
Abstract
The RAS-MAPKinase pathway is a signal transduction cascade which has been studied extensively during the last decades for its role in human oncogenesis. Activation of this cascade is controlled by cycling of the RAS protein between an inactive and an active state and by phosphorylation of downstream proteins. The signalling cascade regulates cell proliferation, differentiation and survival. Disturbed RAS signalling in malignancies is caused by acquired somatic mutations in RAS genes or other components of this pathway. Recently, germline mutations in genes coding for different components of the RAS signalling cascade have been recognized as the cause of several phenotypically overlapping disorders, recently referred to as the neuro-cardio-facial-cutaneous syndromes. Neurofibromatosis type 1, Noonan, LEOPARD, Costello and cardiofaciocutaneous syndromes all present with variable degrees of psychomotor delay, congenital heart defects, facial dysmorphism, short stature, skin abnormalities and a predisposition for malignancy. These findings point to important roles for this evolutionary conserved pathway in oncogenesis, development, cognition and growth. Conclusion: it has become obvious in recent years that the neuro-cardio-facial-cutaneous syndromes all share a common genetic and pathophysiologic basis. Dysregulation of the RAS-MAPKinase pathway is caused by germline mutations in genes involved in this pathway. Undoubtedly more genes causing related syndromes will be discovered in the near future since there are still a substantial number of genes in the pathway that are not yet associated with a known syndrome.
Similar content being viewed by others


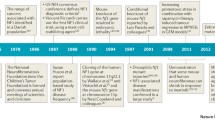
Avoid common mistakes on your manuscript.
Introduction
The “neuro-cardio-facial-cutaneous (NCFC) syndromes” is a term that was recently introduced to group neurofibromatosis type 1 (NF1), LEOPARD syndrome, Noonan syndrome, Costello syndrome and cardio-facio-cutaneous (CFC) syndrome [3]. These disorders all share a variable degree of mental retardation, cardiac defects (often pulmonary valve stenosis or hypertrophic cardiomyopathy), facial dysmorphism, short stature, macrocephaly and skin abnormalities. Moreover, patients with these disorders all have an increased risk for malignancy, except for CFC-syndrome patients. In recent years, a common genetic and pathophysiologic basis for these phenotypically overlapping syndromes has become obvious. The NCFC syndromes result from germline mutations in genes of the evolutionary conserved RAS-MAPKinase pathway. This signal transduction pathway plays a role in growth factor signalling and is a well known factor in oncogenesis. It was already known for some time that inactivating mutations in the NF1 gene, coding for a negative regulator of RAS-signalling, are responsible for neurofibromatosis type 1 (Von Recklinghausen’s disease) [31], and that heterozygous missense mutations in PTPN11, coding for SHP2, which positively modulates RAS signalling, are found in 50% of Noonan syndrome patients [47] and in most LEOPARD syndrome patients [11, 33]. In 2005, de novo heterozygous missense mutations in HRAS were found to be responsible for Costello syndrome [1, 14, 17, 26]. Somewhat later KRAS mutations were discovered in less than 2% of PTPN11-negative Noonan patients [6, 43, 54] and recently mutations in the GEF SOS1 have been reported to account for approximately 20% of PTPN11-negative Noonan syndrome patients [40, 49]. Finally, in CFC syndrome, mutations in KRAS, as well as BRAF, MEK1 and MEK2 were found [38, 41]. BRAF, MEK1 and MEK2 genes code for proteins in the pathway located downstream of the RAS proteins. The scope of this review is to describe the similarities and dissimilarities of the clinical and molecular aspects of these syndromes. An overview of the most typical features of the NCFC syndromes is given in Table 1 and an overview of the RAS-MAPK pathway and associated syndromes is given in Fig. 1. In Fig. 2 pictures of typical face of patients with Costello, Noonan and cardiofaciocutaneous syndromes and skin abnormalities in patients with neurofibromatosis type 1 and LEOPARD syndrome are shown.

