
Abstract
The area of plant and animal genomics covers the entire suite of issues in biology because it aims to determine the structure and function of genetic material. Although specific issues define research advances at an organism level, it is evident that many of the fundamental features of genome structure and the translation of encoded information to function share common ground. The Plant and Animal Genome (PAG) conference held in San Diego (California), in January each year provides an overview across all organisms at the genome level, and often it is evident that investments in the human area provide leadership, applications, and discoveries for researchers studying other organisms. This mini-review utilizes the plenary lectures as a basis for summarizing the trends in the genome-level studies of organisms, and the lectures include presentations by Ewan Birney (EBI, UK), Eric Green (NIH, USA), John Butler (NIST, USA), Elaine Mardis (Washington, USA), Caroline Dean (John Innes Centre, UK), Trudy Mackay (NC State University, USA), Sue Wessler (UC Riverside, USA), and Patrick Wincker (Genoscope, France). The work reviewed is based on published papers. Where unpublished information is cited, permission to include the information in this manuscript was obtained from the presenters.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
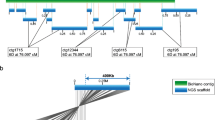
The analysis of genomes has been accelerated by advances in new technologies, such as the high-throughput sequencing of whole genomes, which provides an extensive view of the gene space of organisms. Large-scale sequence-based comparative studies have defined conserved and variable (conditionally dispensable, reviewed in Appels et al. 2013) regions of the genome as well as single nucleotide polymorphisms (SNPs) for molecular markers. The rapid development of DNA sequencing technologies over the last 5 years has meant that large genomes previously out of reach can be subjected to sequencing at a relatively low cost. However, cost-effective and high-throughput techniques for obtaining long-range information about the DNA sequence are still not widely deployed (Selvaraj et al. 2013). Mate–pair libraries can typically be taken up to 20 kb, with 40 and 150 kb inserts possible with fosmid and bacterial artificial chromosome (BAC) clones, respectively. Although physical mapping and assembly of BAC clones still form the backbone of most reference-level assemblies, ordering and orientation of the physical contigs within the genome as a whole remain a challenge (Selvaraj et al. 2013; Burton et al. 2013). The need for long-range mapping of sequences in complex genomes arises because of the need to deal with the ambiguities generated by the presence of extensive tracts of retro-transposable elements. In the genomes of plants such as wheat, the long-range mapping of BAC libraries using KeyGene (van Oeveren et al. 2011; Bayer Crop Science 2013) and BioNano (Lam et al. 2012; Hastie et al. 2013) technologies is providing the basis for more accurate sequence assemblies. The BioNano technology has also been applied to genomic DNA from flow-sorted chromosome arms of wheat 7D (short arm), and it is evident (Fig. 1) that DNA molecules up to and exceeding 500 kbp can be fingerprinted using this approach.

The BioNano technology builds on the earlier optical mapping technologies (Lin et al. 2012) by loading the DNA into nanochannels so that the DNA can be easily scanned to provide DNA fingerprints of molecules at least 500 kb or more in length (bar at the top right is 100 kb). The DNA map is compiled using two nicking enzymes, Nt.BbvCI and Nt.BspQI, followed by labeling the nick motifs (using DNA polymerase) with red and green dyes, respectively. The image, with labeling at the Nt.BspQ1 sites, is unpublished and was kindly provided by H. Van Steenhouse (BioNano, San Diego) based on DNA from the short of wheat chromosome 7D provided by H. Simkova and J. Dolezel (Czech Republic)
High-resolution genetic maps are now also providing genuine contributions to the assembly of complex genomes. In gene-rich regions, the whole genome sequencing of segregating progeny from a cross between two individuals (Mascher et al. 2013) provides thousands of SNPs for the generation of genetic maps with markers spaced less than 10 kb apart (provided the number of segregating individuals analyzed is sufficiently large, Cavanagh et al. 2013; Wang et al. 2014). Even where SNP markers co-segregate, the respective genome sequence contigs that are marked by the SNPs can then be reexamined with the additional knowledge that they are physically close together. In regions where genetic recombination is low (near the centromere), radiation mapping (Feuillet et al. 2012) has provided data contributing to the genome assembly pipeline. The assembly of complex genomes thus integrates diverse sources of data and requires information structure that moves well beyond the requirements of whole genome sequencing per se in the assembly of small genomes. As noted by Burton et al. (2013), these developments for complex genomes follow the model established by the Human Genome Project which integrated a diversity of approaches to achieve the long-range contiguity currently available in the reference genome. The establishment of a good reference genome in model systems, and for the human genome, has provided the foundation for many of the studies reported in this mini-review.
Translating genome and post-genome information to society and industry
Genomic features that were discovered very early and used in diagnostic assays focused on repetitive sequence regions (Appels et al. 1978). The regions appeared to be species-specific in crop plants, and in human forensics, the regions provided the high levels of polymorphism required for distinguishing between members of a family (Jeffreys et al. 1985). The finding of a 33-bp sequence that was repeated in an intron of the myoglobin gene was flanked by a 9-bp direct repeat similar to target site duplications created by transposable elements (Jeffreys et al. 1985) led to the finding that these repeated regions are more widespread in the human genome. Although the direct sequence repeats flanking the short repetitive sequences (mini-satellites) proved not to be a general feature, the mini-satellites are widespread and share a core, conserved, sequence that can be readily assayed. The high levels of recombination at these repetitive sequence loci lead to unequal crossing-over and hence to variation in repeat numbers and levels of polymorphisms that far exceed variation due to mutation alone. In his plenary lecture, John Butler noted that the mini-satellite marker systems have stood the test of time and the high levels of polymorphisms continue to contribute to the DNA profile database that assays a set of 15 simple tandem repeats (STRs). Eight of these STRs overlap with those used in Europe. The database of 13 million DNA profiles (http://www.fbi.gov/anout-us/lab/biometric-analysis) is deployed in crime scene investigations, accident victim and soldier identification, paternal testing, immigration, and missing person investigations as well as providing a reference point for identifying convicted felons (Biesecker et al. 2005).
