
Abstract
The mediastinum is an anatomically defined space in which organs and major blood vessels reside with surrounding soft tissue elements. The thymus is an important organ in the mediastinum, and many of the masses encountered in the mediastinum are related to this organ. Most neoplasms diagnosed in the mediastinum are epithelial tumours (thymomas and thymic carcinomas), lymphomas or germ cell tumours. In contrast, soft tissue tumours of the mediastinum are rare. In 1963, Pachter and Lattes systematically reviewed soft tissue pathology of the mediastinum, covering the hitherto described [2, 226, 227] In this review, based on the 2013 WHO classification of soft tissue tumours and the 2015 WHO classification of tumours of the lung, pleura, thymus and heart, we provide an updated overview of mesenchymal tumours that may be encountered in the mediastinum.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Soft tissue tumours arising in the mediastinum are rare. Their estimated incidence is between 2 and 6 % of mediastinal neoplasms [1–3]. However, this often quoted estimate is based on historical series with relatively small numbers of cases. In addition, previous reviews of mediastinal soft tissue tumours often predate novel typing strategies and current classification schemes.
Most soft tissue tumours described elsewhere in the body have been reported to occur in the mediastinum, of course with the exception of strictly site- or organ-specific neoplasms such as GIST. Because of their rarity, most mesenchymal tumours in the mediastinum have been reported as case reports or small series. Mediastinal sarcomas may either arise de novo or rarely as “somatic-type” malignancy in a mediastinal germ cell tumour (GCT). Development of a sarcomatous component has been reported to occur more frequently in mediastinal GCTs than in other sites [4]. The two most common sarcomas developing in mediastinal GCTs are rhabdomyosarcoma and angiosarcoma. In addition, sarcomatous areas as part of a thymic sarcomatoid carcinoma or pseudosarcomatous stroma in a thymoma may sometimes be a diagnostic consideration [5–7]. In this review, we will focus on de novo primary mesenchymal tumours of the mediastinum. Based on the current WHO classification of soft tissue tumours [8], we will systematically review those entities that have been described in the thymus and mediastinum with an emphasis on their site-specific features.
Adipocytic tumours
Lipomatous tumours are common in the mediastinum and may be located in any compartment. All subtypes of liposarcoma have been reported in the mediastinum.
Lipoma
Lipoma has been frequently reported in the mediastinum and comprises between 1 and 9 % of primary thymic masses [9, 10]. Mediastinal lipoma may arise from connective tissues both of the mediastinum and the thymus gland itself.
Thymolipoma
When a circumscribed mediastinal mass lesion is composed of mature fat and has a distinct component of thymic tissue, it is considered a site-specific tumour and is termed thymolipoma [11]. A definite distinction from lipoma that, by definition, is devoid of thymic tissue may not be possible in small biopsies [10, 12–14]. The nature of thymolipoma is unclear. Histogenetic conceptions postulated that (1) thymolipoma is essentially a lipoma (i.e. an adipocytic neoplasm) with incorporation of normal thymic tissue and it is (2) a combined neoplasm of thymic fat and a neoplastic thymic epithelial component, (3) fatty replacement of a thymoma or (4) fatty replacement of hyperplastic thymic tissue (i.e. not strictly a neoplasm) [15].
Thymolipoma (Fig. 1) is mainly seen in young adults in the second to fourth decade but has been described in all ages (range 3–76 years; median 29 years [12, 14, 16, 17]) with several well-documented paediatric cases [18] and with an equal sex ratio. Thymolipoma may reach a large size (up to several kilograms) and is clinically silent in at least one third of cases but may produce symptoms of breathlessness by compression of lung tissue, recurrent infection or pain [14]. Asymptomatic tumours have been known to be present for considerable time, occasionally being confused with cardiomegaly [14, 19–21], although this misperception is unlikely with modern imaging techniques. Thymolipoma may be associated with autoimmune symptoms, such as anaemia, hypogammaglobulinemia, hyperthyroidism [22] and, most frequently, myasthenia gravis (MG). The incidence of MG in thymolipoma varies considerable in series, ranging from 0 to 50 % [12, 14, 15, 17, 22–24], but is less than 5 % in recent larger series [14, 17, 23]. Patients with thymolipoma-associated MG tend to be older, and their tumours tend to be smaller (Fig. 2) [15, 23]. It may well be that MG-associated cases come to medical attention before these tumours reach a large size [22]. Surgical resection is curative, and malignant transformation does not occur.

Thymolipoma. Thymolipoma in a 43 year-old male, discovered incidentally during routine physical work-related examination. Mature fat with thymic tissue with discernable cortical and medullary compartments and minor cystic change (HE stain)

Histologically, thymolipomas are encapsulated tumours composed of mature fat cells and thymic tissue. The latter consists of epithelial cells and immature TdT-positive lymphocytes (“thymocytes”) and may contain Hassall’s corpuscles. The proportion of fatty tissue may vary considerably (in one series from 30 to 80 %) [14, 17]. Rare occurrence of a thymoma and even thymic carcinoma arising in thymolipoma has been reported [25, 26].
A number of unusual variants of thymolipoma have been described, including one with a prominent vascular component, designated thymohemangiolipoma [27]. However, fatty change is a well-known alteration in soft tissue hemangioma and arterio-venous vascular malformations, and the described tumour could therefore also be considered a thymic vascular tumour with lipomatous stroma. Other variants of thymolipoma contained striated skeletal muscle (“myoid”) cells, similar to the rare myoid cells which occur in the normal thymus [28–30]. Two cases of thymolipoma (one in a 9-year-old girl) with areas of fibrocollagenous tissue were designated as “fibrothymolipoma” [31].
Lipomatosis
Although lipomatosis is considered a non-neoplastic increase of normal mature fat, this non-encapsulated mass lesion is mentioned here for differential diagnostic purposes. In the mediastinum, it is usually detected by imaging studies, where it causes widening of the mediastinum. Patients may complain of dyspnea. It is commonly associated with steroid use, Cushing’s disease or obesity. Idiopathic cases are very rare [32–34].
Lipoblastoma
Lipoblastoma (LPB) is a rare adipocytic tumour composed of fat cells in various stages of maturation, essentially restricted to the paediatric age group with 90 % occurring before 3 years of age without sex predilection. Mediastinal LPB is very rare with approximately 30 cases presented in case reports or small case series [35–66]. Mediastinal LPB may grow to a large size and pack a large volume of the thorax, and the precise origin of the tumour may be difficult to determine in these cases. A significant number of mediastinal LPB extend into the neck. In the series of mediastinal LPB, patients ranged from 6 months to 6.5 years of age (median 19.5 months).
Histologically, LPB is characterized by fatty tissue in varying degrees of maturation. Most cases are circumscribed, but rarely, a diffuse growth is seen (diffuse LPB, lipoblastomatosis). Although LPBs are benign tumours, recurrence may occur [35] and the tumours may envelop vital structures or extend within orifices such as the spinal canal, hampering complete removal [43, 51, 58].
Lipoma variants
Angiolipoma, an adipocytic tumour composed of small, often capillary-sized blood vessels, combined with mature fat cells has been described in the mediastinum (Fig. 3). As the proportion of fat and blood vessels may vary considerably from case to case, it is debatable whether some of these cases could also be considered primary vascular proliferations (capillary hemangiomas) with an adipose component [67–71].

Angiolipoma (lipomatous hemangioma). Tumour in the superior mediastinum in a 67-year-old female who had few complaints but had persistent pleural effusion. The mass was excised, and no recurrence was noted after several years of follow-up. a Axial contrast-enhanced CT image revealing a circumscribed mediastinal mass with variable density. The mass was found to have been present for several years on retrospective evaluation of previous examinations. b Mature fat admixed with thin-walled vessels; there was no atypia of endothelial cells
A few cases of mediastinal spindle cell lipoma with typical histological features including “ropey” dense collagen strands and bland spindle cells have been reported [72, 73]. The immunohistochemical profile with CD34 positivity supported the diagnosis in these cases, but there have been no reports documenting the presence of 16q or 13q chromosomal aberrations that are characteristic of peripheral spindle cell and pleomorphic lipomas [69].
Hibernoma is a rare distinctive benign adipocytic neoplasm composed of brown fat with microvesicular multivacuolated lipocytes, usually in a subpleural site [74] or associated with soft tissue of the chest or extending from the neck [75]. Only very few cases of hibernoma have been reported as mediastinal masses [76–79].
Less than ten cases of myelolipoma have been reported in the mediastinum. Most cases of myelolipoma occurred in adults (sixth to eighth decade) with no gender predilection and were often discovered incidentally or at autopsy where death was due to other causes. Most cases were present in the posterior mediastinum, often in a paravertebral location [69, 80–89]. Similar to in the adrenal gland, the most common site of myelolipoma, hematopoietic tissue and mature fat are combined in varying proportions. In patients with hematologic disease, similar masses can occur as either normal extramedullary hemopoiesis or as extra-medullary extension of the hematologic tumour itself and should not be considered myelolipoma [90].
Liposarcoma
Liposarcoma is a malignant adipocytic tumour with various subtypes which impact on behaviour and prognosis. It is, by far, the most common primary malignant mesenchymal tumour of the mediastinum. The age range is wide. Paediatric cases, even occurring in very young infants, have been reported [16, 91–98]. Although an origin from thymic tissue has been shown in a minority of cases (“thymoliposarcoma”) [93, 95, 99], cases have also been documented in the posterior mediastinum [93, 100–102], suggesting a non-thymic origin. Although numerous case reports and several series [16, 93, 95, 101, 103, 104] of mediastinal liposarcoma have been published, most of these predate the current molecular insights of liposarcoma which may aid in classification. In general, mediastinal liposarcomas are large, and tumours up to 7 kg have been reported [105]. Tumours may remain asymptomatic for long periods. Shortness of breath and pain are the most common symptoms. Despite their large size, vena cava syndrome has only rarely been reported [92, 102].