Overview of the RAS-MAPK pathway and associated NCFC syndromes. Binding of a growth factor to a receptor tyrosine kinase (RTK) activates RAS proteins by the action of GEFs such as SOS which catalyze guanosine nucleotide exchange. RAS-GTP activates RAF and downstream effectors. Signalling is terminated when RAS-GTP is hydrolyzed to RAS-GDP either by the action of GAPs neurofibromin or p120 GAP. Responsible genes for NF1, Noonan, LEOPARD, Costello and CFC syndrome are indicated

Photographs of patients with different NCFC syndromes. a Typical café-au-lait macules in a patient with neurofibromatosis type 1. b Costello syndrome patient with HRAS mutation. c Noonan syndrome patient with PTPN11 mutation. d Noonan syndrome patient with SOS1 mutation. Note the skin abnormalities, the absence of eyebrows and the sparse eyelashes. e Lentigines in LEOPARD syndrome patient. f Cardiofaciocutaneous syndrome patient with BRAF mutation
The RAS-MAPK pathway
RAS genes were first identified as homologues of rodent sarcoma viruses. During the 1960s and 1970s a great deal of research focused on retroviruses that caused tumours in rodents. The goal of this research was to find similar viruses inducing oncogenesis in humans, but this quest turned out to be not very fruitful. However, in 1982 new human DNA sequences homologous to the transforming oncogenes of the v-Harvey (HRAS) and Kirsten rat sarcoma virus (KRAS) were identified that caused oncogenic transformation of mouse fibroblast cells [10]. These DNA sequences were derived from a human bladder cancer and a human lung cancer cell line, respectively. The findings showed that RAS genes could act as oncogenes in human cancer. It seemed that the amino acid substitution of glycine 12 of the RAS protein was responsible for the transforming ability of this mutant RAS. Later on, two other family members, NRAS (neuroblastoma RAS viral oncogene homologue) and ERAS (Embryonic stem cell- expressed RAS), were discovered.
RAS proteins are guanosine nucleotide-bound proteins which cycle between an active GTP-bound and an inactive GDP-bound conformation (Fig. 1). They are key molecules in an evolutionary conserved intracellular signal transduction pathway, called the RAS-MAPK (mitogen activated protein kinase) pathway. Mitogen activated protein (MAP) kinases are proteins that phosphorylate (activate) other proteins as a response to extracellular stimuli (mitogens) and regulate various cellular activities, such as gene expression, mitosis, differentiation, and cell survival/apoptosis (programmed cell death). In the MAPK pathway we find three kinases which sequentially activate each other by phosphorylation of serine/threonine residues. Although several MAPK pathways are known, only the extracellular signal-regulated kinase (ERK) pathway will be discussed here. This pathway is activated by binding of a growth factor (“mitogen”) to a specific receptor (tyrosine kinase type receptor). This causes dimerization of two receptor molecules and activation of their kinase function. Different intracellular proteins (adapter proteins) can bind to the intracytoplasmic portion of these receptors and will activate guanosine nucleotide exchange factors (GEFs) like SOS1. These GEFs then activate membrane-bound RAS by catalyzing exchange of GDP bound to RAS for GTP. RAS-GTP can activate several different signalling molecules of which RAF (MAPKKK=MAPkinasekinasekinase) is the most important for this review. Activated RAF-proteins phosphorylate and activate MEK (MAPKK= MAPkinasekinase), which in turn phosphorylates and activates ERK (=MAPKinase). ERK has more than 70 known substrates, of which include nuclear transcription factors and signalling proteins. The result of signalling through this pathway is a change in the pattern of gene expression which may cause cell proliferation, promote cell survival or control cell differentiation. Signalling through this cascade is terminated when GTP is hydrolyzed to GDP by GTP-ase activating proteins (GAPs), like neurofibromin, the protein product of NF1, or p120 GAP (mutated in capillary malformation-arteriovenous malformation syndrome [13]). RAS-MAPK signalling was reviewed recently [27] as well as RAS GDP-GTP cycling [12, 50]. An overview of the RAS-MAPK pathway and associated syndromes is given in Fig. 1.
RAS genes are proto-oncogenes which are mutated in about 30% of human cancers, most frequently in adenocarcinomas of the pancreas, the colon and the lung, in thyroid cancer and in myeloid leukaemia. RAS proteins in cancer cells are frequently mutated at amino acids G12, G13 and Q61. These mutations impair the GTP-hydrolysis of RAS proteins which results in the accumulation of active GTP-bound RAS and constant activation of downstream proteins of the RAS-MAPK pathway [4]. This leads to continuous proliferation of cells even in the absence of growth factors. The protein immediately downstream of RAS—RAF—is also a proto-oncogene. It has three isoforms of which BRAF is the most effective in activation of the MAPK pathway. The gene coding for BRAF is mutated in 7% of human cancers, most frequently malignant melanoma, thyroid, colon and ovarian carcinomas [15].
Neurofibromatosis type 1 (OMIM 162200)
Neurofibromatis type 1 (NF1) is an autosomal dominant condition characterized by pigmentation abnormalities of the skin (café-au-lait macules, freckling in armpits and groins) (Fig. 2a), Lisch nodules in the iris, and learning and behavioural problems. There is a predisposition for the development of tumours of the peripheral nerves such as benign neurofibromas and malignant peripheral nerve sheet tumours (MPNST). Rhabdomyosarcoma occurs at an increased frequency in children with NF1. In rare situations juvenile myelomonocytic leukaemia (JMML), a myeloproliferative disorder with a severe and often lethal course, is observed. The NF1 protein (neurofibromin) (Fig. 1) is a large protein with multiple functional domains. One important domain is the GAP related domain which regulates RAS by stimulation of its conversion to the inactive GDP-bound form. NF1 mutations disturb this GAP activity of the protein resulting in more active RAS and increased signalling through the RAS-MAPK pathway. NF1 related tumours originate as a result of an acquired inactivation of the only normal NF1 copy in an NF1 patient (Knudsons second hit), and therefore the NF1 gene is a tumour suppressor gene [31, 35, 36]. Somatic NRAS or KRAS mutations are responsible for approximately 25% of sporadic cases of JMML [24], while in NF1 patients loss of the normal copy of the NF1 gene is found [44]. Some NF1 patients also have features of Noonan syndrome and “neurofibromatosis-Noonan syndrome” has been described as a separate entity [9], but NF1 gene mutations represent the major molecular event underlying neurofibromatosis-Noonan syndrome [9].