John Butler noted that although new genome sequence information can provide additional markers for diagnostics (Hares 2012), the value of the current suite of markers is in the legacy information and the ability to relate any new developments to the established suite of markers. In special situations, specific SNPs for distinguishing between identical twins, eye color (Yun et al. 2014), or a new suite of STRs to identify Y chromosomes have been established (PowerPlex Y23, Coble et al. 2013). The importance of multi-allelic markers was the key feature for markers in forensic science, and this has been expanded to plants (Nybom et al. 2014) and domesticated animals (http://www.nfstc.org/pdi/Subject09/pdi_s09_m01_04_a.htm).
In her plenary lecture, Elaine Mardis focused on the cancer genome and the design of diagnostics and vaccines for clinical use (Mardis 2010). The Cancer Genome Atlas (TCGA) network of groups collaborates to compile the molecular details of large numbers of human tumors (TCGA et al. 2013), aiming to have 10,000 specimens analyzed from 25 different tumor types by 2015. The TCGA Pan-Cancer analysis focuses on the details of tumor types at the subtumor/single-cell level in order to define the different consequences of changes in the expression of similar genes and build a gene network-based view of the cancer phenotype. The across-tumor comparative analyses are argued to provide a valuable path into understanding the genetic and epigenetic backgrounds of cancer.
Classical studies on cancers deployed whole chromosome analyses to characterize translocations. In some instances such as chronic myelogenous leukemia (CML), the gene fusion generated as a result of the breakage–rejoining event underpinning the translocation formed a new oncogenic BCR-ABL gene fusion (Melo 1996). Whole genome sequencing provides greater sensitivity for defining translocation events (Chen et al. 2013), and this was illustrated by Welch et al. (2012) in their analysis of a case of acute promyelocytic leukemia (AML) where no chromosomal abnormalities were cytologically visible. The genome-level analysis indicated that a 77-kb segment from chromosome 15 was translocated into the second intron of the RARA gene to create a new fusion gene that was expressed in the leukemia. Importantly, the study allowed new molecular probes to be designed to assay events of this type in other patients. Analysis of AML more widely has also shown that heterogeneity in tumors can provide the basis for relapse due to clonal selection of chemotherapy-resistant cell types resulting from new mutations (Ley et al. 2008; Ding et al. 2012). Remissions or poor response to radiation treatment in acute lymphoblastic leukemia (ALL) can also reflect modified double-strand break repair pathways (Marston et al. 2009). Transcriptome-based studies have provided molecular markers to identify possible pathways that should be inhibited in order to increase the efficacy of radiation treatment (Marston et al. 2009).
Glioblastoma (GBM) was the first cancer type to be systematically studied by TCGA (Brennan et al. 2013). The analysis of the cancer pathways in GBM has been facilitated by whole-exome and transcriptome sequencing and indicated that in 85.3 % of the tumors, the regulation of the p53 pathway was disrupted through mutation/deletion of TP53 (27.9 %), amplification of MDM1/2/4 (15.1 %), and/or deletion of CDKN2A (57.8 %). The depth of analysis also included proteome-level studies and indicated that the impact of genome change on the proteome is not always consistent with expectations and had significant implications for the clinical applications of the knowledge base. The analyses by Brennan et al. (2013) also highlighted the importance of having several time points available in an analysis in order to map the changes in the genome and proteome of different cell types comprising the tumor as it develops over time. A database which complements the outputs from TCGA is DGIdb (Griffith et al. 2013; http://dgidb.org/) and provides the capability of screening gene translations against drug–gene product interactions in order to translate research findings into clinical outputs. The database allows genes of interest to be grouped with genes for which drugs have already been developed. Another important output from proteome-level information is that it provides the capacity to identify unusual antigens that are unique to cells throughout the tumor and bind to MHC molecules. The sequences of these antigens can then be used to design peptides for raising vaccines that target specific tumors (Waldmann 2003; Restifo et al. 2012a, b).
Dynamics of genome change and the distribution of variation
The plenary lecture by Sue Wessler examined the changes to the genome as a result of transposable elements (TEs) with a particular focus on a 430-bp miniature inverted repeat transposable elements (MITEs) called mPing. The mPing elements are a class 2 element (Wicker et al. 2007; Han et al. 2013) in the PIF/Harbinger superfamily, and in some rice cultivars, the element has reached high copy numbers (Naito et al. 2006, 2009). The Ping element contains the genes required for transposition (ORF1 and a Transposase, TPASE; Naito et al. 2009), and while these have been deleted in the mPing element, the genes are still required for mPing transposition. The mPing element was reported to undergo bursts of amplification and, based on the analysis of over 1,700 insertion sites in rice, was preferentially located within 1 kb upstream in the 5′ region of transcription start sites or within 1 kb downstream from the 3′ stop site (Naito et al. 2009). This preferential location close to genes was also found for other MITE families in Brachypodium, sorghum, and maize (Han et al. 2013). In transgenic experiments, the transposition of mPing in Arabidopsis utilized transposases from either autonomous Ping or Pong elements, or the cDNA from a Ping transcript (Yang et al. 2007). Most mPing excision sites are repaired accurately in Arabidopsis (Yang et al. 2007).
