenesis (Rüegg 2006; Shojaei et al. 2008). It has been proposed that inflammatory cells might in fact be responsible for a substantial portion of tumor angiogenesis by acting as initiator of vascularization. Thus antiinflammatory drugs might be suitable tools in anticancer therapy. Innate immune (i.e. macrophages, neutrophils) release a number of factors able to stimulate and activate endothelial cells, such as VEGF, HGF, MMP-2, MMP-9 and IL-8. Neutrophils were largely ignored for a long time in the context of tumor angiogenesis but recent studies have shown their important role in this process (Coussens and Werb 2002). Furthermore, the Human Immunodeficiency Virus (HIV) viral transactivator protein Tat activates neutrophils as part of its angiogenic activity and angiostatin exerts its antiangiogenic activity, at least in part, through its interaction with neutrophils (Benelli et al. 2003). The close relationship between inflammation and angiogenesis observed within tumors is likely due to the natural physiological relationship between these two compartments. Endothelial cells and cells of the immune/inflammatory system are derived from a common hematopoietic precursor cell (Dzierzak and Speck 2008). Endothelial and immune cell are used to interact with each other, for example during leukocyte and lymphocyte trafficking across the endothelium and immune cells can affect vascular functions. Many angiogenic factors influence the activity of immune cells that generally favor tumor survival and tolerance. Thus, in addition to the classical tumor-endothelium axis widely studied in the angiogenesis field, it emerges that infiltrating inflammatory cells favor the cross-talks between tumor cells and endothelial cells, thereby creating a tumor-inflammation-endothelium axis, which may open new therapeutic perspectives (Noonan et al. 2008; Shojaei et al. 2008). In addition, to classical inflammatory cells, bone marrow-derived cells (BMDC) has been recently shown to be mobilized from the bone marrow in response to stimuli originating form the growing tumor, and to recruit at tumor sites to promote tumor angiogenesis and tumor invasion. Furthermore, tumor-mobilized myeloid cells can also be directed to different organs where they can form premetasitatic niches, which favor homing and survival of circulating tumor cells thereby promoting metastasis formation (Kaplan et al. 2006a).
Antiangiogenic therapy is in clinical practice
In experimental models, inhibition of the angiogenic switch prevents progression of hyperplastic foci to in situ carcinoma, decreases cancer insurgence and reduces metastasis. Targeting tumor angiogenesis to inhibit tumor growth has now become a clinical reality. In pathological conditions, such as cancer, endothelial cells are constantly stimulated by angiogenic factors and fail to form a differentiated, mature and stable vascular network (Kerbel 2008). Impinging on factors stimulating endothelial cells, results in the inhibition or even regression of angiogenic vessels (Ferrara and Kerbel 2005). Conceptually, it has been proposed that therapeutic targeting of non-tumoral cells would circumvent the problem of drug resistance and selection of escape variants, since these cells are genetically stable (in contrast to tumor cells). Furthermore, one given antiangiogenic therapy would suit different tumor types. Preclinical experiments and clinical studies demonstrated that reality is far more complex. In fact, the vast majority of drugs that were active in preclinical models failed to show efficacy in clinical trials (Kerbel and Folkman 2002). In spite of some initial negative clinical trials, major progress has been made over the past few years in targeting angiogenesis for human therapy, and inhibition of angiogenesis is now established as a new therapeutic approach to control tumor progression (Kerbel 2008). To date four antiangiogenic drugs are in clinical use after approval by the Food and Drug Administration (FDA): Avastin (bevacizumab) (Ferrara et al. 2005), a humanized anti-VEGF monoclonal antibody, for the treatment of metastatic colorectal cancer, in combination with chemotherapy, Nexavar (sorafenib) (Rini 2006), Sutent (sunitinib) (Motzer et al. 2006), two multi-tyrosine kinase inhibitors, and Torisel (temsirolimus) (Dancey 2005), an inhibitor of the mammalian target of rapamycin (mTOR) kinase. These drugs showed to extend survival of patients with advanced cancers (Sessa et al. 2008). Importantly, these compounds all target the VEGF pathway, either by inhibiting VEGF, its receptors or downstream molecules. Thus, to date the VEGF pathway is the best-characterized and validated therapeutic target to inhibit tumor angiogenesis for renal and gastrointestinal stromal tumors (GIST) (with Sorafenib and Sunitinib) without need for concomitant chemotherapy. In contrast, in lung, colorectal and breast cancers, survival benefit is only observed when antiangiogenic drugs are administered in combination with chemotherapy (Jain et al. 2006; Sessa et al. 2008)
Taken together, these achievements validated the concept that angiogenesis is a relevant therapeutic target in cancer. So far antiangiogenic drugs are being administered to patients with advanced cancers, and the impact on cancer progression and overall survival has remained small. It will be important to test whether antiangiogenic therapy administered in adjuvant settings may have a more significant impact on the survival of patients at risk for relapse. Furthermore, recent experimental evidence indicates that tumors can develop resistance to antiangiogenic therapy (Bergers and Hanahan 2008). This has been shown by inhibiting VEGF pathway and demonstrating that tumors resume angiogenesis by using alternative angiogenic factors, such as FGF-2 (Casanovas et al. 2005). It will be important to identify escape mechanisms of angiogenesis that could be targeted to improve efficacy of current antiangiogenic treatments.