Costello syndrome (OMIM 218040)
Costello syndrome, first described by Costello in 1971 and 1977 [7, 8], is a sporadic (non-familial), occurring disorder characterized by a high birth weight, neonatal feeding problems with subsequent failure to thrive and postnatal growth retardation, redundant skin of the neck, palms and soles, mental retardation, characteristic coarse facial features, relative macrocephaly, cardiac abnormalities and tumour predisposition. Typical facial features in Costello syndrome (Fig. 2b) are a wide forehead, epicanthal folds, depressed nasal bridge, low-set, posteriorly rotated ears with thick lobes, full cheeks and thick lips. Hair is soft, curly and sparsely implanted. Palmar and plantar creases are deep. Benign papillomata in the peri-oral and peri-anal region are a distinct feature. Cardiac defects are found in 63% of patients and include structural heart malformations, most frequently pulmonary valve stenosis, hypertrophic cardiomyopathy and rhythm disturbances, especially atrial tachycardia [34]. Tumour risk in Costello syndrome is estimated to vary from 11% [26] to 17% [18]. The most common tumour observed is rhabdomyosarcoma, followed by neuroblastoma and bladder carcinoma [16]. Mental retardation is mild to moderate with IQ ranging from 25 to 85 [21].
In 2006, Aoki et al. identified missense mutations in codons 12 and 13 of the HRAS gene in 12 individuals with Costello syndrome [1]. The mutations were not present in the parents and occurred “de novo”. This study was the first report of germline RAS mutations as a cause of human disease. Surprisingly, these HRAS heterozygous germline mutations affect the same amino acid residues that are mutated in cancer. Later reports confirmed the presence of HRAS codon 12 and 13 mutations in 85–90% of Costello syndrome patients with G12S being the most prevalent mutation and G12A the second most prevalent [14, 17, 26]. One report described somatic mosaicism for an HRAS codon 12 mutation [19]. As mentioned before, these mutations derange RAS GTP-ase activity and cause activation of the RAS MAPK pathway. Up until now only two patients have been reported with HRAS mutations not affecting aminoacids 12 or 13 of the protein: HRAS K117R [26] and HRAS A146T [52]. These two mutations have never been described in malignancies.
Noonan syndrome (OMIM 163950)
Noonan syndrome is an autosomal dominant disorder which occurs with a frequency of 1/1000–1/2500 live births. Patients have a short stature and specific facial features (Fig. 2c) such as hypertelorism, ptosis, epicanthal folds, low implanted and posteriorly rotated ears and clear blue irises. The neck is broad and often webbed. Congenital heart defects are found in 50–80%. Most frequent defects are pulmonary valve stenosis (20–50%) and hypertrophic cardiomyopathy (20–30%), but also ASD, VSD and Fallot’s tetralogy can be found. Less frequent findings are cryptorchidism in boys and bleeding diathesis with easy bruising and increased risk of bleeding when undergoing surgery. Developmental delay is present in 15–35% of patients and is rather mild. Patients are at risk for development of JMML and rhabdomyosarcoma.
Noonan syndrome has been mapped to a locus on chromosome 12q24.1 by linkage analysis studies [5, 22, 32], and subsequently germline mutations in the PTPN11 gene have been found to account for approximately 50% of Noonan syndrome patients [47]. PTPN11 is a non-receptor protein tyrosine phosphatase encoding the SHP-2 protein which relays signals from activated receptor complexes to downstream signalling molecules, like RAS. PTPN11 mutations occurring in Noonan syndrome are almost always missense mutations and most are gain-of-function mutations. This results in an enhanced phosphatase activity and activation of the RAS-MAPK pathway [46]. Specific germline PTPN11 mutations are found in Noonan syndrome patients with JMML. Acquired PTPN11 mutations, exhibiting stronger activation of SHP-2, were found in about 35% of non-syndromic JMMLs [28, 48].
Genotype-phenotype correlation of Noonan syndrome patients with or without PTPN11 mutations was studied by Zenker et al. in 57 unrelated patients [53]. PTPN11 mutations were significantly associated with the presence of pulmonary valve stenosis, short stature, easy bruising and thorax deformities. PTPN11-negative patients more frequently showed cardiomyopathy and less typical facial features. Most of these were patients without a family history of Noonan syndrome, which has hampered further genetic studies and the identification of additional Noonan syndrome-associated genes for some time.