The preferential insertion of mPing elements into the promoters of rice rather than exons means that their effects on gene expression may be more subtle in modifying gene expression and changing network interactions. The basis for the preferential insertion of mPing elements was investigated further by introducing mPing elements (plus a Ping cDNA containing the ORF1 and TPase coding regions) into soybean (Hancock et al. 2011). The analysis of 72 insertion sites and the determination of their location within 5 kb of a gene annotated in the soybean genome sequence indicated that in rice, the preferential insertion observations were the result of an avoidance of exon insertion. The analysis of the soybean data indicated that mPing did not avoid exon insertions. The mPing element prefers to insert into a 9-bp sequence that is AT-rich, and thus, because rice exons are GC-rich, the avoidance of exon insertions is more obvious than in soybean where the exons are less GC-rich. This means that the avoidance of rice exons can be attributed to their high GC content and probably contributes to the ability of this element to attain high copy numbers despite a preference for inserting near genes. It has been speculated that this preference for insertion near genes may reflect interactions between the transposition machinery and some features of chromatin structure such as compactness/density of nucleosomes (Hancock et al. 2011).
The varieties of rice that contain the most actively transposing element (Naito et al. 2006) are evidently the result of independent and ongoing bursts of mPing amplification for over at least a century. In terms of the dynamics of genome structure, research is ongoing to study how TEs with a preference for inserting near genes can attain high copy number (hundreds, thousands of elements) without killing their host and how these TEs accumulate without being detected by host genome surveillance.
Relating variation in the genome and gene expression to observed phenotypes in populations of Drosophila was addressed by Trudy Mackay in her plenary lecture. A key resource, the Drosophila melanogaster reference panel (DGRP) provided a template for genotype–phenotype mapping (Mackay et al. 2012), and the presentation gave an update on DGRP activity with non-SNP variants being called and genome-prediction methodologies being improved. The panel comprised 192 inbred strains derived by utilizing mated females from an outbred population plus 20 generations of full-sibling inbreeding. The DGRP thus contains a representative sample of genetic variation and, based on the resequencing of 168 lines, has a high-resolution genetic map for locating phenotypic variation in resistance to starvation stress, chill coma recovery time, and startle response.
The resource provided 2,490,165 SNPs and 77,756 microsatellites (minor allele present in at least four lines) for associating chromosome regions to phenotypes of interest. As a proof of concept, the genes Sema-1a and Eip75B were found to be associated with startle response, and pnt with starvation resistance, consistent with previous studies. Against this background, the suite of new candidate genes identified has formed targets for new research (Mackay et al. 2012).
The outputs from the DGRP analysis have been compared to studies carried out on an advanced intercross population derived from 40 DGRP lines randomly mated as a large population size for over 70 generations (Flyland, Huang et al. 2012). The Flyland population was analyzed using extreme pools for starvation resistance, startle response, and chill coma recovery, and among the segregating SNPs in the DGRP, 1,339,448; 1,605,264; and 1,406,458 were segregating in the respective pools from the Flyland population. It was striking that none of the SNP associations in the Flyland population proved to be significant in DGRP (Huang et al. 2012), and further analysis showed that this was due to widespread epistatic interactions affecting all three traits (Mackay 2014), which could be identified because of differences in allele frequency between the DGRP and Flyland populations.
Mutations due to P elements (transposable elements in Drosophila; Magwire et al. 2010) and inversions (Corbett-Detig and Hartl 2012; Mackay et al. 2012) have been characterized in the DGRP as well as in other populations. As many as 40 % of the 1,332 P elements assessed affected traits, one third affecting life span, and among these, 58 were associated with increases in life spans (Magwire et al. 2010). The inversions corresponded to regions of increased diversity (Corbett-Detig and Hartl 2012; Mackay et al. 2012) although in some instances, inversions of 1.7 Mb showed no polymorphisms, probably because they have arisen relatively recently (Corbett-Detig and Hartl 2012). Inversions tended not to disrupt gene sequences. Where the ends of the inversion do interrupt gene/transcribed sequences, it is correlated with the finding that these inversions have short inverted duplications at their ends. It is evident that inversions and transposable elements are a significant source of variation in chromosome structure within a species. The inversion-level structural variation in the genome has a more immediate significance within an organism such as Anopheles gambiae, where the variation due to inversions is considered to be a basis for the success of the mosquito adapting to regions in North Africa (Lee et al. 2013).
The quantitative regulation of gene expression with a focus on individual cells in a tissue was studied at a specific locus in Arabidopsis by Caroline Dean in her plenary lecture. The locus for the primary control of flowering in Arabidopsis (and other flowering plants) is Flowering Locus C (FLC) and codes for a MADS-box domain protein that represses flowering (Song et al. 2013). Inactivation of the repressor by a prolonged period of low temperature (vernalization) allows flowering to occur when the environmental temperature increases. For example, if only 2 weeks of cold was experienced, flowering is later than if 4 weeks of cold occurred (Song et al. 2013). This quantitative response was shown to result from individual cells in the flowering meristematic tissue switching between epistatic states and that it was the proportion of cells that were switched that gave the overall quantitative response.
The mechanism for establishing the level of FLC expression characteristic for a variety adapted to a given environment is based on the conserved chromatin modifiers (reviewed in Crevillén and Dean 2011). The recruitment of the chromatin modifiers to the promoter scaffold of the FLC locus is enhanced by the FRIGIDA protein complex (FRI; Choi et al. 2009) by directly interacting with the nuclear cap-binding complex required for RNA transport/stability. Variation in FRI has been associated with adaption to different environments, and this correlates with FRI influencing the proportion of FLC RNA with a 5′ cap (Geraldo et al. 2009). The chromatin structure at the FLC locus is such that the 5′- and 3′-flanking regions of the gene form a chromatin loop (Crevillén et al. 2013) as part of the active locus conformation. In order to silence the FLC locus, the influence of the chromatin modifiers is reversed by reducing H3 histone methylation at lysine 4 and 36 and increasing methylation at lysine 27. These changes in methylation are carried out in combination with the alternative splicing and polyadenylation of antisense transcripts (COOLAIR) downstream from the polA site and within the chromatin loop (Swiezewski et al. 2007, 2009; Angel et al. 2011; Liu et al. 2007; Crevillén et al. 2013). A key variable in the level of expression of FLC is the level of methylation at H3 histone lysine 27.