The tumor microenvironment as a niche favoring tumor cell survival and metastasis
Normal cells survive and grow within defined environmental niches and are subjected to microenvironmental control. Outside of their specific niche, the tissue environment is hostile to normal cells. Since they lack necessary cell autonomous survival signals, normal cells will not survive an inappropriate microenvironment (Morrison and Spradling 2008). Detachment-induced cell death (anoikis) has been proposed as the mechanism preventing normal cells from leaving their original environment and seeding at inappropriate locations (Chiarugi and Giannoni 2008). In order to evade local tissue control and avoid anoikis during tumor development and progression, malignant cells start interacting with the surrounding extracellular matrix (ECM) (Reddig and Juliano 2005). A bidirectional relationship is initiated between tumor cells and its surrounding stroma as a first step to invasive growth on metastatic spreading (Bidard et al. 2008; Kopfstein and Christofori 2006; Weinberg 2008). Stromal changes sustaining tumor progression include modifications of the extracellular matrix composition, activation of fibroblasts, myoepithelial cells, and the recruitment of pericytes or smooth muscle cells and immune and inflammatory cells (Tlsty and Coussens 2006).
In recent years, evidence accumulated suggesting that a small tumor cell subpopulation in the primary tumor mass might be responsible for tumor initiation, growth, maintenance and spreading. These cells, termed cancer stem cells or cancer initiating cells (CSC/CIC), represent a population with stem cell-like properties, in particular long-term survival, high self-renewal and seeding capacities (Barnhart and Simon 2007; Croker and Allan 2008; Hambardzumyan et al. 2008; Visvader and Lindeman 2008). While the cellular origin of CSC/CIC and the associated molecular pathways are still matter of discussion, the existence of a small tumor cell subpopulation capable of initiating and maintaining tumor growth and initiating metastasis is being increasingly documented and accepted (Rossi et al. 2008; Tan et al. 2006). Furthermore, evidence indicates that CSC/CIC are more resistant to classical therapeutic approaches (i.e. chemotherapy and radiotherapy) compared to the bulk of the tumor cell mass. The mechanism of resistance remains largely elusive and might include the increased expression of multi-drug resistance-type of membrane transporters or a protective effect of the microenvironment. Initial evidence for the existence of CSC/CIC came from the hematopoietic system, where in acute myeloid leukemia rare leukemic (stem) cells with a CD34+/CD38− phenotype were shown to efficiently initiate transplantable experimental tumors in mice (Blair and Pamphilon 2003). Subsequently, it has been demonstrated that tumor cell subpopulations with similar features, but different surface phenotypes, also exist in solid tumors (Ailles and Weissman 2007; Cho and Clarke 2008). In all cases, CSC/CIC are distinguished from the somatic” tumor cell population, by their capacity to efficiently generate new tumors when implanted at low number in mice. Conversely, “somatic” cancer cells are not able to initiate tumor growth, even in high number, in the same in vivo preclinical models.
While the crucial role of the tumor microenvironment in controlling tumor progression and metastasis is now widely accepted, the formation of a specialized environment supporting CSC/CIC survival and growth (the CSC niche), its anatomical organization and the cellular and molecular mediators of these effects, are still under investigation (Sneddon and Werb 2007). The recent identification of human normal brain cells with self-renewal potential (reported as neural stem cells) that populate the subventricular zone, has contributed to the characterization of their niche and to the subsequent identification and characterization of brain tumor CSC/CIC and their niche. Brain tumor CSC/CIC are localized in a vascular niche that is supposed to provide factors promoting their self-renewal (Calabrese et al. 2007; Veeravagu et al. 2008). Perturbation of this niche results in a compromised ability CSC/CIC to promote tumor growth within the brain (Gilbertson and Gutmann 2007; Gilbertson and Rich 2007). These observations demonstrate the relevance of the vascular niche to tumor pathophysiology, and suggest the possibility to target the niche itself, and associated molecular events, to impinge on cancer stem cells for therapeutic purposes (Croker and Allan 2008).
A still open issue in the field is the definition of CSC/CIC through phenotypical markers allowing their accurate identification and isolation from the bulk of the tumor cell population in human and experimental tumors (Cho and Clarke 2008; Visvader and Lindeman 2008). Some markers have been proposed, including CD44+/CD24− in breast cancer, or CD133+ in brain, colon and pancreatic tumors. CD133, however, is also expressed by many normal stem cells in hematopoietic, neural and epithelial tissues (Woodward and Sulman 2008). Isolation of CSC/CIC from specific tumor types would allow their comparison with “somatic” cancer cells and open the way to the identification or specific gene expression profiles responsible for the aggressive CSC/CIC phenotype, including resistance to therapy (Cho and Clarke 2008; Dean et al. 2005).
The premetastatic niche: A niche for CSC/CIC ?
To explain how cancer cells can migrate from a primary site to colonize specific distant sites to grow and form metastases, Stephen Paget proposed the “seed and soil” hypothesis more than a century ago (Ribatti et al. 2006). This theory was based on the observation that tumors do not metastasize randomly but rater at preferred sites. Paget proposed that the formation of metastasis depends both on the properties of the tumor cells (seed) as well as the permissive role of the environment (soil) at the distant site. This hypothesis by which cancer cells are able to survive and proliferate only at specific secondary sites where there is an ideal environment that releases molecular mediators suitable for that type of cancer cells, still represents a main conceptual model of metastasis in modern cancer research (Steeg 2007; Talmadge 2007).