All subtypes of liposarcoma have been reported in the mediastinum. Based on their own series and a comprehensive review of 142 published cases, Boland et al. concluded that the proportions of liposarcoma subtypes in the mediastinum differ from those in other sites [106]. In particular, pleomorphic liposarcoma was considerably more prevalent in the mediastinum. In addition, mediastinal liposarcomas, including the pleomorphic subtype, frequently contained myxoid areas, a finding which has been confirmed in other series [93, 101]. Although the prevalence of mediastinal myxoid liposarcoma was approximately similar to other anatomic locations, a round cell component was not identified in the mediastinal tumours. This was confirmed in other series, in which the presence of a round-cell component was exceptional [93]. The molecular signatures of mediastinal liposarcomas correspond to those of liposarcomas elsewhere, with presence of the t(12;16) FUS-DDIT-3 fusion in myxoid liposarcoma [107] and amplification of mdm-2 in well-differentiated and dedifferentiated liposarcoma [101, 108].
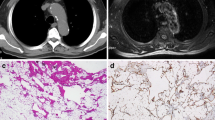
An unusual liposarcoma, termed pleomorphic myxoid liposarcoma (P-MLPS) by Alaggio et al., appears to have a predilection for the mediastinum of young patients [103] (Fig. 4). Five of the 12 cases in their series arose in the mediastinum in patients aged from 13 to 20 years of age. Rare adult cases were described in the series of Boland et al. [106]. In addition to areas similar to conventional low-grade myxoid liposarcoma, foci with increased cellularity, hyperchromatic cells and pleomorphic lipoblasts with increased mitotic activity were observed in areas with loss of the typical capillary vascular pattern and necrosis. These tumours followed an aggressive course, with death occurring within 3 years of diagnosis in three of five patients with available follow-up data. Neither a t(12;16)(q13;p11), the characteristic genetic hallmark of myxoid liposarcoma, nor amplification of mdm-2, a marker for well-differentiated and dedifferentiated liposarcoma, was identified. Other unusual variants of liposarcoma that have been described in the mediastinum contained elements of smooth muscle (lipoleiomyosarcoma) or skeletal muscle [101, 109–111].

Liposarcoma with myxoid change. A posterior mediastinal mass, surrounding the aorta in a 55-year-old male discovered during routine health check (a). The mass was excised (b, c). A recurrence was excised 16 months later; there was no evidence of disease after 1-year follow-up. a Axial noncontrast CT image at the level of the left atrium revealing a smoothly marginated retrocardiac mass with variable attenuation. b Sagittal reformatted CT image showing the retrocardiac mass to extent from the aortic arch to the diaphragm. c Macroscopic image of the transected tumour revealing a fleshy, partially thinly encapsulated mass. d Microscopic image (medium power, HE stain) showing myxocollagenous stroma with scattered atypical mesenchymal cells
The main differential diagnostic considerations of mediastinal liposarcoma are benign lipomatous tumours. Attention to histological features and the use of molecular markers will lead to a correct diagnosis. Dedifferentiated liposarcoma may mimic undifferentiated pleomorphic sarcoma, particularly in biopsy specimens; demonstration of amplification of mdm-2 confirms the diagnosis of liposarcoma. So-called fat-forming solitary fibrous tumour may enter the differential diagnosis of liposarcoma. Mdm-2 amplification is absent in these tumours, and positive staining of STAT-6 on immunohistochemistry may further support a diagnosis of solitary fibrous tumour (SFT) (see below). Liposarcoma as a secondary malignancy developing in a GCT has very rarely been described [112]. Curative surgical resection is the treatment of choice. The mortality of mediastinal liposarcomas ranges from 30 to 50 %.
Fibroblastic/myofibroblastic tumours
Since many of the tumours in the fibroblastic/myofibroblastic category have a predilection for the skin and superficial soft tissues, it is not surprising that only very few of the entities in this category have been reported in the mediastinum. Desmoid tumours (aggressive fibromatosis), SFT and inflammatory myofibroblastic tumour (IMT) are among the more frequently reported types in the mediastinum.
Aggressive fibromatosis /desmoid tumour
Aggressive fibromatosis is a locally invasive (myo)fibroblastic proliferation with bland cytology and without metastatic potential. About 30 cases of primary mediastinal aggressive fibromatosis (AF) have been reported as case reports, many of which were included in reviews by Nakagiri et al. and Bouchikh et al. [113, 114] AF in the mediastinum occurs mainly in younger individuals (age range 3–67 years; median 38 years) [113–119]. Some cases were associated with surgical scars [114]. There are no published data about an association with familial adenomatous polyposis (FAP) or Gardner syndrome. In contrast to non-mediastinal AF, mediastinal AF cases appear to occur slightly more frequently in males [120]. Similar to non-mediastinal AF, mediastinal recurrence is common after surgical removal, which, in this location, is hampered by anatomical constraints. Unresectable AF in the mediastinum may be fatal.
Solitary fibrous tumour
SFT is an uncommon but well-known intrathoracic fibroblastic tumour and often of pleural origin. Since its description, numerous reports of extra-pleural SFT cases have been published with tumours arising in almost all locations in the body [121]. Mediastinal SFTs without a clear connection with the mediastinal pleura have been described in case reports and small series. Hemangiopericytoma, which has also been described in the mediastinum, is now considered identical to SFT.
Mediastinal SFT may arise in any compartment of the mediastinum. A number of mediastinal SFTs had also contact to the epicardium [122], and in such cases, it may be questionable whether these represent true primary mediastinal SFT [123–125]. Thoracic SFTs reach a large size and may extend to the chest wall, protrude in the lung or even invade other structures [124]. SFTs occur over a wide age range but are typically seen in older adults [126–128]. Paediatric cases are very rare [129–131]. There is no sex predilection [122–124, 129–169].
The histomorphology of SFT is characterized by a “patternless” architecture with randomly distributed hypocellular and hypercellular areas, sometimes keloid-like collagen, and thin “stag-horn” branching capillary vessels that may show perivascular hyalinization. Tumour cells are generally spindle shaped with bland cytology and few mitoses. Immunohistochemistry is helpful in the diagnosis of SFT and shows usually strong expression of CD34 [121, 166]. Other markers typically positive in SFT are CD99 and Bcl2 [127]. Variable staining is seen with smooth muscle actin, while cytokeratin positivity is very rare [163], except in variants with epithelioid morphology (see below). Recently, nuclear staining of STAT6 resulting from NAB2-STAT6 gene fusion [170–172] has been shown to be a more specific marker for SFT [173–175], although presence of the NAB2-STAT6 gene fusion has not yet been formally shown for mediastinal SFT.
It has been suggested that mediastinal SFTs more frequently show aggressive behaviour compared to non-mediastinal SFTs [169]. The same criteria predicting malignancy, particularly a high mitotic count (>4 mitoses per 2 mm2), high cellularity, pleomorphism and necrosis apply as outside the mediastinum [127].
So-called fat-forming SFT shows adipocytic differentiation to such an extent that well-differentiated liposarcoma may enter the differential diagnosis [132, 143, 151, 176] Although nuclear STAT6 expression has been shown to be a useful marker for the diagnosis of fat-forming SFT, particularly in CD34 negative cases [177] this has also been demonstrated in a significant proportion of dedifferentiated liposarcomas [108, 178]. However, staining was limited to the non-lipogenic sarcomatous areas. In addition, mdm-2 amplification remains a robust marker for liposarcoma and has not been reported in SFT (Fig. 5).

Fat-forming solitary fibrous tumour. A large tumour in the chest cavity in a 23-year-old female who complained of shortness of breath. The mass was excised but later recurred and resulted in death of the patient. a CT image showing a large tumour that occupies the anterior and posterior mediastinum. b HE stain showing mature fat tissue admixed with ill-defined nodules of spindle cells with elongated nuclei and tapering cytoplasm. c CD34 stain which is diffusely positive in the spindle cells. d STAT-6 immunohistochemistry with diffuse nuclear and cytoplasmic positivity
Other variants of SFT include cases with an epithelioid morphology, either as a focal component in combination with “classical” spindle cells or as tumours that may be exclusively epithelioid [153, 179–185]. This variant may stain for cytokeratin, which may also be seen in the spindle cells. A mediastinal epithelioid SFT was reported by Marchevsky et al., who considered the differential diagnosis of adenomatoid tumour [152]. Epithelioid areas in the highly unusual thymic SFT case reported by Tsubochi et al. showed glandular, neuroepithelial and neuroendocrine morphology in addition to classical SFT features [164]. Immunohistochemistry reflected the diverse histology with STAT6 staining restricted to the classical SFT component.
Mediastinal SFT, in particular malignant cases, may be confused with thymomas with a prominent spindle cell morphology (e.g. type A thymoma), mesothelioma, sarcomatoid carcinoma, synovial sarcoma and malignant peripheral nerve sheath tumour (MPNST). In addition to morphological differences, immunohistochemistry will help to diagnose SFT in most cases. Cytokeratin staining is seen in thymoma, mesothelioma, sarcomatoid carcinoma and synovial sarcoma and is very rare in SFT. In addition, STAT6 is a reliable marker for SFT and has not been described in the other tumours.
Inflammatory myofibroblastic tumour
Inflammatory myofibroblastic tumour (IMT), a tumour composed of (myo)fibroblastic cells and a variably dense non-neoplastic inflammatory component, has been reported in many sites, predominantly in young adults and children [186]. Bona fide mediastinal IMT is rare with less than 20 convincing cases reported in the English literature [186–196]. Similar to IMT in general, which is predominantly a tumour of children and young adults [186], mediastinal IMT appears to arise mostly in young adults (range 13–72; median age 34) with a slight female predominance (M/F = 5:8) [187, 188, 190, 192–196].