At the level of individual cells in the flowering meristem tissue, it is evident that the switch between FLC on or off is not a quantitative effect. Instead, it is the proportion of cells that are switched on which defines the overall response of the meristem to the start of flowering (Rosa et al. 2013). The importance of RNA-based regulation of gene expression, at a more general level, was emphasized by Caroline Dean since 50 % of annotated transcripts have low levels of antisense transcripts which could function in a way that is analogous to the COOLAIR-based modification of chromatin structure at the FLC locus. The sensing mechanisms for cold are still under investigation and are likely to comprise a number of different networks within the plant (Miura and Furumoto 2013).
Integrating large datasets
The requirement for information structure in biology was discussed by Ewan Birney in his plenary lecture and emphasized the importance of integrating diverse datasets as well as the interface between life sciences and industry/environment. His argument was based on cost-effectiveness particularly with respect to the development of diagnostics in health care and agriculture as well as marker technology associated with complex traits in the characterization and breeding of domesticated animals and plants. Information structure can lead to cost-effectiveness both through reduced hardware requirements and through a reduced need for manual labor in data analysis.
The long-term storage of biological datasets has raised questions about efficient algorithms, formats, and methods for data entry, curation, and retrieval (Church et al. 2012; Goldman et al. 2013). Generally, there exists a need to retain large datasets over extended periods of time and data compression formats have been investigated (Fritz et al. 2011). Compression formats can be grouped into lossless formats, from which a perfect copy may be restored, and lossy formats, which achieve better compression at the expense of the ability to restore the original in full detail. The bulk of high-throughput sequencing data, for example, is made up of sequencing reads and the corresponding quality scores, and in the case of lossy compression, it is often the latter that are sacrificed to some degree. In many cases, this has been shown to not compromise the usefulness of the data for subsequent downstream analysis (Popitsch and von Haeseler 2013). Data compression has been studied extensively in the field of computer science; however, in bioinformatics, the challenges are novel because extra assumptions can be made about the data, particularly in using the redundant nature of DNA information. DNA sequences have only four bases, and large sequencing datasets are often highly redundant and compression can be achieved by encoding only their difference when compared to a reference (Fritz et al. 2011).
Widespread access to larger storage capacity (for example ELIXIR, http://www.elixir-europe.org/, with a capacity of 35 PB and transfer speed of 40 GB/s) for a variety of data that needs to be publically available, shared between research groups, and integrated across different projects has made the links between such databases increasingly important. Metadata or “data describing the data” provides the information necessary for relating datasets to each other, to enable the respective dataset to be positioned within a particular research project (Rocca-Serra et al. 2010). The quality of the metadata can determine the possible ways a research project can utilize the data without direct access to the dataset’s original producers. It is now common for large, curated data repositories to standardize the way information is accessed with web services and query languages. The emerging standard for biological experimental metadata is the investigation, study, and assay (ISA) framework (http://www.isacommons.org). The generality and flexibility of tools such as the ISA framework makes it feasible to deal with the dual nature of data in terms of distinguishing between data and metadata. For example, much of the UniProt protein database is an accumulation of experimental data per se, and from this perspective, it is data. However, experimental data being released from other projects can be annotated with the relevant UniProt identifiers in order to provide a context for understanding and integrating new data, and from this perspective, the UniProt information becomes metadata. Thus, the data/metadata distinction is often relative to the main focus of a given experiment.
At the simplest level, two databases can be related to each other when they both reference a common set of biological variables. Some standards have emerged as de facto backbones for reference-based integration. For example, REACTOME cross-references UniProt, ChEBI, and Gene Ontology (GO); the PRIDE database cross-references UniProt and many other protein databases; and the Expression Atlas cross-references Ensembl genes, UniProt, and GO. Therefore, at the most basic level, a biology analyst can manually integrate data from several databases. However, for more complex integration analyses, special-purpose query languages are required to make it more straightforward to carry out the integration process. One approach that has been gaining momentum is the use of linked data/semantic web technologies such as Resource Description Format (RDF), which is often used together with the query language SPARQL. A key benefit of these formats is their open-ended nature and the fact that they are fundamentally designed for meaningful integration of heterogeneous data. Well-established databases providing their data as RDF include GO (http://www.geneontology.org), UniProt (http://beta.spargl.uniprot.org), and Bio2RDF (http://www.bio2rdf.org). In late 2013, EMBL-EBI embraced the RDF format in the EMBL RDF platform (http://www.ebi.ac.uk/rdf/). A closely related trend is the increasing support for programmatic (API) access to integrated databases. One example is the Proteomics Standard Initiative Common QUery InterfaCe (PSICQUIC), developed by the Human Proteomics Organization Proteomics Standards Initiative (HUPO-PSI) as a standard interface to molecular interaction data. This standard is specific to molecular interaction data, unlike RDF, which is fully general.
One of the challenges of effective metadata use is ensuring that related fields are comparable between experiments. For example, precisely defining the tissue type of a biological sample is nontrivial and definitions may differ between countries. The use of controlled ontologies solves this problem and provides a way for domain experts to describe their knowledge in a way that can be applied automatically for the purpose of integrating datasets. The GO is perhaps the most well known; other examples include the Plant Ontology (PO) and the Environmental Ontology (EnvO). The OWL framework, which is an extension built on top of RDF, has become one of the de facto standards for ontology development and use. The Open Biomedical Ontology (OBO) project has played a leading role in advancing the development of ontologies (http://www.obofoundry.org).