Metastasis formation itself is a multi-step process that require tumor cells to escape from the primary site, intravasate into the hematic or lymphatic circulation, migrate and extravasate into secondary organs (Christofori 2006; Steeg 2006). Recent work has shed new light on the genetic, molecular and cellular basis of metastasis (Gupta and Massague 2006; Nguyen and Massague 2007). CSC/CIC might represent the unique sub-population of cells with the potential to successfully form metastasis in a distant organ (Li et al. 2007; Wicha 2006). Metastasis formation, however, is a rather inefficient process, mostly due to the need for a cancer cells to find a proper microenvironment for initiating tumor growth in a secondary organ. It has been proposed that in order to form metastases, primary tumors might produce factors that induce the formation of a suitable and appropriate environment in the organ where metastasis will be seeded. This has lead to the concept of the premetastatic niche, whereby a special, permissive microenvironment in secondary target organs is induced over distance by the primary tumor (Kaplan et al. 2006b). In this sense, there is a striking parallel between CSC/CIC maintenance and expansion in the primary tumor and formation of metastases in a distant organ. Both events require a particular niche or microenvironment and might share many cues promoting self-renewal ability, migration and invasion, resistance to apoptosis, and increased resistance to cytotoxic drugs. The first in vivo experimental evidence that metastatic seeding requires the formation of a niche was the discovery that VEGFR-1+ BMDC colonize target organs to form tumor-specific premetastatic sites, before the arrival of the metastatic tumor cells themselves (Kaplan et al. 2005). In these sites BMDC express several hematopoietic markers, such as CD34, CD116, c-kit, Sca-1, as well as integrins (e.g. α4β1), chemokines and chemokine receptors (e.g. CXCL12/CXCR4), promoting either their homing to the target tissue or recruitment and attachment of tumor metastatic cells, or both.
Among the chemokines involved in tumor metastasis, the CXCL12 (also known as SDF-1 or stromal-derived factor)/CXCR4 axis plays a critical role in stem cell migration (Petit et al. 2007). Activation of CXCR4 induces motility, chemotactic response, adhesion, secretion of MMPs and release of angiogenic factors, such as VEGF-A. Interestingly this premetastatic niche, like the normal niche, is characterized by the presence of specific ECM proteins, such as fibronectin, a ligand for α4β1 expressed on VEGFR-1+ cells. Thus, it appears that in order to survive at distant sites, disseminating tumor cells need to recreate a supportive microenvironment similar to the one formed in the primary tumor. One efficient way to do it is by directing BMDC or immune/inflammatory cells to these distant sites of future metastasis (DeNardo et al. 2008). The chemoattractant proteins S100A8 and S100A9 were the first factors shown to instruct the formation of the premetastatic niche (Hiratsuka et al. 2008; Kaplan et al. 2006b). Additional studies are needed to further elucidate the biological mechanisms involved in the formation of this premetastatic niche, in particular regarding the identification of the molecular mechanism responsible for the development of clinically relevant metastases from the initial seeds. The unraveling of such mechanisms could be useful for the development of a more accurate knowledge about the metastatic process, with major clinical implications to the prevention, monitoring and management of metastatic tumor spreading (Steeg and Theodorescu 2008).
Therapy-induced modification of the tumor environment impacts tumor progression
It is generally assumed that tumor escape and progression toward metastasis during or after therapy is due to the appearance of resistant tumor cells through a combination of therapy-induced genetic instability and subsequent clonal selection of the most fitted cell. This assumption appears particularly relevant to radiotherapy, since the antitumor effect of radiotherapy on tumor cells involve double strand DNA breaks (Gudkov and Komarova 2003). Most cells with damaged DNA undergo p53-mediated apoptosis, mitotic cell death or senescence-like irreversible growth arrest. However, surviving cells might have increased genomic instability and mutations due to mismatched DNA repair and have the potential to rapidly evolve toward a more aggressive phenotype. Recent findings indicate that radiotherapy also rapidly and persistently alters the tissue microenvironment. These modifications affect cell phenotype, tissue metabolism, bidirectional interactions and signaling events between cells (Barcellos-Hoff et al. 2005). While there is evidence indicating that these changes might contribute to the antitumor effects of radiotherapy, for example by creating a fibrotic scar tissue restraining tumor invasion, there are also clinical and experimental observations indicating that irradiated stroma might exert tumor-promoting effects (Barcellos-Hoff et al. 2005). Experimentally, tumors growing within a preirradiated stroma have reduced growth but show more invasive and metastatic phenotype, an effect known as the tumor bed effect (Milas et al. 1987, 1988). Clinically, while adjuvant radiotherapy significantly improves local tumor control (Bartelink et al. 2001), tumor recurrences within a preirradiated field are associated with higher risk of local invasion and metastasis and poor prognosis compared to recurrences outside the irradiated area (O’Brien et al. 1986; Suit 1992; Vicini et al. 2003; Vikram et al. 1984). We have recently reported that tumors growing in a preirradiated bed have reduced angiogenesis and are more hypoxic compared to control tumors (Monnier et al. 2008). Tumor cells derived from these hypoxic tumors retain an invasive behavior in vitro and metastatic capacities in vivo, even after prolonged in vitro culture. A similar phenotype was observed when tumor cells were selected through repeated cycles of hypoxia in vitro, demonstrating that hypoxia was sufficient for the selection of resistant cells. Through gene expression profiling and gain and loss of function experiments, we identified the matricellular protein CYR61 and the integrin adhesion receptor αVβ5, as proteins cooperating in mediating survival under hypoxic condition, invasion and metastasis. Importantly, pharmacological inhibition of αVβ5 with a small molecular αV integrin inhibitor EMD121974 (cilengitide) (Smith 2003) and an anti-αV antibody, suppressed invasion and metastasis induced by CYR61 and attenuated metastasis of tumors growing within a preirradiated field in a cell autonomous manner. Based on these findings, we propose a model in which irradiation impairs the ability of the tumor stroma to elicit an effective angiogenic response, and that tumors growing within this angiogenesis-deficient stroma develop sustained hypoxia, which in turn, acts as a strong factor to select for hypoxia resistant, invasive and metastatic cells. Irradiated stroma itself might also actively contribute to create a microenvironment favoring tumor cell survival, invasion and metastasis. The radiation therapy response of the tumor microenvironment comprises an increased secretion or activation of many antiapoptotic (e.g. insulin-like growth factor-1) and pro-inflammatory cytokines (e.g. GM-CSF), or tumor promoting factors, such as EGF, G-CSF, FGF or TGF-β. Some of the tumor-promoting effects of irradiated stroma may be indirect though the recruitment of BMDC from peripheral blood, further boosting and sustaining the microenvironment itself.