In 2006, KRAS mutations were found in less than 2% of Noonan syndrome patients [6, 43, 54]. The phenotype associated with KRAS mutations seems to have a broad range varying from mild to severe Noonan syndrome with features of CFC syndrome and Costello syndrome. One patient with a JMML-like disorder has been described [43].
In contrast with HRAS mutations in Costello syndrome, overlap between germline KRAS mutations in Noonan syndrome and tumour specific mutations is more exception than rule. Only two mutations that were previously found in cancer cells have been described in Noonan syndrome patients (K5N, Q22R) [54]. In functional assays Noonan syndrome associated-mutations have less pronounced stimulating effects than known cancer associated mutations [43]. One hypothesis to explain these findings is that KRAS has important roles during embryogenesis, and therefore strong activating KRAS mutations, like the ones found in cancer, would not be tolerated in the germline and result in embryonic lethality [29]. This idea is reflected by the fact that mice with inactivated KRAS genes die in utero, whereas inactivation of HRAS or NRAS genes is not lethal in mice [23].
The quest for Noonan syndrome-associated genes very recently revealed another new player in the field; mutations in SOS1, an important GEF (activator of RAS proteins), account for 10–20% of Noonan syndrome patients [40, 49]. Phenotypically these patients often present with ectodermal abnormalities (dry scaly skin, absent eyebrows and eyelashes) (Fig. 2d), but development and growth are often normal.
LEOPARD syndrome (OMIM 151100)
LEOPARD syndrome is an acronym which refers to the combination of multiple Lentigines, ECG abnormalities, Ocular hypertelorism, Pulmonary valve stenosis, Abnormalities of the genitalia, Retardation of growth and sensorineural Deafness. It is inherited as an autosomal dominant disorder. Most typical features are multiple lentigines, a “Noonan-like” facial appearance and congenital cardiac abnormalities. Apart from pulmonic stenosis, also subvalvular aortic stenosis and progressive cardiomyopathy are found. Mental retardation is usually mild, if present. Specific heterozygous missense mutations in PTPN11 are found in these patients [11, 33].
Cardiofaciocutaneous syndrome (OMIM 115150)
Cardiofaciocutaneous (CFC) syndrome is a rare sporadic (non-familial) occurring disorder. Previously, much debate has arisen whether it is a separate condition or a severe variant of Noonan syndrome. Mutation analysis however did not reveal PTPN11 mutations in typical CFC syndrome, which points to the distinct aetiology of the syndrome [25]. Affected individuals often present with polyhydramnios during pregnancy and postnatal failure to thrive. Typical facial findings (Fig. 2e) are a high forehead with bitemporal constriction, downslanting palpebral fissures, a depressed nasal bridge and posteriorly rotated ears with prominent helices. Differential diagnosis with other NCFC syndromes like Costello syndrome and severe Noonan syndrome is mainly based on the typical ectodermal abnormalities: dry, hyperkeratotic, scaly skin and sparse, curly and friable scalp hair. Ulerythaema ophryogenes (absent eyebrows with hyperkeratosis) is a distinct feature. At an older age palmoplantar hyperkeratosis and lymphoedema may be observed. Psychomotor delay in these patients is moderate to severe. One or more cardiac abnormalities are found in 75% of individuals, with pulmonary valve stenosis in 45%, atrial septum defect (ASD) in 22% and some form of myocardial disease (most often hypertrophic cardiomyopathy) in 40% [39]. In contrast to the other NCFC syndromes, CFC syndrome is believed to have no increased malignancy risk. The observation however of acute lymphoblastoid leukaemia (ALL) in one patient with CFC syndrome [38] raises questions about this matter, although the possibility exists that this finding is due to a coincidence. BRAF mutations (Fig. 1) are found in 40% [38] to 78 [41]% of CFC patients. The most prevalent mutation (Q257R) is in the cysteine-rich domain (in exon 6) and another cluster of mutations is located in the protein kinase domain (mutations in exons 11, 12, 14 and 15). Most of the CFC associated-BRAF mutations are not found in cancer. Mutations in the proteins immediately downstream in the pathway, MEK1 and MEK2, have also been found in CFC individuals [41, 37]. KRAS mutations have been reported in four individuals with a clinical diagnosis of CFC syndrome [38, 43]. Phenotypically these patients had less typical skin abnormalities. Since evidence is emerging that Noonan syndrome is caused by mutations in KRAS and upstream signalling molecules, while CFC syndrome is caused by mutant downstream molecules of RAS, it seems reasonable to classify all individuals with germline KRAS mutations as having Noonan syndrome [42].