Procedures for data release are now starting to reflect the importance of data structure as exemplified by the open journal GigaScience (http://www.gigasciencejournal.com/), closely affiliated with BGI-Shenzhen (Ling 2013). GigaScience has a special category of paper, the “Data note,” which allows for the release of significant datasets. This ties in with the overall goals of DataCite (http://www.datacite.org), an international consortium of organizations aiming to support the use (and re-use) of public scientific data and to promote data as a research output in and of itself (Brase et al. 2009). It has been argued that publications for which data is made publicly available have a higher citation rate on average (Piwowar and Vision 2013). Well-established journals such as Nature are beginning to adopt data-publishing platforms as a core element of the publishing process. Importantly, digital object identifiers (DOIs), which have a long history of use in the context of scientific articles, are now being used to identify published datasets. It is now considered good practice for journal articles to cite the dataset used in the study in the references.
Metagenomics in the discovery of new life-forms
The integration of high-throughput sequencing into semi- or fully-automated data acquisition methods, new bioinformatics tools, and standardized data organization forms the basis for metagenomic projects that investigate the life-forms populating different parts of our environment. The data outputs can be stored in a structured format so that access is relatively simple (https://www.ebi.ac.uk/metagenomics/). The plenary lecture by Patrick Wincker described the Tara Oceans project (Karsenti et al. 2011; Hingamp et al. 2013) in which approximately 27,800 biological samples were collected from 153 ocean stations at three depths that were defined by approximately 13,000 measurements for detailed analysis. The DNA analysis of the metagenomes was carried out on organisms 0.2 to 1.6 um in size, from 17 samples collected at 13 sites. The focus for the study was on the abundance of nucleocytoplasmic large DNA viruses (NCLDVs) in the marine environment, and to achieve this, 16 NCLDV marker genes and 35 cellular marker genes (including the 18S rDNA variable region) were used to align the Roche-454 reads produced from DNA extracted from the samples. The metagenomic reads were utilized without prior assembly software analysis by the COMPAREADS software (Maillet et al. 2012).
The NCLDVs constitute a group of eukaryotic viruses that have an ecological role in the sea in contributing to the turnover of their unicellular hosts as well as causing diseases in animals. The data presented by Patrick Wincker showed that metagenomic sequence analyses can guide the discovery process for new marine viruses and their host interaction in future research.
Directions for genomic research
The use of DNA as a storage medium for digital information as a replacement for hard drives is a future possibility being actively investigated, as discussed by Ewan Birney in his plenary lecture. Recently, a scalable method that encodes nontrivial amounts of information was described (Goldman et al. 2013). DNA is an attractive medium because of its proven durability and very high storage density that is several orders of magnitude greater than any existing commercial storage devices (Church et al. 2012). However, applications will not be economical until the cost of sequencing and synthesizing arbitrary DNA sequences has been greatly reduced.
The plenary lecture by Eric Green provided an overview of the broad area of genomics with a particular focus on the human genome (Green and Guyer 2011; McCarthy et al. 2013). It was evident that advancing genomics to attain complete reference genomes in order to define functional elements in the proteome was a top priority. The large scale of genomics required attention to organizational structure because international consortia are usually involved, data standards to minimize errors and maximize utility, and computational procedures. The rapid release of the large data catalogs was a priority but needed to respect the ownership by researchers and initiatives such as DataCite (Brase et al. 2009), discussed in this review and which introduced digital object identifiers (DOIs), were beginning to deal with the need for structure in the information becoming available. The requirement for low-cost data production (DNA, RNA sequencing, proteomics, and metabolomics) was closely linked to the translation of research outputs to society more broadly and the development of appropriate policies. Many of the concepts and challenges in human genomics also apply to plants and animals as discussed in this review.
In the human genomic area, application in cancer pharmacogenomics, the diagnosis of rare disorders, and the tracking of disease outbreaks are new applications that build on the developing expertise (McCarthy et al. 2013). Some breakthroughs in defining the molecular biology of well-known disorders have been provided in this review. In addition, the role of the host microbiome (through metagenomic capabilities) and identification of drug response biomarkers in order to facilitate the application of existing drugs for new purposes are targets for a broader translation into society. The application of DNA sequencing to providing a noninvasive test for prenatal screening (Allison 2013) is an example of a particularly rapid uptake. The approach is based on the discovery that cell-free fetal DNA (cffDNA) is released when placental cells break down, and this cffDNA can comprise 5–10 % of the genetic material in a pregnant woman’s bloodstream. The placental source of the DNA means false positives for trisomy of chromosomes are possible (since this tissue is not necessarily identical to that of the fetus), and the data is generally treated as an indicator of risk only (Allison 2013).
A corollary of the speed at which data acquisition is occurring is the need for education programs to keep up and provide the required training for the next generation of researchers, policy makers, and participants in industry and society more broadly. The new technologies need to be matched by a biological understanding within the broad range of researchers and clinicians (McCarthy et al. 2013) as well as individuals involved in extension and consulting activity in agriculture.
For the use of databases, Ewan Birney noted that EMBL/EBI had upgraded its infrastructure for online training (https://www.ebi.ac.uk/training/). In the medical area, both Eric Green and Elaine Mardis emphasized the requirements for clinicians to understand genomic-based information, as well as the importance of having access to the research outputs (http://www.iccg.org/; https://www.ncbi.nlm.nih.gov/clinvar/). In the plant and animal areas, innovation in undergraduate-level teaching (Burnette and Wessler 2013) is deploying the research outputs in the transposable element area as a model for students in biology to experience first-hand the analysis of phenotypes and genotypes. This provides the basis for correlating changes in genotypes using marker polymorphisms, for understanding the fundamentals of biology, as well as for appreciating the nature of research and applying knowledge from one organism to another.