Furthermore, there are emerging data on CSC/CIC and resistance to radiation therapy: this radioresistant sub-population within the tumor mass shows a 50% lower dose-dependent formation of reactive oxygen species in response to radiation and reduced or even absent double-strand DNA breaks. In a breast cancer experimental model, CSC/CIC (as defined by the CD44+/CD24− phenotype) derived from MCF-7 and MDA-MB-231 human cancer lines were isolated and exposed to a single dose of radiation. These cells were more resistant to radiation therapy, had few or no double-strand DNA breaks, and a 50% decrease in dose-dependent formation of reactive oxygen species, compared to the bulk of cancer cell population (Phillips et al. 2006). In another study, CD133+ CSC/CIC isolated from glioma showed increased survival after radiation treatment as compared to the CD133− cells, preserving the tumor-forming ability and aggressive phenotype of the non-irradiated CD133+ cells (Chiou et al. 2008). CSC/CIC appear to survive better and to repair DNA more efficiently compared to the bulk of the tumor cell population, and this in turn could make the tumor more resistant to radiotherapy. Thus, radiotherapy might modify the microenvironment in a way to favor the survival of CSC/CIC, thereby explaining the increased aggressive behavior of tumors recurring within a preirradiated bed. The contribution of tumor microenvironment to promote survival of CSC/CIC could represent a novel therapeutic target to prevent or manage cancer recurrences after radiotherapy.
Conclusions
In recent years the study of tumor microenvironment, its cellular and molecular components, and how they can affect neoplastic progression, has become an emerging topic in cancer research. Factors released by the tumor cells themselves, in particular pro-/antiinflammatory molecules or pro-/antiangiogenic mediators, and factors polarizing immune surveillance toward a tumor-permissive or tumor-rejecting phenotype contribute in creating an environment mostly friendly and sometimes hostile to the tumor. Importantly, events and molecules implicated in this cross talk within the tumor microenvironment have emerged as attractive targets in anticancer therapeutic intervention.
The introduction in the clinic of antiangiogenic drugs has been widely recognized as a major breakthrough in biomedical research and clinical oncology. This represents a proof-of-concept indicating that targeting stromal events can impact tumor progression. More recent works suggest that stromal events may also modulate the response of cancer cells to therapeutic interventions targeting the tumor cells themselves, in particular chemotherapy, and may contribute to the emergence of resistance to therapy. While several mechanisms of resistance to antiangiogenic treatments have been proposed, there is a limited knowledge on how tumor cells and the microenvironment respond, adapt and evolve under this therapeutic approach.
Taken together, these findings emphasize the need to further unravel the complex molecular networks and cross talk between different components of the tumor microenvironment and the tumor cells itself. A deepened knowledge on mechanisms involved in tumor progression and invasiveness toward metastasis could be essential to improve efficacy of current therapeutic interventions with significant clinical impact.
References
Aggarwal BB, Shishodia S, Sandur SK, Pandey MK, Sethi G (2006) Inflammation and cancer: how hot is the link? Biochem Pharmacol 72:1605–1621
Ailles LE, Weissman IL (2007) Cancer stem cells in solid tumors. Curr Opin Biotechnol 18:460–466
Albini A, Sporn MB (2007) The tumour microenvironment as a target for chemoprevention. Nat Rev Cancer 7:139–147
Alitalo K, Carmeliet P (2002) Molecular mechanisms of lymphangiogenesis in health and disease. Cancer Cell 1:219–227
Alitalo K, Tammela T, Petrova TV (2005) Lymphangiogenesis in development and human disease. Nature 438:946–953
Allavena P, Sica A, Garlanda C, Mantovani A (2008a) The Yin-Yang of tumor-associated macrophages in neoplastic progression and immune surveillance. Immunol Rev 222:155–161
Allavena P, Sica A, Solinas G, Porta C, Mantovani A (2008b) The inflammatory micro-environment in tumor progression: the role of tumor-associated macrophages. Crit Rev Oncol Hematol 66:1–9
Avraamides CJ, Garmy-Susini B, Varner JA (2008) Integrins in angiogenesis and lymphangiogenesis. Nat Rev Cancer 8:604–617
Balkwill F (2004) Cancer and the chemokine network. Nat Rev Cancer 4:540–550
Balkwill F, Mantovani A (2001) Inflammation and cancer: back to Virchow? Lancet 357:539–545
Barcellos-Hoff MH, Park C, Wright EG (2005) Radiation and the microenvironment—tumorigenesis and therapy. Nat Rev Cancer 5:867–875