Histologically, IMT is an infiltrative tumour composed of (myo)fibroblastic cells admixed with an inflammatory infiltrate of lymphocytes, plasma cells and eosinophils. Three histological patterns (granulation tissue-like/compact fascicular spindle cell pattern with marked inflammation/low cellularity scar-like pattern) that can all occur within a single tumour have been described but do not portend a specific biological behaviour [186].
Multifocality is not uncommon in IMT. Cytoplasmic expression of ALK protein is detectable in about half of cases and correlates well with presence of ALK gene rearrangements. ALK aberrations have been reported in a single mediastinal case [192].
The biological behaviour of IMT is variable and recurrences are common, but distant metastasis is rare.
Similar to non-mediastinal sites, treatment rests mainly on complete local excision. Patients with ALK-rearranged IMT are potential candidates for treatment with specific inhibitors, although this has so far not been described for mediastinal cases.
Other fibroblastic/myofibroblastic tumours
Calcifying fibrous pseudotumour, a very rare soft tissue tumour with proposed but unconfirmed relationship to IMT, has been reported in the mediastinum [197–200]. The tumours all occurred in the mediastinum of adults. No evidence of disease was recorded during 11–49-month follow-up after surgery in three patients.
Reports of fibrosarcoma arising in the mediastinum predate the current concept that adult fibrosarcoma is a very rare tumour. The most recent report of an adult mediastinal fibrosarcoma dates from 1995 [201]. While in older reports, fibrosarcoma was one of the more common reported soft tissue tumours, it is likely that these would now be diagnosed as dedifferentiated liposarcoma, MPNST or synovial sarcoma. Infantile fibrosarcoma has not been reported in the mediastinum.
Three cases of low-grade fibromyxoid sarcoma have been reported in the mediastinum, among which one case was attached to the epicardium [202–204]. All cases occurred in middle-aged patients, and one patient had a late recurrence 9 years following surgery, which was resected; metastases were not recorded.
A single case of infantile myofibromatosis with multifocal masses in the lung and mediastinal soft tissue in a 4-week-old male infant has been reported. Five years after surgical resection and pneumonectomy, no disease activity was recorded [205].
A single case of mediastinal giant cell angiofibroma was reported in a 62-year-old female [206]. However, giant cell angiofibroma, which is most commonly described in the orbit, is now considered a variant of SFT [207].
Fibrohistiocytic tumours
Fibrohistiocytic tumours are mainly encountered in superficial soft tissues or joints. Two cases of benign fibrous histiocytoma in the mediastinum have been reported [208, 209]. Despite its terminology, angiomatoid fibrous histiocytoma is commonly included among tumours of uncertain origin and is not considered a variant of benign fibrous histiocytoma. Two cases of angiomatoid fibrous histiocytoma have been reported in the mediastinum [210, 211]. Both tumours were resected without evidence of recurrence or metastases.
Giant cell tumour of soft tissue (GCT) is most commonly observed in superficial soft tissue of the extremities, trunk and head and neck. Five primary mediastinal cases have been reported, occurring in individuals aged 18–53 years (in three female and two male patients) [212–214]. The tumours were almost all located in the posterior mediastinum and ranged from 2.5 to 15 cm, filling the hemithorax. No recurrences were noted, but the follow-up period was not stated in all published cases.
Malignant fibrohistiocytic tumours, previously referred to as malignant fibrous histiocytoma (MFH), are now classified as undifferentiated high-grade pleomorphic sarcomas and are not considered of true histiocytic lineage (see part 2 of this review). So-called MFH cases have been recorded in the mediastinum with generally poor prognosis.
Secondary mediastinal mesenchymal tumours in germ cell tumours
When a mediastinal sarcoma is diagnosed, the possibility must be considered that sarcomatous tissue remains as the sole component in a long-standing primary mediastinal GCT, particularly in younger male patients [215]. Likewise, a mediastinal sarcoma could be the metastatic vestige of a burnt-out primary gonadal GCT. Demonstration of a germ cell (GC) origin of a mediastinal sarcoma may be accomplished by thorough sampling to reveal GCT remnants. Serological investigations (β-HCG, AFP) may reveal a cryptic GCT. The presence of isochromosome 12p (i(12p)) is a characteristic abnormality of type II GCTs [216, 217] which persists in the somatic elements when the typical GCT components are no longer recognizable [218–220]. Thus, identifying an i12p in the mesenchymal tissue could confirm the origin of a mediastinal sarcoma from a GCT. Development of a sarcomatous component in a GCT occurs more frequently in mediastinal GCTs than in other sites [4]. It has been suggested that this results from the fact that GCTs in this location grow undetected over longer time periods, are larger and may acquire malignant somatic tumour subclones more frequently [215]. The two most common sarcomas developing in mediastinal GCTs are rhabdomyosarcoma and angiosarcoma [4, 215, 221, 222]. Less commonly reported sarcomas arising in GCTs are leiomyosarcoma, MPNST and, rarely, liposarcoma [4, 215, 222, 223]. Although most secondary sarcomas in GCTs develop in the anterior mediastinum, rare cases arise in the posterior mediastinum. Similarly and in analogy to somatic (teratomatous) elements becoming the dominant tissue in GCTs treated by chemotherapy, sarcomatous tissue may be the only residual tissue left after chemotherapeutically treated GCTs [224, 225]. Treatment should be tailored to the somatic component rather than to its germ cell origin [223]. Nevertheless, similar to non-germ-cell-related sarcoma, the prognosis of a mediastinal sarcoma arising in a GCT is poor.
References
Dubashi B, Cyriac S, Tenali SG (2009) Clinicopathological analysis and outcome of primary mediastinal malignancies—a report of 91 cases from a single institute. Annal Thor Med 4:140–142. doi:10.4103/1817-1737.53354
Pachter MR, Lattes R (1963) Mesenchymal tumors of the mediastinum. I. Tumors of fibrous tissue, adipose tissue, smooth muscle, and striated muscle. Cancer 16:74–94
Wychulis AR, Payne WS, Clagett OT, Woolner LB (1971) Surgical treatment of mediastinal tumors: a 40 year experience. J Thorac Cardiovasc Surg 62:379–392
Malagon HD, Valdez AM, Moran CA, Suster S (2007) Germ cell tumors with sarcomatous components: a clinicopathologic and immunohistochemical study of 46 cases. Am J Surg Pathol 31:1356–1362. doi:10.1097/PAS.0b013e318033c7c4
Eimoto T, Kitaoka M, Ogawa H, Niwa H, Murase T, Tateyama H, Inagaki H, Soji T, Wang HJ (2002) Thymic sarcomatoid carcinoma with skeletal muscle differentiation: report of two cases, one with cytogenetic analysis. Histopathology 40:46–57
Okudela K, Nakamura N, Sano J, Ito T, Kitamura H (2001) Thymic carcinosarcoma consisting of squamous cell carcinomatous and embryonal rhabdomyosarcomatous components. Report of a case and review of the literature. Pathol Res Pract 197:205–210
Suster S, Moran CA, Chan JK (1997) Thymoma with pseudosarcomatous stroma: report of an unusual histologic variant of thymic epithelial neoplasm that may simulate carcinosarcoma. Am J Surg Pathol 21:1316–1323
Fletcher C, Bridge J, Hogendoorn P, Mertens F (2013) WHO classification of tumours of soft tissue and bone. IARC Press, Lyon
Shub C, Parkin TW, Lie JT (1979) An unusual mediastinal lipoma simulating cardiomegaly. Mayo Clin Proc 54:60–62
Teplick JG, Nedwich A, Haskin ME (1973) Roentgenographic features of thymolipoma. Am J Roentgenol Radium Ther Nucl Med 117:873–880