References
Allison M (2013) Genomic testing reaches into the womb. Nat Biotechnol 31:595–601
Angel A, Song J, Dean C, Howard M (2011) A Polycomb-based switch underlying quantitative epigenetic memory. Nature 476:105–108. doi:10.1038/nature10241
Appels R, Driscoll C, Peacock WJ (1978) Heterochromatin and highly repeated DNA sequences in rye (Secale cereale). Chromosoma (Berl) 70:67–89
Appels R, Barrero R, Bellgard M (2013) Advances in biotechnology and informatics to link variation in the genome to phenotypes in plants and animals. Funct Integr Genomics 13(1):1–9. doi:10.1007/s10142-013-0319-2
Bayer CropScience (2013) Shaping wheat for the future. www.cropscience.bayer.com/en/Magazine/Shaping-Wheat-for-the-Future.aspx
Biesecker LG, Bailey-Wilson JE, Ballantyne J, Baum H, Bieber FR, Brenner C, Budowle B, Butler JM, Carmody G, Conneally PM, Duceman B, Eisenberg A, Forman L, Kidd KK, Leclair B, Niezgoda S, Parsons TJ, Pugh E, Shaler R, Sherry ST, Sozer A, Walsh A (2005) DNA identifications after the 9/11 World Trade Center attack. Science 310(5751):1122–1123
Brase J, Farquhar A, Gastl A, Gruttemeier H, Heijne M, Heller A, Piguet A, Rombouts J, Sandfaer M, Sens I (2009) Approach for a joint global registration agency for research data. Inf Serv Use 29:13–27. doi:10.3233/ISU-2009-0595
Brennan CW, Verhaak RG, McKenna A, Campos B, Noushmehr H, Salama SR, Zheng S, Chakravarty D, Sanborn JZ, Berman SH, Beroukhim R, Bernard B, Wu CJ, Genovese G, Shmulevich I, Barnholtz-Sloan J, Zou L, Vegesna R, Shukla SA, Ciriello G, Yung WK, Zhang W, Sougnez C, Mikkelsen T, Aldape K, Bigner DD, Van Meir EG, Prados M, Sloan A, Black KL, Eschbacher J, Finocchiaro G, Friedman W, Andrews DW, Guha A, Iacocca M, O’Neill BP, Foltz G, Myers J, Weisenberger DJ, Penny R, Kucherlapati R, Perou CM, Hayes DN, Gibbs R, Marra M, Mills GB, Lander E, Spellman P, Wilson R, Sander C, Weinstein J, Meyerson M, Gabriel S, Laird PW, Haussler D, Getz G, Chin L, TCGAResearch Network (2013) The somatic genomic landscape of glioblastoma. Cell 155:462–477. doi:10.1016/j.cell.2013.09.034
Burnette JM 3rd, Wessler SR (2013) Transposing from the laboratory to the classroom to generate authentic research experiences for undergraduates. Genetics 193:367–375. doi:10.1534/genetics.112.147355
Burton JN, Adey A, Patwardhan RP, Ruolan Qiu R, Kitzman JO, Shendure J (2013) Chromosome-scale scaffolding of de novo genome assemblies based on chromatin interactions. Nat Biotechnol 31:1119–1125. doi:10.1038/nbt.2727
Cavanagh CR, Chao S, Wang S, Huang BE, Stephen S, Kiani S, Forrest K, Saintenac C, Brown-Guedira GL, Akhunova A, See D, Bai G, Pumphrey M, Tomar L, Wong D, Kong S, Reynolds M, da Silva ML, Bockelman H, Talbert L, Anderson JA, Dreisigacker S, Baenziger S, Carter A, Korzun V, Morrell PL, Dubcovsky J, Morell MK, Sorrells ME, Hayden MJ, Akhunov E (2013) Genome-wide comparative diversity uncovers multiple targets of selection for improvement in hexaploid wheat landraces and cultivars. Proc Natl Acad Sci U S A 110:8057–8062. doi:10.1073/pnas.1217133110
Chen K, Navin NE, Wang Y, Schmidt HK, Wallis JW, Niu B, Fan X, Zhao H, McLellan MD, Hoadley KA, Mardis ER, Ley TJ, Perou CM, Wilson RK, Ding L (2013) BreakTrans: uncovering the genomic architecture of gene fusions. Genome Biol 14(8):R87
Choi J, Hyun Y, Kang MJ, In Yun H, Yun JY, Lister C, Dean C, Amasino RM, Noh B, Noh YS, Choi Y (2009) Resetting and regulation of Flowering Locus C expression during Arabidopsis reproductive development. Plant J 57:918–931. doi:10.1111/j.1365-313X.2008.03776.x
Church GM, Gao Y, Kosuri S (2012) Next-generation digital information storage in DNA. Science 337:1628
Coble MD, Hill CR, Butler JM (2013) Haplotype data for 23 Y-chromosome markers in four U.S. population groups. Forensic Sci Int Genet 7:e66–e68
Corbett-Detig RB, Hartl DL (2012) Population genomics of inversion polymorphisms in Drosophila melanogaster. PLoS Genet 8(12):e1003056. doi:10.1371/journal.pgen.1003056
Crevillén P, Dean C (2011) Regulation of the floral repressor gene FLC: the complexity of transcription in a chromatin context. Curr Opin Plant Biol 14:38–44. doi:10.1016/j.pbi.2010.08.015
Crevillén P, Sonmez C, Wu Z, Dean C (2013) A gene loop containing the floral repressor FLC is disrupted in the early phase of vernalization. EMBO J 32:140–148. doi:10.1038/emboj.2012.324