Barnhart BC, Simon MC (2007) Metastasis and stem cell pathways. Cancer Metastasis Rev 26:261–271
Bartelink H, Horiot JC, Poortmans P, Struikmans H, Van den Bogaert W, Barillot I, Fourquet A, Borger J, Jager J, Hoogenraad W, Collette L, Pierart M (2001) Recurrence rates after treatment of breast cancer with standard radiotherapy with or without additional radiation. N Engl J Med 345:1378–1387
Benelli R, Morini M, Brigati C, Noonan DM, Albini A (2003) Angiostatin inhibits extracellular HIV-Tat-induced inflammatory angiogenesis. Int J Oncol 22:87–91
Benelli R, Lorusso G, Albini A, Noonan DM (2006) Cytokines and chemokines as regulators of angiogenesis in health and disease. Curr Pharm Des 12:3101–3115
Bergers G, Benjamin LE (2003) Tumorigenesis and the angiogenic switch. Nat Rev Cancer 3:401–410
Bergers G, Hanahan D (2008) Modes of resistance to anti-angiogenic therapy. Nat Rev Cancer 8:592–603
Bidard FC, Pierga JY, Vincent-Salomon A, Poupon MF (2008) A “class action” against the microenvironment: do cancer cells cooperate in metastasis? Cancer Metastasis Rev 27:5–10
Bierie B, Moses HL (2006a) TGF-beta and cancer. Cytokine Growth Factor Rev 17:29–40
Bierie B, Moses HL (2006b) Tumour microenvironment: TGFbeta: the molecular Jekyll and Hyde of cancer. Nat Rev Cancer 6:506–520
Bissell MJ, Radisky DC, Rizki A, Weaver VM, Petersen OW (2002) The organizing principle: microenvironmental influences in the normal and malignant breast. Differentiation 70:537–546
Blair A, Pamphilon DH (2003) Leukaemic stem cells. Transfus Med 13:363–375
Calabrese C, Poppleton H, Kocak M, Hogg TL, Fuller C, Hamner B, Oh EY, Gaber MW, Finklestein D, Allen M, Frank A, Bayazitov IT, Zakharenko SS, Gajjar A, Davidoff A, Gilbertson RJ (2007) A perivascular niche for brain tumor stem cells. Cancer Cell 11:69–82
Carmeliet P (2000) Mechanisms of angiogenesis and arteriogenesis. Nat Med 6:389–395
Carmeliet P (2005) Angiogenesis in life, disease and medicine. Nature 438:932–936
Carmeliet P, Jain RK (2000) Angiogenesis in cancer and other diseases. Nature 407:249–257
Casanovas O, Hicklin DJ, Bergers G, Hanahan D (2005) Drug resistance by evasion of antiangiogenic targeting of VEGF signaling in late-stage pancreatic islet tumors. Cancer Cell 8:299–309
Chiarugi P, Giannoni E (2008) Anoikis: a necessary death program for anchorage-dependent cells. Biochem Pharmacol. Published online: 25 July 2008. doi:10.1016/j.bcp.2008.07.023
Chiou SH, Kao CL, Chen YW, Chien CS, Hung SC, Lo JF, Chen YJ, Ku HH, Hsu MT, Wong TT (2008) Identification of CD133-positive radioresistant cells in atypical teratoid/rhabdoid tumor. PLoS ONE 3:e2090
Cho RW, Clarke MF (2008) Recent advances in cancer stem cells. Curr Opin Genet Dev 18:48–53
Christofori G (2006) New signals from the invasive front. Nature 441:444–450
Coussens LM, Werb Z (2002) Inflammation and cancer. Nature 420:860–867
Coussens LM, Tinkle CL, Hanahan D, Werb Z (2000) MMP-9 supplied by bone marrow-derived cells contributes to skin carcinogenesis. Cell 103:481–490
Croker AK, Allan AL (2008) Cancer stem cells: implications for the progression and treatment of metastatic disease. J Cell Mol Med 12:374–390
Dancey JE (2005) Inhibitors of the mammalian target of rapamycin. Expert Opin Investig Drugs 14:313–328
Dean M, Fojo T, Bates S (2005) Tumour stem cells and drug resistance. Nat Rev Cancer 5:275–284
DeNardo DG, Coussens LM (2007) Inflammation and breast cancer. Balancing immune response: crosstalk between adaptive and innate immune cells during breast cancer progression. Breast Cancer Res 9:212
DeNardo DG, Johansson M, Coussens LM (2008) Immune cells as mediators of solid tumor metastasis. Cancer Metastasis Rev 27:11–18
Dogne JM, Hanson J, Supuran C, Pratico D (2006) Coxibs and cardiovascular side-effects: from light to shadow. Curr Pharm Des 12:971–975
Drake CG, Jaffee E, Pardoll DM (2006) Mechanisms of immune evasion by tumors. Adv Immunol 90:51–81
Dzierzak E, Speck NA (2008) Of lineage and legacy: the development of mammalian hematopoietic stem cells. Nat Immunol 9:129–136
Ferrara N (2005) The role of VEGF in the regulation of physiological and pathological angiogenesis. EXS 94:209–231
Ferrara N, Kerbel RS (2005) Angiogenesis as a therapeutic target. Nature 438:967–974
Ferrara N, Hillan KJ, Novotny W (2005) Bevacizumab (Avastin), a humanized anti-VEGF monoclonal antibody for cancer therapy. Biochem Biophys Res Commun 333:328–335
Folkman J (1995) Angiogenesis in cancer, vascular, rheumatoid and other disease. Nat Med 1:27–31
Gerber HP, Ferrara N (2003) The role of VEGF in normal and neoplastic hematopoiesis. J Mol Med 81:20–31
Gilbertson RJ, Gutmann DH (2007) Tumorigenesis in the brain: location, location, location. Cancer Res 67:5579–5582