Hall GF (1949) A case of thymolipoma with observations on a possible relationship to intrathoracic lipomata. Br J Surg 36:321–324
Damadoglu E, Salturk C, Takir HB, Ertugrul M, Yilmaz A, Atasalihi A (2007) Mediastinal thymolipoma: an analysis of 10 cases. Respirology 12:924–927. doi:10.1111/j.1440-1843.2007.01150.x
Gamanagatti S, Sharma R, Hatimota P, Guleria R, Arvind S (2005) Giant thymolipoma. AJR Am J Roentgenol 185:283–284
Rosado-de-Christenson ML, Pugatch RD, Moran CA, Galobardes J (1994) Thymolipoma: analysis of 27 cases. Radiology 193:121–126
Pan CH, Chiang CY, Chen SS (1988) Thymolipoma in patients with myasthenia gravis: report of two cases and review. Acta Neurol Scand 78:16–21
Cicciarelli FE, Soule EH, McGoon DC (1964) Lipoma and liposarcoma of the mediastinum: a report of 14 tumors including one lipoma of the thymus. J Thorac Cardiovasc Surg 47:411–429
Moran CA, Rosado-de-Christenson M, Suster S (1995) Thymolipoma: clinicopathologic review of 33 cases. Mod Pathol 8:741–744
Kitano Y, Yokomori K, Ohkura M, Kataoka T, Narita M, Takemura T (1993) Giant thymolipoma in a child. J Pediatr Surg 28:1622–1625
Almog C, Weissberg D, Herczeg E, M P (1977) Thymolipoma simulating cardiomegaly: a clinicopathological rarity. Thorax 32:116–120
Casullo J, Palayew MJ, Lisbona A (1992) General case of the day. Thymolip Radiograph 12:1250–1254. doi:10.1148/radiographics.12.6.1439025
Rieker RJ, Schirmacher P, Schnabel PA, Moser K, Hoffmann H, Dienemann H, Pfannschmidt J (2010) Thymolipoma. A report of nine cases, with emphasis on its association with myasthenia gravis. Surg Today 40:132–136. doi:10.1007/s00595-009-4042-5
Le Marc’hadour F, Pinel N, Pasquier B, Dieny A, Stoebner P, Couderc P (1991) Thymolipoma in association with myasthenia gravis. Am J Surg Pathol 15:802–809
Huang CS, Li WY, Lee PC, Kao KP, Chou TY, Wu MH, Hsu HS, Wu YC, Hsu WH, Huang BS (2014) Analysis of outcomes following surgical treatment of thymolipomatous myasthenia gravis: comparison with thymomatous and non-thymomatous myasthenia gravis. Interact Cardiovasc Thorac Surg 18:475–481. doi:10.1093/icvts/ivt531
Otto HF, Loning T, Lachenmayer L, Janzen RW, Gurtler KF, Fischer K (1982) Thymolipoma in association with myasthenia gravis. Cancer 50:1623–1628
Argani P, de Chiocca IC, Rosai J (1998) Thymoma arising with a thymolipoma. Histopathology 32:573–574
Haddad H, Joudeh A, El-Taani H, Mansour A, Morcos B, Fayoumi S, Sughayer M (2009) Thymoma and thymic carcinoma arising in a thymolipoma: report of a unique case. Int J Surg Pathol 17:55–59. doi:10.1177/1066896908315822
Ogino S, Franks TJ, Deubner H, Koss MN (2000) Thymohemangiolipoma, a rare histologic variant of thymolipoma: a case report and review of the literature. Ann Diagn Pathol 4:236–239. doi:10.1053/adpa.2000.8131
Gannon BR, Dexter DF, Petsikas D, Isotalo PA (2007) Mediastinal thymolipoma: a rare occurrence with striated myoid cells. Tumori 93:198–200
Iseki M, Tsuda N, Kishikawa M, Shimada O, Hayashi T, Kawahara K, Tomita M (1990) Thymolipoma with striated myoid cells. Histological, immunohistochemical, and ultrastructural study. Am J Surg Pathol 14:395–398
Pai RK, Irvine R (2004) Pathologic quiz case: giant mediastinal mass in a 69-year-old man. Thymolipoma. Arch Pathol Lab Med 128:e159–e160. doi:10.1043/1543-2165(2004)128<e159:PQCGMM>2.0.CO;2
Moran CA, Zeren H, Koss MN (1994) Thymofibrolipoma. A histologic variant of thymolipoma. Arch Pathol Lab Med 118:281–282
Dhawan SS, Khouzam R (2007) Atypical mediastinal lipomatosis. Heart Lung J Crit Care 36:223–225. doi:10.1016/j.hrtlng.2006.08.010
Homer MJ, Wechsler RJ, Carter BL (1978) Mediastinal lipomatosis CT confirmation of a normal variant. Radiology 128:657–661. doi:10.1148/128.3.657
Nguyen KQ, Hoeffel C, Le LH, Phan HT (1998) Mediastinal lipomatosis. South Med J 91:1169–1172
Amra NK, Amr SS (2009) Mediastinal lipoblastomatosis: report of a case with complex karyotype and review of the literature. Pediat Develop Pathol Offic J Soc Pediat Pathol Paediat Pathol Soc 12:469–474. doi:10.2350/08-09-0525.1
Benato C, Falezza G, Lonardoni A, Magnanelli G, Ricci M, Gilioli E, Calabro F (2011) Acute respiratory distress caused by a giant mediastinal lipoblastoma in a 16-month-old boy. Ann Thorac Surg 92:e119–e120. doi:10.1016/j.athoracsur.2011.06.019
Broeders A, Smet MH, Breysem L, Marchal G (2000) Lipoblastoma: a rare mediastinal tumour in a child. Pediatr Radiol 30:580. doi:10.1007/s002470000247
Cacciaguerra S, Lebet M, Di Cataldo A, Milone P, Lanzafame S, Petrillo G, Di Benedetto A (1995) An unusual intrathoracic tumor: giant lipoblastoma. Eur J Pediat Surg Offic J Aust Assoc Pediat Surg… [et al.]=Zeitschrift fur Kinderchirurgie 5:40–42. doi: 10.1055/s-2008-1066161
Canonico F, Patassini M, Malaspina C (2011) Nonmyxoid mediastinal lipoblastoma in a 2-year-old girl: case report with US, CT, and MRI findings. J Ultrasound 14:14–17. doi:10.1016/j.jus.2011.01.002
Ching AS, Lee SF, Chan YL (2002) Diagnosing paediatric mediastinal lipoblastoma using ultrasound-guided percutaneous needle biopsy: review and report. Clin Imaging 26:23–26
Chung EB, Enzinger FM (1973) Benign lipoblastomatosis. An analysis of 35 cases. Cancer 32:482–492
Dishop MK, O’Connor WN, Abraham S, Cottrill (2001) Primary cardiac lipoblastoma. Pediat Develop Pathol Offic J Soc Pediat Pathol Paediat Pathol Soc 4:276–280
Dogan R, Kara M, Firat P, Gedikoglu G (2007) An unusual tumor of the neck and mediastinum: lipoblastomatosis resulting in paraparesis. Eur J Cardiothorac Surg 31:325–327. doi:10.1016/j.ejcts.2006.11.012
Federici S, Cuoghi D, Sciutti R (1992) Benign mediastinal lipoblastoma in a 14-months-old infant. Pediatr Radiol 22:150–151
Geramizadeh B, Javadi F, Foroutan HR (2011) Intrathoracic lipoblastoma in a 15-month-old infant. Rare Tumor 3:e51. doi:10.4081/rt.2011.e51
Guillen-Quesada A, Costa-Clara JM (2007) Mediastinal lipoblastoma with intramedullary extension. Rev Neurol 44:745–746
Hanafiah M, Noryati M, Arni T (2013) Mediastinal lipoblastoma: unexpected finding of a chest infection. BMJ Case Reports. doi:10.1136/bcr-2013-009879
Irgau I, McNicholas KW (1998) Mediastinal lipoblastoma involving the left innominate vein and the left phrenic nerve. J Pediatr Surg 33:1540–1542
Jung SM, Chang PY, Luo CC, Huang CS, Lai JY, Hsueh C (2005) Lipoblastoma/lipoblastomatosis: a clinicopathologic study of 16 cases in Taiwan. Pediatr Surg Int 21:809–812. doi:10.1007/s00383-005-1502-x
Kerkeni Y, Sahnoun L, Ksia A, Hidouri S, Chahed J, Krichen I, Mekki M, Belghith M, Nouri A (2014) Lipoblastoma in childhood: about 10 cases. Afric J Paediat Surg AJPS 11:32–34. doi:10.4103/0189-6725.129210
Ko SF, Shieh CS, Shih TY, Hsiao CC, Ng SH, Lee TY, Wan YL, Chen WJ (1998) Mediastinal lipoblastoma with intraspinal extension: MRI demonstration. Magn Reson Imaging 16:445–448
Kucera A, Snajdauf J, Vyhnanek M, Moravek J, Kodet R, Stejskalova E, Dvorakova M (2008) Lipoblastoma in children: an analysis of 5 cases. Acta Chir Belg 108:580–582
Mahour GH, Bryan BJ, Isaacs H Jr (1988) Lipoblastoma and lipoblastomatosis—a report of six cases. Surgery 104:577
Moaath A, Raed E, Mohammad R, Mohammad S (2009) Lipoblastoma: a rare mediastinal tumor. Ann Thorac Surg 88:1695–1697. doi:10.1016/j.athoracsur.2009.03.033
Moholkar S, Sebire NJ, Roebuck DJ (2006) Radiological-pathological correlation in lipoblastoma and lipoblastomatosis. Pediatr Radiol 36:851–856. doi:10.1007/s00247-006-0175-5
Mutlu M, Yars N, Imamoglu M, Kosucu P, Turgutalp H (2009) Mediastinal lipoblastoma causing diaphragmatic eventration: a case report and review of the literature. J Pediatr Hematol Oncol 31:346–348. doi:10.1097/MPH.0b013e31818e5354
Nordin AB, Fallon SC, Jea A, Kim ES (2013) The use of spinal angiography in the management of posterior mediastinal tumors: case series and review of the literature. J Pediatr Surg 48:1871–1877. doi:10.1016/j.jpedsurg.2013.04.029