Ding L, Ley TJ, Larson DE, Miller CA, Koboldt DC, Welch JS, Ritchey JK, Young MA, Lamprecht T, McLellan MD, McMichael JF, Wallis JW, Lu C, Shen D, Harris CC, Dooling DJ, Fulton RS, Fulton LL, Chen K, Schmidt H, Kalicki-Veizer J, Magrini VJ, Cook L, McGrath SD, Vickery TL, Wendl MC, Heath S, Watson MA, Link DC, Tomasson MH, Shannon WD, Payton JE, Kulkarni S, Westervelt P, Walter MJ, Graubert TA, Mardis ER, Wilson RK, DiPersio JF (2012) Clonal evolution in relapsed acute myeloid leukaemia revealed by whole-genome sequencing. Nature 481:506–510. doi:10.1038/nature10738
Feuillet C, Stein N, Rossini L, Praud S, Mayer K, Schulman A, Eversole K, Appels R (2012) Integrating cereal genomics to support innovation in the Triticeae. Funct Integr Genomics 12:573–583. doi:10.1007/s10142-012-0300-5
Fritz MHY, Leinonen R, Cochrane G, Birney E (2011) Efficient storage of high throughput DNA sequencing data using reference-based compression. Genome Res 21:734–740. doi:10.1101/gr.114819.110
Geraldo N, Bäurle I, Kidou S, Hu X, Dean C (2009) FRIGIDA delays flowering in Arabidopsis via a cotranscriptional mechanism involving direct interaction with the nuclear cap-binding complex. Plant Physiol 150:1611–1618. doi:10.1104/pp. 109.137448
Goldman N, Bertone P, Chen S, Dessimoz C, LeProust EM, Sipos B, Birney E (2013) Toward practical high-capacity low-maintenance storage of digital information in synthesised DNA. Nature 494(7435):77–80. doi:10.1038/nature11875
Green ED, Guyer MS (2011) Charting a course for genomic medicine from base pairs to bedside. Nature 470:204–213. doi:10.1038/nature09764
Griffith M, Griffith OL, Coffman AC, Weible JV, McMichael JF, Spies NC, Koval J, Das I, Callaway MB, Eldred JM, Miller CA, Subramanian J, Govindan R, Kumar RD, Bose R, Ding L, Walker JR, Larson DE, Dooling DJ, Smith SM, Ley TJ, Mardis ER, Wilson RK (2013) DGIdb: mining the druggable genome. Nat Methods 10(12):1209–1210. doi:10.1038/nmeth.2689
Han Y, Qin S, Wessler SR (2013) Comparison of class 2 transposable elements at superfamily resolution reveals conserved and distinct features in cereal grass genomes. BMC Genomics 14:71. doi:10.1186/1471-2164-14-71
Hancock CN, Zhang F, Floyd K, Richardson AO, Lafayette P, Tucker D, Wessler SR, Parrott WA (2011) The rice miniature inverted repeat transposable element mPing is an effective insertional mutagen in soybean. Plant Physiol 157:552–562. doi:10.1104/pp. 111.181206
Hares DR (2012) Expanding the CODIS core loci in the United States. FSI Genet 6:e52–e54
Hastie A, Dong L, Smith A, Finklestein J, Lam ET, Huo N, Cao H, Kwok P-Y, Deal KR, Dvorak J, Luo M-C, Gu Y, Xiao M (2013) Rapid genome mapping in nanochannel arrays for highly complete and accurate de novo sequence assembly of the complex Aegilops tauschii genome. PLoS One 8:e55864. doi:10.1371/journal.pone.0055864
Hingamp P, Grimsley N, Acinas SG, Clerissi C, Subirana L, Poulain J, Ferrera I, Sarmento H, Villar E, Lima-Mendez G, Faust K, Sunagawa S, Claveriel J-M, Moreau H, Desdevises Y, Bork P, Raes J, de Vargas C, Karsenti E, Kandels-Lewis S, Jaillon O, Not F, Pesant S, Wincker P, Ogata H (2013) Exploring nucleo-cytoplasmic large DNA viruses in Tara Oceans microbial metagenomes. ISME J 7:1678–1695
Huang W, Richards S, Carbone MA, Zhu D, Anholt RR, Ayroles JF, Duncan L, Jordan KW, Lawrence F, Magwire MM, Warner CB, Blankenburg K, Han Y, Javaid M, Jayaseelan J, Jhangiani SN, Muzny D, Ongeri F, Perales L, Wu YQ, Zhang Y, Zou X, Stone EA, Gibbs RA, Mackay TF (2012) Epistasis dominates the genetic architecture of Drosophila quantitative traits. Proc Natl Acad Sci U S A 109:15553–15559
Jeffreys AJ, Wilson V, Thein SL (1985) Hypervariable “minisatellite” regions in human DNA. Nature 314:67–73
Karsenti E, Acinas SG, Bork P, Bowler C, De Vargas C et al (2011) A holistic approach to marine eco-systems biology. PLoS Biol 9:e1001177. doi:10.1371/journal.pbio.1001177
Lam ET, Hastie A, Lin C, Ehrlich D, Das SK et al (2012) Genome mapping on nanochannel arrays for structural variation analysis and sequence assembly. Nat Biotechnol 30:771–776
Lee Y, Collier TC, Sanford MR, Marsden CD, Fofana A, Cornel AJ, Lanzaro GC (2013) Chromosome inversions, genomic differentiation and speciation in the African malaria mosquito Anopheles gambiae. PLoS One 8(3):e57887. doi:10.1371/journal.pone.0057887
Ley TJ, Mardis ER, Ding L, Fulton B, McLellan MD, Chen K, Dooling D, Dunford-Shore BH, McGrath S, Hickenbotham M, Cook L, Abbott R, Larson DE, Koboldt DC, Pohl C, Smith S, Hawkins A, Abbott S, Locke D, Hillier LW, Miner T, Fulton L, Magrini V, Wylie T, Glasscock J, Conyers J, Sander N, Shi X, Osborne JR, Minx P, Gordon D, Chinwalla A, Zhao Y, Ries RE, Payton JE, Westervelt P, Tomasson MH, Watson M, Baty J, Ivanovich J, Heath S, Shannon WD, Nagarajan R, Walter MJ, Link DC, Graubert TA, DiPersio JF, Wilson RK (2008) DNA sequencing of a cytogenetically normal acute myeloid leukaemia genome. Nature. 456:66–72. doi:10.1038/nature07485