Gilbertson RJ, Rich JN (2007) Making a tumour’s bed: glioblastoma stem cells and the vascular niche. Nat Rev Cancer 7:733–736
Gilboa E (1999) How tumors escape immune destruction and what we can do about it. Cancer Immunol Immunother 48:382–385
Gudkov AV, Komarova EA (2003) The role of p53 in determining sensitivity to radiotherapy. Nat Rev Cancer 3:117–129
Gupta GP, Massague J (2006) Cancer metastasis: building a framework. Cell 127:679–695
Haase VH (2006) The VHL/HIF oxygen-sensing pathway and its relevance to kidney disease. Kidney Int 69:1302–1307
Hagemann T, Balkwill F, Lawrence T (2007) Inflammation and cancer: a double-edged sword. Cancer Cell 12:300–301
Hambardzumyan D, Becher OJ, Holland EC (2008) Cancer stem cells and survival pathways. Cell Cycle 7:1371–1378
Hanahan D, Folkman J (1996) Patterns and emerging mechanisms of the angiogenic switch during tumorigenesis. Cell 86:353–364
Hanahan D, Weinberg RA (2000) The hallmarks of cancer. Cell 100:57–70
Harmey JH, Bucana CD, Lu W, Byrne AM, McDonnell S, Lynch C, Bouchier-Hayes D, Dong Z (2002) Lipopolysaccharide-induced metastatic growth is associated with increased angiogenesis, vascular permeability and tumor cell invasion. Int J Cancer 101:415–422
Hiratsuka S, Watanabe A, Sakurai Y, Akashi-Takamura S, Ishibashi S, Miyake K, Shibuya M, Akira S, Aburatani H, Maru Y (2008) The S100A8-serum amyloid A3-TLR4 paracrine cascade establishes a pre-metastatic phase. Nat Cell Biol. Published online: 28 September 2008. doi:10.1038/ncb1794
Jain RK (2003) Molecular regulation of vessel maturation. Nat Med 9:685–693
Jain RK, Duda DG, Clark JW, Loeffler JS (2006) Lessons from phase III clinical trials on anti-VEGF therapy for cancer. Nat Clin Pract Oncol 3:24–40
Ji H, Houghton AM, Mariani TJ, Perera S, Kim CB, Padera R, Tonon G, McNamara K, Marconcini LA, Hezel A, El-Bardeesy N, Bronson RT, Sugarbaker D, Maser RS, Shapiro SD, Wong KK (2006) K-ras activation generates an inflammatory response in lung tumors. Oncogene 25:2105–2112
Johansson M, Denardo DG, Coussens LM (2008) Polarized immune responses differentially regulate cancer development. Immunol Rev 222:145–154
Kalluri R, Zeisberg M (2006) Fibroblasts in cancer. Nat Rev Cancer 6:392–401
Kaplan RN, Riba RD, Zacharoulis S, Bramley AH, Vincent L, Costa C, MacDonald DD, Jin DK, Shido K, Kerns SA, Zhu Z, Hicklin D, Wu Y, Port JL, Altorki N, Port ER, Ruggero D, Shmelkov SV, Jensen KK, Rafii S, Lyden D (2005) VEGFR1-positive haematopoietic bone marrow progenitors initiate the pre-metastatic niche. Nature 438:820–827
Kaplan RN, Psaila B, Lyden D (2006a) Bone marrow cells in the ‘pre-metastatic niche’: within bone and beyond. Cancer Metastasis Rev 25:521–529
Kaplan RN, Rafii S, Lyden D (2006b) Preparing the “soil”: the premetastatic niche. Cancer Res 66:11089–11093
Karamysheva AF (2008) Mechanisms of angiogenesis. Biochemistry (Mosc) 73:751–762
Karin M (2006) Nuclear factor-kappaB in cancer development and progression. Nature 441:431–436
Karin M, Lawrence T, Nizet V (2006) Innate immunity gone awry: linking microbial infections to chronic inflammation and cancer. Cell 124:823–835
Keith B, Simon MC (2007) Hypoxia-inducible factors, stem cells, and cancer. Cell 129:465–472
Kerbel RS (2008) Tumor angiogenesis. N Engl J Med 358:2039–2049
Kerbel R, Folkman J (2002) Clinical translation of angiogenesis inhibitors. Nat Rev Cancer 2:727–739
Kopfstein L, Christofori G (2006) Metastasis: cell-autonomous mechanisms versus contributions by the tumor microenvironment. Cell Mol Life Sci 63:449–468
Kuilman T, Michaloglou C, Vredeveld LC, Douma S, van Doorn R, Desmet CJ, Aarden LA, Mooi WJ, Peeper DS (2008) Oncogene-induced senescence relayed by an interleukin-dependent inflammatory network. Cell 133:1019–1031
Lee JM, Dedhar S, Kalluri R, Thompson EW (2006) The epithelial–mesenchymal transition: new insights in signaling, development, and disease. J Cell Biol 172:973–981
Li F, Tiede B, Massague J, Kang Y (2007) Beyond tumorigenesis: cancer stem cells in metastasis. Cell Res 17:3–14
Lin EY, Pollard JW (2004) Role of infiltrated leucocytes in tumour growth and spread. Br J Cancer 90:2053–2058
Lin EY, Nguyen AV, Russell RG, Pollard JW (2001) Colony-stimulating factor 1 promotes progression of mammary tumors to malignancy. J Exp Med 193:727–740
Mantovani A, Schioppa T, Porta C, Allavena P, Sica A (2006) Role of tumor-associated macrophages in tumor progression and invasion. Cancer Metastasis Rev 25:315–322
Mantovani A, Allavena P, Sica A, Balkwill F (2008a) Cancer-related inflammation. Nature 454:436–444
Mantovani A, Romero P, Palucka AK, Marincola FM (2008b) Tumour immunity: effector response to tumour and role of the microenvironment. Lancet 371:771–783