Raman Sharma R, Mahapatra AK, Pawar SJ, Sousa J, Musa MM (2002) An unusual posterior mediastinal lipoblastoma with spinal epidural extension presenting as a painful suprascapular swelling: case report and a brief review of the literature. J Clin Neurosci Offic J Neurosurg Soc Austral 9:204–207. doi:10.1054/jocn.2001.0946
Rao KL, Eradi B, Marwaha RK, Trehan A, Saxena AK, Menon P (2003) Dumbbell tumour of the mediastinum. Arch Dis Child 88:117
Salem R, Zohd M, Njim L, Maazoun K, Jellali MA, Zrig A, Mnari W, Harzallah W, Nouri A, Zakhama A, Golli M (2011) Lipoblastoma: a rare lesion in the differential diagnosis of childhood mediastinal tumors. J Pediatr Surg 46:e21–e23. doi:10.1016/j.jpedsurg.2011.01.030
Seidel FG, Magill HL, Burton EM, Boulden TF, Brooks MT, Hanna SL (1990) Cases of the day. Pediatric. Lipoblastom Radiograph 10:728–731. doi:10.1148/radiographics.10.4.2377767
Stringel G, Shandling B, Mancer K, Ein SH (1982) Lipoblastoma in infants and children. J Pediatr Surg 17:277–280
Tabrisky J, Rowe JH, Christine SG, Weinstein ED, Asch M (1974) Benign mediastinal lipoblastomatosis. J Pediatr Surg 9:399–401
Tahbaz O, Bakhshayesh Karam M, Zahirifard S, Kaynama K, Haghighi S (2005) Mediastinal lipoblastoma: report of a case. Iran J Radiol 2 (3,4):177–120
Thakur B, Shan ZC (2006) Giant mediastinal lipoblastoma: a case report and review of the literature. Indian J Surg 68:108–110
Whyte AM, Powell N (1990) Mediastinal lipoblastoma of infancy. Clin Radiol 42:205–206
Choi JY, Goo JM, Chung MJ, Kim HC, Im JG (2000) Angiolipoma of the posterior mediastinum with extension into the spinal canal: a case report. Korea J Radiol Offic J Korea Radiol Soc 1:212–214
Gamez Garcia P, de Pablo Gafas A, Salas Anton C, Santolaya Cohen R, Madrigal Royo L, Varela de Ugarte A (2002) Mediastinal dumbbell angiolipoma. Arch Bronconeumol 38:545–546
Kim K, Koo BC, Davis JT, Franco-Saenz R (1984) Primary myelolipoma of mediastinum. J Comput Tomogr 8:119–123
Kline ME, Patel BU, Agosti SJ (1990) Noninfiltrating angiolipoma of the mediastinum. Radiology 175:737–738. doi:10.1148/radiology.175.3.2343124
Negri G, Regolo P, Gerevini S, Arrigoni G, Zannini P (2000) Mediastinal dumbbell angiolipoma. Ann Thorac Surg 70:957–958
La Mantia E, Franco R, Rocco R, Rocco G (2013) Spindle cell lipoma: a rare tumor of the mediastinum. J Thorac Dis 5:E152–E154. doi:10.3978/j.issn.2072-1439.2013.07.01
Oaks J, Margolis DJ (2007) Spindle cell lipoma of the mediastinum: a differential consideration for liposarcoma. J Thorac Imaging 22:355–357. doi:10.1097/01.rti.0000179474.92618.79
Hertoghs M, Van Schil P, Rutsaert R, Van Marck E, Vallaeys J (2009) Intrathoracic hibernoma: report of two cases. Lung Cancer 64:367–370. doi:10.1016/j.lungcan.2008.11.003
Santambrogio L, Cioffi U, De Simone M, Nosotti M, Pavoni G, Caputo V, Bonavina L (1997) Cervicomediastinal hibernoma. Ann Thorac Surg 64:1160–1162
Ahn C, Harvey JC (1990) Mediastinal hibernoma, a rare tumor. Ann Thorac Surg 50:828–830
Arauzo Alvarez E, Gomez Roman J, Fernandez Martinez De Septien C, Calvo Castillo I (2014) Mediastinal hibernoma: singular appearance on computed tomography. Radiologia 56:e50–e53. doi:10.1016/j.rx.2012.03.005
Baldi A, Santini M, Mellone P, Esposito V, Groeger AM, Caputi M, Baldi F (2004) Mediastinal hibernoma: a case report. J Clin Pathol 57:993–994. doi:10.1136/jcp.2004.017897
Udwadia ZF, Kumar N, Bhaduri AS (1999) Mediastinal hibernoma. Eur J Cardiothorac Surg 15:533–535
Ema T, Kawano R (2013) Myelolipoma of the posterior mediastinum: report of a case. Gen Thorac Cardiovasc Surg. doi:10.1007/s11748-013-0230-8
Foster JB (1958) Primary thoracic myelolipoma: case report. AMA Arch Pathol 65:295–297
Franiel T, Fleischer B, Raab BW, Fuzesi (2004) Bilateral thoracic extraadrenal myelolipoma. Eur J Cardiothorac Surg 26:1220–1222. doi:10.1016/j.ejcts.2004.08.024
Gao B, Sugimura H, Sugimura S, Hattori Y, Iriyama T, Kano H (2002) Mediastinal myelolipoma. Asian Cardiovasc Thorac Ann 10:189–190
Koizumi J, Harada H, Yamamoto N, Shiiku C, Ogasa T, Takahashi M, Abe T, Suzuki T (1999) A case of mediastinal myelolipoma. Kyobu Geka 52:869–871
Krag D, Reich SB (1972) Heterotopic bone marrow (myelolipoma) of the mediastinum. Chest 61:514–515
Saleeby ER (1925) Heterotopia of the bone marrow without apparent cause. Am J Pathol 1(69–76):63
Strimlan CV, Khasnabis S (1993) Primary mediastinal myelolipoma. Cleve Clin J Med 60:69–71
Suzuki T, Mushiaki T, Hori G, Tonozuka H, Suzuki H, Noguchi H, Sagawa F, Mitsuya T (1988) A case of primary myelolipoma of the posterior mediastinum. Nihon Kyobu Shikkan Gakkai Zasshi 26:1318–1322
Vaziri M, Sadeghipour A, Pazooki A, Shoolami LZ (2008) Primary mediastinal myelolipoma. Ann Thorac Surg 85:1805–1806. doi:10.1016/j.athoracsur.2007.11.023
Litwer H (1960) Myelolipoma of the mediastinum. Radiology 74:471–473. doi:10.1148/74.3.471
Anand Rajan KD, Subbarao KC, Agarwala S, Gupta SD (2010) Mediastinal liposarcoma of mixed type in childhood: a report of a case with unusual histologic features. Indian J Pathol Microbiol 53:525–528. doi:10.4103/0377-4929.68297
Chiyo M, Fujisawa T, Yasukawa T, Shiba M, Shibuya K, Sekine Y, Hiroshima K, Ohwada (2001) Successful resection of a primary liposarcoma in the anterior mediastinum in a child: report of a case. Surg Today 31:230–232
Hahn HP, Fletcher CD (2007) Primary mediastinal liposarcoma: clinicopathologic analysis of 24 cases. Am J Surg Pathol 31:1868–1874. doi:10.1097/PAS.0b013e318093f925
Kauffman SL, Stout AP (1959) Lipoblastic tumors of children. Cancer 12:912–925
Klimstra DS, Moran CA, Perino G, Koss MN, Rosai J (1995) Liposarcoma of the anterior mediastinum and thymus. A clinicopathologic study of 28 cases. Am J Surg Pathol 19:782–791
Mikkilineni RS, Bhat S, Cheng AW, Prevosti LG (1994) Liposarcoma of the posterior mediastinum in a child. Chest 106:1288–1289
Saeed M, Plett S, Kim GE, Daldrup-Link H, Courtier J (2010) Radiological-pathological correlation of pleomorphic liposarcoma of the anterior mediastinum in a 17-year-old girl. Pediatr Radiol 40(Suppl 1):S68–S70. doi:10.1007/s00247-010-1797-1
Wilson JR, Bartley TD (1964) Liposarcoma of the mediastinum. Report of a case in a child and review of the literature. J Thorac Cardiovasc Surg 48:486–490
Sung MT, Ko SF, Hsieh MJ, Chen YJ, Chen WJ, Huang HY (2003) Thymoliposarcoma. Ann Thorac Surg 76:2082–2085
Marulli G, Rea F, Feltracco P, Calabrese F, Giacometti C, Rizzardi G, Vincenzo L, Sartori F (2007) Successful resection of a giant primary liposarcoma of the posterior mediastinum. J Thorac Oncol Offic Pub Int Assoc Stud Lung Canc 2:453–455. doi:10.1097/01.JTO.0000268681.10367.cf
Ortega P, Suster D, Falconieri G, Zambrano E, Moran CA, Morrison C, Suster S (2014) Liposarcomas of the posterior mediastinum: clinicopathologic study of 18 cases. Mod Pathol. doi:10.1038/modpathol.2014.152
Schweitzer DL, Aguam AS (1977) Primary liposarcoma of the mediastinum. Report of a case and review of the literature. J Thorac Cardiovasc Surg 74:83–97
Alaggio R, Coffin CM, Weiss SW, Bridge JA, Issakov J, Oliveira AM, Folpe AL (2009) Liposarcomas in young patients: a study of 82 cases occurring in patients younger than 22 years of age. Am J Surg Pathol 33:645–658. doi:10.1097/PAS.0b013e3181963c9c
Chen M, Yang J, Zhu L, Zhou C, Zhao H (2014) Primary intrathoracic liposarcoma: a clinicopathologic study and prognostic analysis of 23 cases. J Cardiothorac Surg 9:119. doi:10.1186/1749-8090-9-119
Uner R, Balim AI, Oktem K (1963) Mediastinal liposarcoma. Report of a case. Dis Chest 43:103–105
Boland JM, Colby TV, Folpe AL (2012) Liposarcomas of the mediastinum and thorax: a clinicopathologic and molecular cytogenetic study of 24 cases, emphasizing unusual and diverse histologic features. Am J Surg Pathol 36:1395–1403. doi:10.1097/PAS.0b013e3182562bc1
Bonnette P, Jouan J, Colchen A, Epardeau B, Grapin D, Grapin JP (2000) Myxoid liposarcoma of the mediastinum. Rev Mal Respir 17:109–111