Conclusions
Recently, germline mutations in genes of the RAS-MAPK pathway have been discovered as the cause of the NCFC syndromes. Common features in these disorders are variable degrees of learning difficulties, facial dysmorphism, short stature, and tumour predisposition pointing to the importance of RAS-MAPK signalling in oncogenesis as well as in cognition, development and postnatal growth. Studies in other species show that these different functions of the RAS signalling pathway are evolutionary conserved, e.g. learning and memory are affected in mice [45] as well as in drosophila models [20] for NF1. NF1 also seems to be important for growth in Drosophila [51]. Inactivation of Nf1 in the murine central nervous system results in optic glioma formation [55]. Myeloproliferative disease develops in mice with inactivation of Nf1 in haematopoietic cells [30], as well as in mice heterozygous for the activating D61G PTPN11 mutation [2].
Still, a lot of questions do remain. How mutations in different components of the same signalling pathway result in different yet overlapping phenotypes is not fully understood. There are still many (especially Noonan syndrome) patients in whom no mutation in any of the known genes is found. It is very likely that other genes of the same pathway are mutated in these patients and in other NCFC syndromes. Undoubtedly more data on these issues will be discovered in the near future.
Abbreviations
- BRAF:
-
V-raf murine sarcoma viral oncogene homologue B1
- CFC syndrome:
-
Cardio-facio-cutaneous syndrome
- ERAS:
-
Embryonic stem cell-expressed rat sarcoma viral oncogene homologue
- ERK:
-
Extracellular signal-regulated kinase
- GAP:
-
GTP-ase activating protein
- GDP:
-
Guanosine diphosphate
- GEF:
-
Guanosine nucleotide exchange factor
- GTP:
-
Guanosine triphosphate
- HRAS:
-
v-Harvey rat sarcoma viral oncogene homologue
- JMML:
-
Juvenile myelomonocytic leukaemia
- KRAS:
-
Kirsten rat sarcoma viral oncogene homologue
- MAPK:
-
Mitogen activated protein kinase
- MEK1:
-
MAPK kinase/ERK kinase 1
- MEK2:
-
MAPK kinase/ERK kinase 2
- MPNST:
-
Malignant peripheral nerve sheet tumour
- NCFCsyndrome:
-
Neuro-cardio-facial-cutaneous syndrome
- NF1:
-
Neurofibromatosis type 1
- NRAS:
-
Neuroblastoma rat sarcoma viral oncogene homologue
- PTPN11:
-
Protein-tyrosine phosphatase, nonreceptor-type 11
- RAF:
-
Murine sarcoma viral oncogene homologue
- RAS:
-
Rat sarcoma viral oncogene homologue
- SHP-2:
-
Src homology protein 2
- SOS1:
-
Son of sevenless, drosophila, homologue 1
References
Aoki Y, Niihori T, Kawame H, Kurosawa K, Ohashi H, Tanaka Y, Filocamo M, Kato K, Suzuki Y, Kure S, Matsubara Y (2005) Germline mutations in HRAS proto-oncogene cause Costello syndrome. Nat Genet 37:1038–1040
Araki T, Mohi MG, Ismat FA, Bronson RT, Williams IR, Kutok JL, Yang W, Pao LI, Gilliland DG, Epstein JA, Neel BG (2004) Mouse model of Noonan syndrome reveals cell type- and gene dosage-dependent effects of Ptpn11 mutation. Nat Med 10:849–857
Bentires-Alj M, Kontaridis MI, Neel BG (2006) Stops along the RAS pathway in human genetic disease. Nat Med 12:283–285
Bos JL (1989) ras oncogenes in human cancer: a review. Cancer Res 49:4682–4689
Brady AF, Jamieson CR, van der Burgt I, Crosby A, van Reen M, Kremer H, Mariman E, Patton MA, Jeffery S (1997) Further delineation of the critical region for Noonan syndrome on the long arm of chromosome 12. Eur J Hum Genet 5:336–337
Carta C, Pantaleoni F, Bocchinfuso G, Stella L, Vasta I, Sarkozy A, Digilio C, Palleschi A, Pizzuti A, Grammatico P, Zampino G, Dallapiccola B, Gelb BD, Tartaglia M (2006) Germline missense mutations affecting KRAS isoform B are associated with a severe Noonan syndrome phenotype. Am J Hum Genet 79:129–135
Costello JM (1971) A new syndrome. NZ Med J 74:374
Costello JM (1977) A new syndrome: mental subnormality and nasal papillomata. Aust Paediatr J 13:114–118