McDonald DM, Choyke PL (2003) Imaging of angiogenesis: from microscope to clinic. Nat Med 9:713–725
Milas L, Hunter N, Peters LJ (1987) The tumor bed effect: dependence of tumor take, growth rate, and metastasis on the time interval between irradiation and tumor cell transplantation. Int J Radiat Oncol Biol Phys 13:379–383
Milas L, Hirata H, Hunter N, Peters LJ (1988) Effect of radiation-induced injury of tumor bed stroma on metastatic spread of murine sarcomas and carcinomas. Cancer Res 48:2116–2120
Mittelman A, Lucchese A, Sinha AA, Kanduc D (2002) Monoclonal and polyclonal humoral immune response to EC HER-2/NEU peptides with low similarity to the host’s proteome. Int J Cancer 98:741–747
Mocellin S, Marincola FM, Young HA (2005) Interleukin-10 and the immune response against cancer: a counterpoint. J Leukoc Biol 78:1043–1051
Monnier Y, Farmer P, Bieler G, Imaizumi N, Sengstag T, Alghisi GC, Stehle JC, Ciarloni L, Andrejevic-Blant S, Moeckli R, Mirimanoff RO, Goodman SL, Delorenzi M, Rüegg C (2008) CYR61 and alphaVbeta5 integrin cooperate to promote invasion and metastasis of tumors growing in preirradiated stroma. Cancer Res 68:7323–7331
Morrison SJ, Spradling AC (2008) Stem cells and niches: mechanisms that promote stem cell maintenance throughout life. Cell 132:598–611
Motzer RJ, Michaelson MD, Redman BG, Hudes GR, Wilding G, Figlin RA, Ginsberg MS, Kim ST, Baum CM, DePrimo SE, Li JZ, Bello CL, Theuer CP, George DJ, Rini BI (2006) Activity of SU11248, a multitargeted inhibitor of vascular endothelial growth factor receptor and platelet-derived growth factor receptor, in patients with metastatic renal cell carcinoma. J Clin Oncol 24:16–24
Naugler WE, Karin M (2008) NF-kappaB and cancer-identifying targets and mechanisms. Curr Opin Genet Dev 18:19–26
Neufeld G, Kessler O (2008) The semaphorins: versatile regulators of tumour progression and tumour angiogenesis. Nat Rev Cancer 8:632–645
Nguyen DX, Massague J (2007) Genetic determinants of cancer metastasis. Nat Rev Genet 8:341–352
Noonan DM, De Lerma Barbaro A, Vannini N, Mortara L, Albini A (2008) Inflammation, inflammatory cells and angiogenesis: decisions and indecisions. Cancer Metastasis Rev 27:31–40
O’Brien CJ, Smith JW, Soong SJ, Urist MM, Maddox WA (1986) Neck dissection with and without radiotherapy: prognostic factors, patterns of recurrence, and survival. Am J Surg 152:456–463
Orimo A, Weinberg RA (2006) Stromal fibroblasts in cancer: a novel tumor-promoting cell type. Cell Cycle 5:1597–1601
Orimo A, Gupta PB, Sgroi DC, Arenzana-Seisdedos F, Delaunay T, Naeem R, Carey VJ, Richardson AL, Weinberg RA (2005) Stromal fibroblasts present in invasive human breast carcinomas promote tumor growth and angiogenesis through elevated SDF-1/CXCL12 secretion. Cell 121:335–348
Ostrand-Rosenberg S (2008) Immune surveillance: a balance between protumor and antitumor immunity. Curr Opin Genet Dev 18:11–18
Petit I, Jin D, Rafii S (2007) The SDF-1-CXCR4 signaling pathway: a molecular hub modulating neo-angiogenesis. Trends Immunol 28:299–307
Phillips TM, McBride WH, Pajonk F (2006) The response of CD24(-/low)/CD44+ breast cancer-initiating cells to radiation. J Natl Cancer Inst 98:1777–1785
Piancatelli D, Romano P, Sebastiani P, Adorno D, Casciani CU (1999) Local expression of cytokines in human colorectal carcinoma: evidence of specific interleukin-6 gene expression. J Immunother 22:25–32
Pollard JW (2004) Tumour-educated macrophages promote tumour progression and metastasis. Nat Rev Cancer 4:71–78
Prescott SM, Fitzpatrick FA (2000) Cyclooxygenase-2 and carcinogenesis. Biochim Biophys Acta 1470:M69–M78
Pugh CW, Ratcliffe PJ (2003) Regulation of angiogenesis by hypoxia: role of the HIF system. Nat Med 9:677–684
Punnonen R, Teisala K, Kuoppala T, Bennett B, Punnonen J (1998) Cytokine production profiles in the peritoneal fluids of patients with malignant or benign gynecologic tumors. Cancer 83:788–796
Reddig PJ, Juliano RL (2005) Clinging to life: cell to matrix adhesion and cell survival. Cancer Metastasis Rev 24:425–439
Ribatti D, Mangialardi G, Vacca A (2006) Stephen Paget and the ‘seed and soil’ theory of metastatic dissemination. Clin Exp Med 6:145–149
Rini BI (2006) Sorafenib. Expert Opin Pharmacother 7:453–461
Robinson SC, Coussens LM (2005) Soluble mediators of inflammation during tumor development. Adv Cancer Res 93:159–187
Rollins BJ (2006) Inflammatory chemokines in cancer growth and progression. Eur J Cancer 42:760–767
Rossi DJ, Jamieson CH, Weissman IL (2008) Stems cells and the pathways to aging and cancer. Cell 132:681–696
Rüegg C (2006) Leukocytes, inflammation, and angiogenesis in cancer: fatal attractions. J Leukoc Biol 80:682–684