Doyle LA, Tao D, Marino-Enriquez A (2014) STAT6 is amplified in a subset of dedifferentiated liposarcoma. Mod Pathol 27:1231–1237. doi:10.1038/modpathol.2013.247
Folpe AL, Weiss SW (2002) Lipoleiomyosarcoma (well-differentiated liposarcoma with leiomyosarcomatous differentiation): a clinicopathologic study of nine cases including one with dedifferentiation. Am J Surg Pathol 26:742–749
Gomez-Roman JJ, Val-Bernal JF (1997) Lipoleiomyosarcoma of the mediastinum. Pathology 29:428–430
Weissferdt A, Moran CA (2014) Lipomatous tumors of the anterior mediastinum with muscle differentiation: a clinicopathological and immunohistochemical study of three cases. Virchows Arch 464:489–493. doi:10.1007/s00428-014-1556-z
Moran CA, Suster S (1997) Primary germ cell tumors of the mediastinum: I. Analysis of 322 cases with special emphasis on teratomatous lesions and a proposal for histopathologic classification and clinical staging. Cancer 80:681–690
Bouchikh M, Arame A, Riquet M, Le Pimpec-Barthes F (2013) Cardiac failure due to a giant desmoid tumour of the posterior mediastinum. Eur J Cardiothorac Surg 44:1137–1139. doi:10.1093/ejcts/ezt214
Nakagiri T, Koseki M, Nakamoto K, Taniyama K (2007) Paraesophageal mediastinal desmoid tumor: case report. Gen Thorac Cardiovasc Surg 55:125–129. doi:10.1007/s11748-006-0068-4
Fujinaga K, Sakamoto S, Sawada Y, Tanaka J, Mizumoto T (2012) Recurrent mediastinal desmoid tumor treated by surgical resection; report of a case. Kyobu Geka 65:252–254
Ko SF, Ng SH, Hsiao CC, Hsieh CS, Lin JW, Huang CC, Shih TY (1996) Juvenile fibromatosis of the posterior mediastinum with intraspinal extension AJNR. Am J Neuroradiol 17:522–524
Torii Y, Sasano S, Obara T (2005) A case of desmoid tumor involving the posterior mediastinum. Nihon Kokyuki Gakkai Zasshi 43:613–617
Xie Y, Xie K, Gou Q, He J, Zhong L, Wang Y (2014) Recurrent desmoid tumor of the mediastinum: a case report. Oncol Lett 8:2276–2278. doi:10.3892/ol.2014.2431
Yoshida S, Kimura H, Iwai N, Yasufuku K, Yamaguchi Y, Takahara Y (1997) A surgical case of aggressive fibromatosis. Nihon Kyobu Geka Gakkai Zasshi 45:2016–2020
van Broekhoven DLM, Grünhagen DJ, den Bakker MA, van Dalen T, Verhoef C (2015) Time trends in the incidence and treatment of extra-abdominal and abdominal aggressive fibromatosis: a population based study. Ann Surg Oncol 22(9):2817–2823. doi:10.1245/s10434-015-4632-y
Fletcher C, Bridge J, Lee J-C (2013) Extra-pleural solitary fibrous tumour WHO classification of tumours of soft tissue and bone, 4th edn. IARC Press, Lyon, pp 80–82
Qedra N, Kadry M, Ivanitskaia-Kuhn E, Buz S, Meyer R, Laube H, Hetzer R (2010) Solitary fibrous mediastinal tumor with coronary vascular supply: an unusual case. J Thorac Cardiovasc Surg 139:e23–e25. doi:10.1016/j.jtcvs.2008.08.038
Goto Y, Sakurada T, Suzuki I, Nanjo H, Masuda H (1997) A localized fibrous tumor (mesothelioma) in the mediastinum: report of a case. Surg Today 27:871–873
Xue X, Chen J, Ma W, Zhu D, Zhang W, Chen G, Wei S, Zhou Q (2009) Mediastinal solitary fibrous tumor with right diaphragm invasion: report of a case. Surg Today 39:332–334. doi:10.1007/s00595-008-3841-4
Yashiro T, Shiba M, Sato K, Matsuzaki O, Iizasa T, Otsuji M, Fujisawa T (2004) Huge localized fibrous tumor of the pleura resembling a mediastinal tumor: report of a case. Surg Today 34:58–61. doi:10.1007/s00595-003-2648-6
Briselli M, Mark EJ, Dickersin GR (1981) Solitary fibrous tumors of the pleura: eight new cases and review of 360 cases in the literature. Cancer 47:2678–2689
de Perrot M, Fischer S, Brundler MA, Sekine Y, Keshavjee S (2002) Solitary fibrous tumors of the pleura. Ann Thorac Surg 74:285–293
England DM, Hochholzer L, McCarthy MJ (1989) Localized benign and malignant fibrous tumors of the pleura. A clinicopathologic review of 223 cases. Am J Surg Pathol 13:640–658
Atkinson JB, Mahour GH, Isaacs H Jr, Ortega JA (1984) Hemangiopericytoma in infants and children. A report of six patients. Am J Surg 148:372–374
Horikawa-Kyo Y, Tanaka T, Tanano H, Kitayama Y, Karakawa S, Taniyama K (2009) Mediastinal hemangiopericytoma. Pediatr Blood Cancer 53:206–207. doi:10.1002/pbc.22054
Simonton SC, Swanson PE, Watterson J, Priest JR (1995) Primary mediastinal hemangiopericytoma with fatal outcome in a child. Arch Pathol Lab Med 119:839–841
Amonkar GP, Deshpande JR, Kandalkar BM (2006) An unusual lipomatous hemangiopericytoma. J Postgrad Med 52:71–72
Bicakcioglu P, Aydin E, Celik A, Demirag F, Karaoglanoglu N (2012) Primary classical hemangiopericytomas of thorax. Ann Thorac Surg 94:255–259. doi:10.1016/j.athoracsur.2012.03.063
Cakir E, Findik G, Hosgun D, Demirag F (2010) Primary mediastinal haemangiopericytoma—an unusual cause of massive haemoptysis in a young woman. Acta Chir Belg 110:235–237
Chnaris A, Barbetakis N, Efstathiou A, Fessatidis I (2006) Primary mediastinal hemangiopericytoma. World J Surg Oncol 4:23. doi:10.1186/1477-7819-4-23
De Raet J, Sacre R, Hoorens A, Fletcher C, Lamote J (2008) Malignant giant solitary fibrous tumor of the mediastinum SFT. J Thorac Oncol Offic Pub Int Assoc Stud Lung Canc 3:1068–1070. doi:10.1097/JTO.0b013e318183af5d
Deodato G, Messina S, Nicolosi M, Chisari A, Piccirillo P, Compagnone S, Tornambene F, Torre T (1991) Intrathoracic hemangiopericytoma localized in the mediastinum. Minerva Chir 46:1205–1215
Eguchi T, Ito N, Makiuchi A, Yoshida K (2010) A solitary fibrous tumor arising from the thymus. Interact Cardiovasc Thorac Surg 11:362–363. doi:10.1510/icvts.2010.237461
Feldman F, Seaman WB (1964) Primary thoracic hemangiopericytoma. Radiology 82:998–1009. doi:10.1148/82.6.998
Fukushima K, Yamaguchi T, Take A, Ohara T, Hasegawa T, Mochizuki M (1992) A case report of so-called solitary fibrous tumor of the mediastinum. Nihon Kyobu Geka Gakkai Zasshi 40:978–982
Gannon BR, O’Hara CD, Reid K, Isotalo PA (2007) Solitary fibrous tumor of the anterior mediastinum: a rare extrapleural neoplasm. Tumori 93:508–510
Goodlad JR, Fletcher CD (1991) Solitary fibrous tumour arising at unusual sites: analysis of a series. Histopathology 19:515–522
Guillou L, Gebhard S, Coindre JM (2000) Lipomatous hemangiopericytoma: a fat-containing variant of solitary fibrous tumor? Clinicopathologic, immunohistochemical, and ultrastructural analysis of a series in favor of a unifying concept. Hum Pathol 31:1108–1115. doi:10.1053/hupa.2000.9777
Hanau CA, Miettinen M (1995) Solitary fibrous tumor: histological and immunohistochemical spectrum of benign and malignant variants presenting at different sites. Hum Pathol 26:440–449
Hayashi A, Takamori S, Tayama K, Mitsuoka M, Tamura K, Shirouzu K, Fujimoto K, Watanabe J (1998) Primary hemangiopericytoma of the superior mediastinum: a case report. Ann Thorac Cardiovasc Surg 4:283–285
Hiraki A, Murakami T, Aoe K, Matsuda E, Maeda T, Uemori Y, Ueoka H (2006) Recurrent superior mediastinal primary hemangiopericytoma 23 years after the complete initial excision: a case report. Acta Med Okayama 60:197–200
Iwata T, Nishiyama N, Izumi N, Tsukioka T, Suehiro S (2007) Solitary fibrous tumor of the thymus with local invasiveness and pleural dissemination: report of a case. Ann Thorac Cardiovasc Surg 13:198–202
Khalifa MA, Montgomery EA, Azumi N, Gomes MN, Zeman RK, Min KW, Lack EE (1997) Solitary fibrous tumors: a series of lesions, some in unusual sites. South Med J 90:793–799
Koo BC, Smith RR, Morgan RJ, Gohara AF (1985) Primary mediastinal hemangiopericytoma: computed tomography correlation. J Comput Tomogr 9:253–256
Kulshreshtha P, Kannan N, Bhardwaj R, Batra S, Gupta S (2014) Primary mediastinal hemangiopericytoma treated with preoperative embolization and surgery. Ann Thorac Surg 97:335–338. doi:10.1016/j.athoracsur.2013.04.128
Liu X, Zhang HY, Bu H, Meng GZ, Zhang Z, Ke Q (2007) Fat-forming variant of solitary fibrous tumor of the mediastinum. Chin Med J (Engl) 120