De Luca A, Bottillo I, Sarkozy A, Carta C, Neri C, Bellacchio E, Schirinzi A, Conti E, Zampino G, Battaglia A, Majore S, Rinaldi MM, Carella M, Marino B, Pizzuti A, Digilio MC, Tartaglia M, Dallapiccola B (2005) NF1 gene mutations represent the major molecular event underlying neurofibromatosis-Noonan syndrome. Am J Hum Genet 77:1092–1101
Der CJ, Krontiris TG, Cooper GM (1982) Transforming genes of human bladder and lung carcinoma cell lines are homologous to the ras genes of Harvey and Kirsten sarcoma viruses. Proc Natl Acad Sci U S A 79:3637–3640
Digilio MC, Conti E, Sarkozy A, Mingarelli R, Dottorini T, Marino B, Pizzuti A, Dallapiccola B (2002) Grouping of multiple-lentigines/LEOPARD and Noonan syndromes on the PTPN11 gene. Am J Hum Genet 71:389–394
Donovan S, Shannon KM, Bollag G (2002) GTPase activating proteins: critical regulators of intracellular signaling. Biochim Biophys Acta 1602:23–45
Eerola I, Boon LM, Mulliken JB, Burrows PE, Dompmartin A, Watanabe S, Vanwijck R, Vikkula M (2003) Capillary malformation-arteriovenous malformation, a new clinical and genetic disorder caused by RASA1 mutations. Am J Hum Genet 73:1240–1249
Estep AL, Tidyman WE, Teitell MA, Cotter PD, Rauen KA (2006) HRAS mutations in Costello syndrome: detection of constitutional activating mutations in codon 12 and 13 and loss of wild-type allele in malignancy. Am J Med Genet A 140:8–16
Garnett MJ, Marais R (2004) Guilty as charged: B-RAF is a human oncogene. Cancer Cell 6:313–319
Gripp KW (2005) Tumor predisposition in Costello syndrome. Am J Med Genet C Semin Med Genet 137:72–77
Gripp KW, Lin AE, Stabley DL, Nicholson L, Scott CI Jr, Doyle D, Aoki Y, Matsubara Y, Zackai EH, Lapunzina P, Gonzalez-Meneses A, Holbrook J, Agresta CA, Gonzalez IL, Sol-Church K (2006) HRAS mutation analysis in Costello syndrome: genotype and phenotype correlation. Am J Med Genet A 140:1–7
Gripp KW, Scott CI Jr, Nicholson L, McDonald-McGinn DM, Ozeran JD, Jones MC, Lin AE, Zackai EH (2002) Five additional Costello syndrome patients with rhabdomyosarcoma: proposal for a tumor screening protocol. Am J Med Genet 108:80–87
Gripp KW, Stabley DL, Nicholson L, Hoffman JD, Sol-Church K (2006) Somatic mosaicism for an HRAS mutation causes Costello syndrome. Am J Med Genet A 140:2163–2169
Guo HF, Tong J, Hannan F, Luo L, Zhong Y (2000) A neurofibromatosis-1-regulated pathway is required for learning in Drosophila. Nature 403:895–898
Hennekam RC (2003) Costello syndrome: an overview. Am J Med Genet C Semin Med Genet 117:42–48
Jamieson CR, van der Burgt I, Brady AF, van Reen M, Elsawi MM, Hol F, Jeffery S, Patton MA, Mariman E (1994) Mapping a gene for Noonan syndrome to the long arm of chromosome 12. Nat Genet 8:357–360
Johnson L, Greenbaum D, Cichowski K, Mercer K, Murphy E, Schmitt E, Bronson RT, Umanoff H, Edelmann W, Kucherlapati R, Jacks T (1997) K-ras is an essential gene in the mouse with partial functional overlap with N-ras. Genes Dev 11:2468–2481
Kalra R, Paderanga DC, Olson K, Shannon KM (1994) Genetic analysis is consistent with the hypothesis that NF1 limits myeloid cell growth through p21ras. Blood 84:3435–3439
Kavamura MI, Pomponi MG, Zollino M, Lecce R, Murdolo M, Brunoni D, Alchorne MM, Opitz JM, Neri G (2003) PTPN11 mutations are not responsible for the Cardiofaciocutaneous (CFC) syndrome. Eur J Hum Genet 11:64–68
Kerr B, Delrue MA, Sigaudy S, Perveen R, Marche M, Burgelin I, Stef M, Tang B, Eden OB, O’Sullivan J, Sandre-Giovannoli A, Reardon W, Brewer C, Bennett C, Quarell O, M’Cann E, Donnai D, Stewart F, Hennekam R, Cave H, Verloes A, Philip N, Lacombe D, Levy N, Arveiler B, Black G (2006) Genotype-phenotype correlation in Costello syndrome: HRAS mutation analysis in 43 cases. J Med Genet 43:401–405
Kolch W (2005) Coordinating ERK/MAPK signalling through scaffolds and inhibitors. Nat Rev Mol Cell Biol 6:827–837