Rüegg C, Mariotti A (2003) Vascular integrins: pleiotropic adhesion and signaling molecules in vascular homeostasis and angiogenesis. Cell Mol Life Sci 60:1135–1157
Semenza GL (2001) HIF-1 and mechanisms of hypoxia sensing. Curr Opin Cell Biol 13:167–171
Semenza GL (2002) HIF-1 and tumor progression: pathophysiology and therapeutics. Trends Mol Med 8:S62–S67
Semenza GL (2007) Vasculogenesis, angiogenesis, and arteriogenesis: mechanisms of blood vessel formation and remodeling. J Cell Biochem 102:840–847
Sessa C, Guibal A, Del Conte G, Ruegg C (2008) Biomarkers of angiogenesis for the development of antiangiogenic therapies in oncology: tools or decorations? Nat Clin Pract Oncol 5:378–391
Shibuya M (2001) Structure and dual function of vascular endothelial growth factor receptor-1 (Flt-1). Int J Biochem Cell Biol 33:409–420
Shojaei F, Zhong C, Wu X, Yu L, Ferrara N (2008) Role of myeloid cells in tumor angiogenesis and growth. Trends Cell Biol 18:372–378
Smith JW (2003) Cilengitide Merck. Curr Opin Investig Drugs 4:741–745
Sneddon JB, Werb Z (2007) Location, location, location: the cancer stem cell niche. Cell Stem Cell 1:607–611
Steeg PS (2006) Tumor metastasis: mechanistic insights and clinical challenges. Nat Med 12:895–904
Steeg PS (2007) Cancer: micromanagement of metastasis. Nature 449:671–673
Steeg PS, Theodorescu D (2008) Metastasis: a therapeutic target for cancer. Nat Clin Pract Oncol 5:206–219
Strom TB, Anderson PL, Rubin-Kelley VE, Williams DP, Kiyokawa T, Murphy JR (1990) Immunotoxins and cytokine toxin fusion proteins. Semin Immunol 2:467–479
Suit HD (1992) Local control and patient survival. Int J Radiat Oncol Biol Phys 23:653–660
Talmadge JE (2007) Clonal selection of metastasis within the life history of a tumor. Cancer Res 67:11471–11475
Tan BT, Park CY, Ailles LE, Weissman IL (2006) The cancer stem cell hypothesis: a work in progress. Lab Invest 86:1203–1207
Taylor DD, Gercel-Taylor C (1998) Tumor-reactive immunoglobulins in ovarian cancer: diagnostic and therapeutic significance? Oncol Rep 5:1519–1524
Teicher BA (2001) Malignant cells, directors of the malignant process: role of transforming growth factor-beta. Cancer Metastasis Rev 20:133–143
Tlsty TD, Coussens LM (2006) Tumor stroma and regulation of cancer development. Annu Rev Pathol 1:119–150
van der Woude CJ, Kleibeuker JH, Jansen PL, Moshage H (2004) Chronic inflammation, apoptosis and (pre-)malignant lesions in the gastro-intestinal tract. Apoptosis 9:123–130
Veeravagu A, Bababeygy SR, Kalani MY, Hou LC, Tse V (2008) The cancer stem cell–vascular niche complex in brain tumor formation. Stem Cells Dev 17:859–867
Verdegaal EM, Huinink DB, Hoogstraten C, Marijnissen AK, Gorsira MB, Claas FH, Osanto S (1999) Isolation of broadly reactive, tumor-specific, HLA Class-I restricted CTL from blood lymphocytes of a breast cancer patient. Hum Immunol 60:1195–1206
Vicini FA, Kestin L, Huang R, Martinez A (2003) Does local recurrence affect the rate of distant metastases and survival in patients with early-stage breast carcinoma treated with breast-conserving therapy? Cancer 97:910–919
Vikram B, Strong EW, Shah JP, Spiro R (1984) Failure at distant sites following multimodality treatment for advanced head and neck cancer. Head Neck Surg 6:730–733
Visvader JE, Lindeman GJ (2008) Cancer stem cells in solid tumours: accumulating evidence and unresolved questions. Nat Rev Cancer 8:755–768
Weinberg RA (2008) Coevolution in the tumor microenvironment. Nat Genet 40:494–495
Wicha MS (2006) Cancer stem cells and metastasis: lethal seeds. Clin Cancer Res 12:5606–5607
Woodward WA, Sulman EP (2008) Cancer stem cells: markers or biomarkers? Cancer Metastasis Rev 27:459–470
Xu XC (2002) COX-2 inhibitors in cancer treatment and prevention, a recent development. Anticancer Drugs 13:127–137
Yu P, Fu YX (2006) Tumor-infiltrating T lymphocytes: friends or foes? Lab Invest 86:231–245
Acknowledgments
Research in our laboratory is supported by grants from the European Union 7th Framework programme (FP7) on Tumor metastasis integrated concept (TuMIC), Swiss National Science Foundation (FNS), the Cooperative Cancer Research Program of Oncosuisse, the National Center for Competence in Research (NCCR) Molecular Oncology, a research instrument of the FNS, Oncosuisse and the Medic foundation. We are grateful to F. Kuonen and N. Vannini for critical reading of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License ( https://creativecommons.org/licenses/by-nc/2.0 ), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Lorusso, G., Rüegg, C. The tumor microenvironment and its contribution to tumor evolution toward metastasis. Histochem Cell Biol 130, 1091–1103 (2008). https://doi.org/10.1007/s00418-008-0530-8
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00418-008-0530-8