Marchevsky AM, Varshney D, Fuller C (2003) Mediastinal epithelioid solitary fibrous tumor. Arch Pathol Lab Med 127:e212–e215. doi:10.1043/0003-9985(2003)127<e212:MESFT>2.0.CO;2
Martorell M, Perez-Valles A, Gozalbo F, Garcia-Garcia JA, Gutierrez J, Gaona J (2007) Solitary fibrous tumor of the thigh with epithelioid features: a case report. Diagn Pathol 2:19. doi:10.1186/1746-1596-2-19
Morandi U, Stefani A, De Santis M, Paci M, Lodi R (2000) Preoperative embolization in surgical treatment of mediastinal hemangiopericytoma. Ann Thorac Surg 69:937–939
Mori M, Nakanishi N, Furuya K (1994) Hemangiopericytoma of the mediastinum causing spontaneous hemothorax. Ann Thorac Surg 58:1525–1527
Morimitsu Y, Nakajima M, Hisaoka M, Hashimoto H (2000) Extrapleural solitary fibrous tumor: clinicopathologic study of 17 cases and molecular analysis of the p53 pathway. APMIS 108:617–625
Nejjar M, Hidraoui K, Mihradi W, Ridai M, Zerouali NO, El Attar H, Azzouzi S, Iraqi A (2004) Solitary fibrous mediastinal tumor. A case report. Rev Pneumol Clin 60:235–238
Ohno K, Takai M, Obata G et al (1988) A case report of mediastinal hemangiopericytoma. J Jpn Soc Clin Surg 49:1008–1012 [Japanese]
Osanai T, Kanazawa T, Nakamura K, Tsushima T, Kimura M, Onodera K (1994) A case of primary cystic mediastinal hemangiopericytoma. Arch Pathol Lab Med 118:575–577
Plones T, Kayser G, Passlick B (2013) Solitary malignant fibrous tumor of the mediastinum. Asian Cardiovasc Thorac Ann 21:491–492. doi:10.1177/0218492312458596
Puleo S, Di Cataldo A, Li Destri G, La Greca G, Castobello C, Bartoloni G, Privitera G, Scandurra G, Latteri F, Rodolico G (1988) Radio-hyperthermia for subcutaneous metastasis of hemangiopericytoma of the mediastinum: case report. Tumori 74:485–488
Shiraishi T, Hirayama S, Hiratsuka M, Iwasaki A, Makimoto Y, Iwasaki H, Kawahara K, Shirakusa T (2004) Mediastinal solitary fibrous tumor: report of a case with direct invasion to the trachea. Thorac Cardiovasc Surg 52:110–112. doi:10.1055/s-2004-817808
Suehisa H, Yamashita M, Komori E, Sawada S, Teramoto N (2010) Solitary fibrous tumor of the mediastinum. Gen Thorac Cardiovasc Surg 58:205–208. doi:10.1007/s11748-009-0510-5
Tsubochi H, Endo T, Sogabe M, Endo S, Morinaga S, Dobashi Y (2014) Solitary fibrous tumor of the thymus with variegated epithelial components. Int J Clin Exp Pathol 7:7477–7484
Tzeng Y, Lan G, Chung M, Fu J (2001) Primary mediastinal hemangiopericytoma: a case report. Chin J Radiol 26:91–96
van de Rijn M, Lombard CM, Rouse RV (1994) Expression of CD34 by solitary fibrous tumors of the pleura, mediastinum, and lung. Am J Surg Pathol 18:814–820
Vanfleteren LE, Peulen HM, Creytens DH, Smulders NM, Utama I, de Ruysscher DK, ten Velde GP (2009) Complete metabolic remission of an irresectable mediastinal solitary fibrous tumour with concurrent chemoradiation. Thorax 64:822–823. doi:10.1136/thx.2008.109561
Weidner N (1991) Solitary fibrous tumor of the mediastinum. Ultrastruct Pathol 15:489–492
Witkin GB, Rosai J (1989) Solitary fibrous tumor of the mediastinum. A report of 14 cases. Am J Surg Pathol 13:547–557
Chmielecki J, Crago AM, Rosenberg M, O’Connor R, Walker SR, Ambrogio L, Auclair D, McKenna A, Heinrich MC, Frank DA, Meyerson M (2013) Whole-exome sequencing identifies a recurrent NAB2-STAT6 fusion in solitary fibrous tumors. Nat Genet 45:131–132. doi:10.1038/ng.2522
Mohajeri A, Tayebwa J, Collin A, Nilsson J, Magnusson L, von Steyern FV, Brosjo O, Domanski HA, Larsson O, Sciot R, Debiec-Rychter M, Hornick JL, Mandahl N, Nord KH, Mertens F (2013) Comprehensive genetic analysis identifies a pathognomonic NAB2/STAT6 fusion gene, nonrandom secondary genomic imbalances, and a characteristic gene expression profile in solitary fibrous tumor. Genes Chromosomes Cancer 52:873–886. doi:10.1002/gcc.22083
Robinson DR, Wu YM, Kalyana-Sundaram S, Cao X, Lonigro RJ, Sung YS, Chen CL, Zhang L, Wang R, Su F, Iyer MK, Roychowdhury S, Siddiqui J, Pienta KJ, Kunju LP, Talpaz M, Mosquera JM, Singer S, Schuetze SM, Antonescu CR, Chinnaiyan AM (2013) Identification of recurrent NAB2-STAT6 gene fusions in solitary fibrous tumor by integrative sequencing. Nat Genet 45:180–185. doi:10.1038/ng.2509
Cheah AL, Billings SD, Goldblum JR, Carver P, Tanas MZ, Rubin BP (2014) STAT6 rabbit monoclonal antibody is a robust diagnostic tool for the distinction of solitary fibrous tumour from its mimics. Pathology 46:389–395. doi:10.1097/PAT.0000000000000122
Doyle LA, Vivero M, Fletcher CD, Mertens F, Hornick JL (2014) Nuclear expression of STAT6 distinguishes solitary fibrous tumor from histologic mimics. Mod Pathol 27:390–395. doi:10.1038/modpathol.2013.164
Yoshida A, Tsuta K, Ohno M, Yoshida M, Narita Y, Kawai A, Asamura H, Kushima R (2014) STAT6 immunohistochemistry is helpful in the diagnosis of solitary fibrous tumors. Am J Surg Pathol 38:552–559. doi:10.1097/PAS.0000000000000137
Lee JC, Fletcher CD (2011) Malignant fat-forming solitary fibrous tumor (so-called “lipomatous hemangiopericytoma”): clinicopathologic analysis of 14 cases. Am J Surg Pathol 35:1177–1185. doi:10.1097/PAS.0b013e318219cd0b
Creytens D, Ferdinande L (2014) Diagnostic utility of STAT6 immunohistochemistry in the diagnosis of fat-forming solitary fibrous tumors. Appl Immunohistochem Mol Morphol. doi:10.1097/PAI.0000000000000166
Creytens D, Libbrecht L, Ferdinande L (2014) Nuclear expression of STAT6 in dedifferentiated liposarcomas with a solitary fibrous tumor-like morphology: a diagnostic pitfall. Appl Immunohistochem Mol Morphol. doi:10.1097/PAI.0000000000000081
Awasthi R, O’Neill JK, Keen CE, Sarsfield PT, Devaraj VS, Stone CA, Smith ME (2006) Biphasic solitary fibrous tumour: a report of two cases with epithelioid features. Virchows Arch 448:306–310. doi:10.1007/s00428-005-0099-8
Barak S, Wang Z, Miettinen M (2012) Immunoreactivity for calretinin and keratins in desmoid fibromatosis and other myofibroblastic tumors: a diagnostic pitfall. Am J Surg Pathol 36:1404–1409. doi:10.1097/PAS.0b013e3182556def
Fu J, Zhang R, Zhang H, Bu H, Chen H, Yin X, Zhang Z, Wei B (2012) Epithelioid solitary fibrous tumor of the central nervous system. Clin Neurol Neurosurg 114:72–76. doi:10.1016/j.clineuro.2011.07.021
Mourra N, Lewin M, Sautet A, Parc R, Flejou JF (2005) Epithelioid solitary fibrous tumor in the ischioanal fossa. Virchows Arch 446:674–676. doi:10.1007/s00428-005-1255-x
Parrozzani R, Fusetti S, Montesco C, Favero V, Midena E (2013) Biphasic solitary fibrous tumor of the orbit with distant metastases. Int Ophthalmol 33:701–705. doi:10.1007/s10792-012-9706-2
Warraich I, Dunn DM, Oliver JW (2006) Solitary fibrous tumor of the orbit with epithelioid features. Arch Pathol Lab Med 130:1039–1041. doi:10.1043/1543-2165(2006)130[1039:SFTOTO]2.0.CO;2
Yan B, Raju GC, Salto-Tellez M (2008) Epithelioid, cytokeratin expressing malignant solitary fibrous tumour of the pleura. Pathology 40:98–99. doi:10.1080/00313020701716417
Coffin CM, Watterson J, Priest JR, Dehner LP (1995) Extrapulmonary inflammatory myofibroblastic tumor (inflammatory pseudotumor). A clinicopathologic and immunohistochemical study of 84 cases. Am J Surg Pathol 19:859–872
Chen CH, Lin RL, Liu HC, Hung TT, Huang WC (2008) Inflammatory myofibroblastic tumor mimicking anterior mediastinal malignancy. Ann Thorac Surg 86:1362–1364. doi:10.1016/j.athoracsur.2008.03.031
Crespo C, Navarro M, Gonzalez I, Lorente MF, Gonzalez R, Mayol (2001) Intracranial and mediastinal inflammatory myofibroblastic tumour. Pediatr Radiol 31:600–602. doi:10.1007/s002470100489
Ghandi N, Ghanadan A, Azizian MR, Hejazi P, Aghazadeh N, Tavousi P, Daneshpazhooh M (2014) Paraneoplastic pemphigus associated with inflammatory myofibroblastic tumour of the mediastinum: a favourable response to treatment and review of the literature. Australas J Dermatol. doi:10.1111/ajd.12264
Guan Y, Chen G, Zhang W, Chen H, He J (2012) Computed tomography appearance of inflammatory myofibroblastic tumor in the mediastinum. J Comput Assist Tomogr 36:654–658. doi:10.1097/RCT.0b013e31826801ba