Kratz CP, Niemeyer CM, Castleberry RP, Cetin M, Bergstrasser E, Emanuel PD, Hasle H, Kardos G, Klein C, Kojima S, Stary J, Trebo M, Zecca M, Gelb BD, Tartaglia M, Loh ML (2005) The mutational spectrum of PTPN11 in juvenile myelomonocytic leukemia and Noonan syndrome/myeloproliferative disease. Blood 106:2183–2185
Kratz CP, Schubbert S, Bollag G, Niemeyer CM, Shannon KM, Zenker M (2006) Germline mutations in components of the ras signaling pathway in noonan syndrome and related disorders. Cell Cycle 5:1607–1611
Le DT, Kong N, Zhu Y, Lauchle JO, Aiyigari A, Braun BS, Wang E, Kogan SC, Le Beau MM, Parada L, Shannon KM (2004) Somatic inactivation of Nf1 in hematopoietic cells results in a progressive myeloproliferative disorder. Blood 103:4243–4250
Legius E, Marchuk DA, Collins FS, Glover TW (1993) Somatic deletion of the neurofibromatosis type 1 gene in a neurofibrosarcoma supports a tumour suppressor gene hypothesis. Nat Genet 3:122–126
Legius E, Schollen E, Matthijs G, Fryns JP (1998) Fine mapping of Noonan/cardio-facio cutaneous syndrome in a large family. Eur J Hum Genet 6:32–37
Legius E, Schrander-Stumpel C, Schollen E, Pulles-Heintzberger C, Gewillig M, Fryns JP (2002) PTPN11 mutations in LEOPARD syndrome. J Med Genet 39:571–574
Lin AE, Grossfeld PD, Hamilton RM, Smoot L, Gripp KW, Proud V, Weksberg R, Wheeler P, Picker J, Irons M, Zackai E, Marino B, Scott CI Jr, Nicholson L (2002) Further delineation of cardiac abnormalities in Costello syndrome. Am J Med Genet 111:115–129
Maertens O, Brems H, Vandesompele J, De Raedt T, Heyns I, Rosenbaum T, De Schepper S, De Paepe A, Mortier G, Janssens S, Speleman F, Legius E, Messiaen L (2006) Comprehensive NF1 screening on cultured Schwann cells from neurofibromas. Hum Mutat 27:1030–1040
Maertens O, Prenen H, Debiec-Rychter M, Wozniak A, Sciot R, Pauwels P, De Wever I, Vermeesch JR, De Raedt T, De Paepe A, Speleman F, van Oosterom A, Messiaen L, Legius E (2006) Molecular pathogenesis of multiple gastrointestinal stromal tumors in NF1 patients. Hum Mol Genet 15:1015–1023
Narumi Y, Aoki Y, Niihori T, Neri G, Cave H, Verloes A, Nava C, Kavamura MI, Okamoto N, Kurosawa K, Hennekam RC, Wilson LC, Gillessen-Kaesbach G, Wieczorek D, Lapunzina P, Ohashi H, Makita Y, Kondo I, Tsuchiya S, Ito E, Sameshima K, Kato K, Kure S, Matsubara Y (2007) Molecular and clinical characterization of cardio-facio-cutaneous (CFC) syndrome: overlapping clinical manifestations with Costello syndrome. Am J Med Genet A 143:799–807
Niihori T, Aoki Y, Narumi Y, Neri G, Cave H, Verloes A, Okamoto N, Hennekam RC, Gillessen-Kaesbach G, Wieczorek D, Kavamura MI, Kurosawa K, Ohashi H, Wilson L, Heron D, Bonneau D, Corona G, Kaname T, Naritomi K, Baumann C, Matsumoto N, Kato K, Kure S, Matsubara Y (2006) Germline KRAS and BRAF mutations in cardio-facio-cutaneous syndrome. Nat Genet 38:294–296
Roberts A, Allanson J, Jadico SK, Kavamura MI, Noonan J, Opitz JM, Young T, Neri G (2006) The cardiofaciocutaneous syndrome. J Med Genet 43:833–842
Roberts AE, Araki T, Swanson KD, Montgomery KT, Schiripo TA, Joshi VA, Li L, Yassin Y, Tamburino AM, Neel BG, Kucherlapati RS (2007) Germline gain-of-function mutations in SOS1 cause Noonan syndrome. Nat Genet 39:70–74
Rodriguez-Viciana P, Tetsu O, Tidyman WE, Estep AL, Conger BA, Cruz MS, McCormick F, Rauen KA (2006) Germline mutations in genes within the MAPK pathway cause cardio-facio-cutaneous syndrome. Science 311:1287–1290
Schubbert S, Bollag G, Shannon K (2007) Deregulated ras signaling in developmental disorders: new tricks for an old dog. Curr Opin Genet Dev 17:15–22
Schubbert S, Zenker M, Rowe SL, Boll S, Klein C, Bollag G, van der Burgt I, Musante L, Kalscheuer V, Wehner LE, Nguyen H, West B, Zhang KY, Sistermans E, Rauch A, Niemeyer CM, Shannon K, Kratz CP (2006) Germline KRAS mutations cause Noonan syndrome. Nat Genet 38:331–336
Shannon KM, O’Connell P, Martin GA, Paderanga D, Olson K, Dinndorf P, McCormick F (1994) Loss of the normal NF1 allele from the bone marrow of children with type 1 neurofibromatosis and malignant myeloid disorders. N Engl J Med 330:597–601