Karnak I, Senocak ME, Ciftci AO, Caglar M, Bingol-Kologlu M, Tanyel FC, Buyukpamukcu N (2001) Inflammatory myofibroblastic tumor in children: diagnosis and treatment. J Pediatr Surg 36:908–912. doi:10.1053/jpsu.2001.23970
Makimoto Y, Nabeshima K, Iwasaki H, Ishiguro A, Miyoshi T, Shiraishi T, Iwasaki A, Shirakusa T (2005) Inflammatory myofibroblastic tumor of the posterior mediastinum: an older adult case with anaplastic lymphoma kinase abnormalities determined using immunohistochemistry and fluorescence in situ hybridization. Virchows Arch 446:451–455. doi:10.1007/s00428-004-1170-6
Meng X, Wang R (2013) Inflammatory myofibroblastic tumor occurs in the mediastinum. J Cancer Res Ther 9:721–723. doi:10.4103/0973-1482.126467
Ramachandra S, Hollowood K, Bisceglia M, Fletcher CD (1995) Inflammatory pseudotumour of soft tissues: a clinicopathological and immunohistochemical analysis of 18 cases. Histopathology 27:313–323
Sugiyama K, Nakajima K, Nakajima Y (2008) Inflammatory myofibroblastic tumor in the mediastinum mimicking a malignant tumor. Diagn Interv Radiol 14:197–199
Yamaguchi M, Yoshino I, Osoegawa A, Kameyama T, Tagawa T, Fukuyama S, Oda Y, Tsuneyoshi M, Maehara Y (2003) Inflammatory myofibroblastic tumor of the mediastinum presenting as superior vena cava syndrome. J Thorac Cardiovasc Surg 126:870–872
Chang JW, Kim JH, Maeng YH (2001) Calcifying fibrous pseudotumor of the anterior mediastinum. Korean J Thorac Cardiovasc Surg 44(4):318–320. doi:10.5090/kjtcs.2011.44.4.318
Dumont P, de Muret A, Skrobala D, Robin P, Toumieux B (1997) Calcifying fibrous pseudotumor of the mediastinum. Ann Thorac Surg 63:543–544
Jeong HS, Lee GK, Sung R, Ahn JH, Song HG (1997) Calcifying fibrous pseudotumor of mediastinum—a case report. J Korean Med Sci 12:58–62
Nascimento AF, Ruiz R, Hornick JL, Fletcher CD (2002) Calcifying fibrous ‘pseudotumor’: clinicopathologic study of 15 cases and analysis of its relationship to inflammatory myofibroblastic tumor. Int J Surg Pathol 10:189–196
Okubo K, Kuwabara M, Ito K, Matsuoka K (1995) A case of mediastinal fibrosarcoma. Nihon Kyobu Geka Gakkai Zasshi 43:221–225
Galetta D, Cesario A, Margaritora S, Granone P (2004) Primary mediastinal hyalinizing spindle cell tumor with giant rosettes. Ann Thorac Surg 77:2206–2209. doi:10.1016/S0003-4975(03)01388-2
Jakowski JD, Wakely PE Jr (2008) Primary intrathoracic low-grade fibromyxoid sarcoma. Hum Pathol 39:623–628. doi:10.1016/j.humpath.2007.08.017
Takanami I, Takeuchi K, Naruke M (1999) Low-grade fibromyxoid sarcoma arising in the mediastinum. J Thorac Cardiovasc Surg 118:970–971
Short M, Dramis A, Ramani P, Parikh DH (2008) Mediastinal and pulmonary infantile myofibromatosis: an unusual surgical presentation. J Pediatr Surg 43:e29–e31. doi:10.1016/j.jpedsurg.2008.06.046
Fukunaga M, Ushigome S (1998) Giant cell angiofibroma of the mediastinum. Histopathology 32:187–189
Furusato E, Valenzuela IA, Fanburg-Smith JC, Auerbach A, Furusato B, Cameron JD, Rushing EJ (2011) Orbital solitary fibrous tumor: encompassing terminology for hemangiopericytoma, giant cell angiofibroma, and fibrous histiocytoma of the orbit: reappraisal of 41 cases. Hum Pathol 42:120–128. doi:10.1016/j.humpath.2010.05.021
Annam V, Krishna AT, Annam V (2010) Deep benign fibrous histiocytoma in the posterior mediastinum. Indian J Cancer 47:231–233. doi:10.4103/0019-509X.63018
Sood N, Singhal VS (1992) Benign fibrous histiocytoma of the mediastinum. Indian J Chest Dis Allied Sci 34:33–37
Asakura S, Tezuka N, Inoue S, Kihara N, Fujino S (2001) Angiomatoid fibrous histiocytoma in mediastinum. Ann Thorac Surg 72:283–285
Davies KA, Cope AP, Schofield JB, Chu CQ, Mason JC, Krausz T, Smith P, Denman AM, Walport MJ (1995) A rare mediastinal tumour presenting with systemic effects due to IL-6 and tumour necrosis factor (TNF) production. Clin Exp Immunol 99:117–123
Fu K, Moran CA, Suster S (2002) Primary mediastinal giant cell tumors: a clinicopathologic and immunohistochemical study of two cases. Ann Diagn Pathol 6:100–105
Goldberg J, Azizad S, Bandovic J, Khan A (2009) Primary mediastinal giant cell tumor. Rare Tumor 1:e45. doi:10.4081/rt.2009.e45
Jain D, Arava S, Mishra B, Sharma S, Sharma R, Parshad R (2015) Soft tissue giant cell tumor of low malignant potential of mediastinum: a rare case report. Int J Surg Pathol 23:71–74. doi:10.1177/1066896914540937
Manivel C, Wick MR, Abenoza P, Rosai J (1986) The occurrence of sarcomatous components in primary mediastinal germ cell tumors. Am J Surg Pathol 10:711–717
Bosl GJ, Ilson DH, Rodriguez E, Motzer RJ, Reuter VE, Chaganti RS (1994) Clinical relevance of the i(12p) marker chromosome in germ cell tumors. J Natl Cancer Inst 86:349–355
Oosterhuis JW, Looijenga LH (2005) Testicular germ-cell tumours in a broader perspective. Nat Rev Cancer 5:210–222. doi:10.1038/nrc1568
Motzer RJ, Amsterdam A, Prieto V, Sheinfeld J, Murty VV, Mazumdar M, Bosl GJ, Chaganti RS, Reuter VE (1998) Teratoma with malignant transformation: diverse malignant histologies arising in men with germ cell tumors. J Urol 159:133–138
Motzer RJ, Rodriguez E, Reuter VE, Bosl GJ, Mazumdar M, Chaganti RS (1995) Molecular and cytogenetic studies in the diagnosis of patients with poorly differentiated carcinomas of unknown primary site. J Clin Oncol 13:274–282
Wehle D, Yonescu R, Long PP, Gala N, Epstein J, Griffin CA (2008) Fluorescence in situ hybridization of 12p in germ cell tumors using a bacterial artificial chromosome clone 12p probe on paraffin-embedded tissue: clinical test validation. Cancer Genet Cytogenet 183:99–104. doi:10.1016/j.cancergencyto.2008.02.012
Caballero C, Gomez S, Matias-Guiu X, Prat J (1992) Rhabdomyosarcomas developing in association with mediastinal germ cell tumours. Virchows Arch A Pathol Anat Histopathol 420:539–543
Contreras AL, Punar M, Tamboli P, Tu SM, Pisters L, Moran C, Czerniak BA, Guo CC (2010) Mediastinal germ cell tumors with an angiosarcomatous component: a report of 12 cases. Hum Pathol 41:832–837. doi:10.1016/j.humpath.2009.11.008
Gonzalez-Vela JL, Savage PD, Manivel JC, Torkelson JL, Kennedy BJ (1990) Poor prognosis of mediastinal germ cell cancers containing sarcomatous components. Cancer 66:1114–1116
McCartney AC, Paradinas FJ, Newlands ES (1984) Significance of the ‘maturation’ of metastases from germ cell tumours after intensive chemotherapy. Histopathology 8:457–467
Oosterhuis JW, Suurmeyer AJ, Sleyfer DT, Koops HS, Oldhoff J, Fleuren G (1983) Effects of multiple-drug chemotherapy (cis-diamminedichloroplatinum, bleomycin, and vinblastine) on the maturation of retroperitoneal lymph node metastases of nonseminomatous germ cell tumors of the testis. No evidence for de novo induction of differentiation. Cancer 51:408–416
Pachter MR, Lattes R (1963) Mesenchymal tumors of the mediastinum II. Tumors of Blood Vascular Origin. Cancer 16:95–107
Pachter MR, Lattes R (1963) Mesenchymal tumors of the mediastinum. III. Tumors of lymph vascular origin. Cancer 16:108–117
Acknowledgments
The authors wish to thank Dr. Satoshi Kaneda (Department of Radiology, Saiseikai Central Hospital, Tokyo, Japan) for providing radiological images and Dr. Ieneke Hartmann (Department of Radiology, Maasstad Ziekenhuis, Rotterdam, The Netherlands) for reviewing the radiological images and providing suggestions for the figure legends.
Compliance with ethical standards
ᅟ
Conflict of interest
The work of this manuscript work does not result in potential conflicts of interest; there was no research involving human participants and/or animals performed for this manuscript
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
den Bakker, M.A., Marx, A., Mukai, K. et al. Mesenchymal tumours of the mediastinum—part I. Virchows Arch 467, 487–500 (2015). https://doi.org/10.1007/s00428-015-1830-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00428-015-1830-8