Silva AJ, Frankland PW, Marowitz Z, Friedman E, Laszlo GS, Cioffi D, Jacks T, Bourtchuladze R (1997) A mouse model for the learning and memory deficits associated with neurofibromatosis type I. Nat Genet 15:281–284
Tartaglia M, Martinelli S, Stella L, Bocchinfuso G, Flex E, Cordeddu V, Zampino G, Burgt I, Palleschi A, Petrucci TC, Sorcini M, Schoch C, Foa R, Emanuel PD, Gelb BD (2006) Diversity and functional consequences of germline and somatic PTPN11 mutations in human disease. Am J Hum Genet 78:279–290
Tartaglia M, Mehler EL, Goldberg R, Zampino G, Brunner HG, Kremer H, van der Burgt I, Crosby AH, Ion A, Jeffery S, Kalidas K, Patton MA, Kucherlapati RS, Gelb BD (2001) Mutations in PTPN11, encoding the protein tyrosine phosphatase SHP-2, cause Noonan syndrome. Nat Genet 29:465–468
Tartaglia M, Niemeyer CM, Fragale A, Song X, Buechner J, Jung A, Hahlen K, Hasle H, Licht JD, Gelb BD (2003) Somatic mutations in PTPN11 in juvenile myelomonocytic leukemia, myelodysplastic syndromes and acute myeloid leukemia. Nat Genet 34:148–150
Tartaglia M, Pennacchio LA, Zhao C, Yadav KK, Fodale V, Sarkozy A, Pandit B, Oishi K, Martinelli S, Schackwitz W, Ustaszewska A, Martin J, Bristow J, Carta C, Lepri F, Neri C, Vasta I, Gibson K, Curry CJ, Siguero JP, Digilio MC, Zampino G, Dallapiccola B, Bar-Sagi D, Gelb BD (2007) Gain-of-function SOS1 mutations cause a distinctive form of Noonan syndrome. Nat Genet 39:75–79
Vetter IR, Wittinghofer A (2001) The guanine nucleotide-binding switch in three dimensions. Science 294:1299–1304
Walker JA, Tchoudakova AV, McKenney PT, Brill S, Wu D, Cowley GS, Hariharan IK, Bernards A (2006) Reduced growth of Drosophila neurofibromatosis 1 mutants reflects a non-cell-autonomous requirement for GTPase-activating protein activity in larval neurons. Genes Dev 20:3311–3323
Zampino G, Pantaleoni F, Carta C, Cobellis G, Vasta I, Neri C, Pogna EA, De Feo E, Delogu A, Sarkozy A, Atzeri F, Selicorni A, Rauen KA, Cytrynbaum CS, Weksberg R, Dallapiccola B, Ballabio A, Gelb BD, Neri G, Tartaglia M (2007) Diversity, parental germline origin, and phenotypic spectrum of de novo HRAS missense changes in Costello syndrome. Hum Mutat 28:265–272
Zenker M, Buheitel G, Rauch R, Koenig R, Bosse K, Kress W, Tietze HU, Doerr HG, Hofbeck M, Singer H, Reis A, Rauch A (2004) Genotype-phenotype correlations in Noonan syndrome. J Pediatr 144:368–374
Zenker M, Lehmann K, Schulz AL, Barth H, Hansmann D, Koenig R, Korinthenberg R, Kreiss-Nachtsheim M, Meinecke P, Morlot S, Mundlos S, Quante AS, Raskin S, Schnabel D, Wehner LE, Kratz CP, Horn D, Kutsche K (2007) Expansion of the genotypic and phenotypic spectrum in patients with KRAS germline mutations. J Med Genet 44:131–135
Zhu Y, Harada T, Liu L, Lush ME, Guignard F, Harada C, Burns DK, Bajenaru ML, Gutmann DH, Parada LF (2005) Inactivation of NF1 in CNS causes increased glial progenitor proliferation and optic glioma formation. Development 132:5577–5588
Acknowledgements
ED is a predoctoral researcher of the Fonds voor Wetenschappelijk Onderzoek-Vlaanderen and EL is a part-time clinical researcher of the Fonds voor Wetenschappelijk Onderzoek Vlaanderen (FWO-Vlaanderen). This work is supported by a Concerted Action Grant from the KU Leuven.
Author information
Authors and Affiliations
Corresponding author
Additional information
Human genes and proteins are written in capital letters; murine genes and proteins in small letters. Genes are written in italic, proteins are written straight.
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License ( https://creativecommons.org/licenses/by-nc/2.0 ), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Denayer, E., Legius, E. What’s new in the neuro-cardio-facial-cutaneous syndromes?. Eur J Pediatr 166, 1091–1098 (2007). https://doi.org/10.1007/s00431-007-0535-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00431-007-0535-7