Lin HC, Goldstein S, Mendelowitz L, Zhou S, Wetzel J et al (2012) AGORA: assembly guided by optical restriction alignment. BMC Bioinforma 13:189
Ling H–Q (2013) Genomic data from Triticum urartu—the progenitor of wheat A genome. GigaScience. doi:10.5524/100050
Liu F, Quesada V, Crevillén P, Bäurle I, Swiezewski S, Dean C (2007) The Arabidopsis RNA-binding protein FCA requires a lysine-specific demethylase 1 homolog to downregulate FLC. Mol Cell 28:398–407
Mackay TF (2014) Epistasis and quantitative traits: using model organisms to study gene–gene interactions. Nat Rev Genet 15:22–33. doi:10.1038/nrg3627
Mackay TF, Richards S, Stone EA, Barbadilla A, Ayroles JF, Zhu D, Casillas S, Han Y, Magwire MM, Cridland JM, Richardson MF, Anholt RR, Barrón M, Bess C, Blankenburg KP, Carbone MA, Castellano D, Chaboub L, Duncan L, Harris Z, Javaid M, Jayaseelan JC, Jhangiani SN, Jordan KW, Lara F, Lawrence F, Lee SL, Librado P, Linheiro RS, Lyman RF, Mackey AJ, Munidasa M, Muzny DM, Nazareth L, Newsham I, Perales L, Pu LL, Qu C, Ràmia M, Reid JG, Rollmann SM, Rozas J, Saada N, Turlapati L, Worley KC, Wu YQ, Yamamoto A, Zhu Y, Bergman CM, Thornton KR, Mittelman D, Gibbs RA (2012) The Drosophila melanogaster genetic reference panel. Nature 482:173–178. doi:10.1038/nature10811
Magwire MM, Yamamoto A, Carbone MA, Roshina NV, Symonenko AV, Pasyukova EG, Morozova TV, Mackay TF (2010) Quantitative and molecular genetic analyses of mutations increasing Drosophila life span. PLoS Genet 6:e1001037. doi:10.1371/journal.pgen.1001037
Maillet N, Lemaitre C, Chikhi R, Lavenier D, Peterlongo (2012) Compareads: comparing huge metagenomic experiments. BMC Bioinforma 13(Suppl 19):S10
Mardis ER (2010) Cancer genomics identifies determinants of tumor biology. Genome Biol 11:211
Marston E, Weston V, Jesson J, Maina E, McConville C, Agathanggelou A, Skowronska A, Mapp K, Sameith K, Powell JE, Lawson S, Kearns P, Falciani F, Taylor M, Stankovic T (2009) Stratification of pediatric ALL by in vitro cellular responses to DNA double-strand breaks provides insight into the molecular mechanisms underlying clinical response. Blood 113:117–126. doi:10.1182/blood-2008-03-142950
Mascher M, Muehlbauer GJ, Rokhsar DS, Chapman J, Schmutz J, Barry K, Muñoz-Amatriaín M, Close TJ, Wise RP, Schulman AH, Himmelbach A, Mayer KFX, Scholz U, Poland JA, Stein N, Waugh R (2013) Anchoring and ordering NGS contig assemblies by population sequencing (POPSEQ). Plant J 76:718–727. doi:10.1111/tpj.12319
McCarthy JJ, McLeod HL, Ginsburg GS (2013) Genomic medicine: a decade of successes, challenges, and opportunities. Sci Transl Med 5(189):189sr4. doi:10.1126/scitranslmed.3005785
Melo JV (1996) The molecular biology of chronic myeloid leukaemia. Leukemia: official journal of the Leukemia Society of America, Leukemia Research Fund, UK. Nature 456:66–72. doi:10.1038/nature07485
Miura K, Furumoto T (2013) Cold signaling and cold response in plants. Int J Mol Sci 14:5312–5337. doi:10.3390/ilms14035312
Naito K, Cho E, Yang G, Campbell MA, Yano K, Okumoto Y, Tanisaka T, Wessler SR (2006) Dramatic amplification of a rice transposable element during recent domestication. Proc Natl Acad Sci U S A 103:17620–17625
Naito K, Zhang F, Tsukiyama T, Saito H, Hancock CN, Richardson AO, Okumoto Y, Tanisaka T, Wessler SR (2009) Unexpected consequences of a sudden and massive transposon amplification on rice gene expression. Nature 461:1130–1134. doi:10.1038/nature08479
Nybom H, Weising K, Björn Rotter B (2014) DNA fingerprinting in botany: past, present, future. Investig Genet 5:1
Piwowar HA, Vision TJ (2013) Data reuse and the open data citation advantage. Peer J 1:e175. doi:10.7717/peerj.175.eCollection 2013
Popitsch M, von Haeseler A (2013) NGC: lossless and lossy compression of aligned high throughput sequencing data. Nucleic Acids Res 41(1):e27
Restifo NP, Dudley ME, Steven A (2012a) Rosenberg adoptive immunotherapy for cancer: harnessing the T cell response. Nat Rev Immunol 12:269–280
Restifo NP, Dudley ME, Rosenberg SA (2012b) Adoptive immunotherapy for cancer: harnessing the T cell response. Nat Rev Immunol 12:269–281
Rocca-Serra P et al (2010) ISA software suite: supporting standards-compliant experimental annotation and enabling curation at the community level. Bioinformatics 26(18)
Rosa S, De Lucia F, Mylne JS, Zhu D, Ohmido N, Pendle A, Kato N, Shaw P, Dean C (2013) Physical clustering of FLC alleles during Polycomb-mediated epigenetic silencing in vernalization. Genes Dev 27(17):1845–1850. doi:10.1101/gad.221713.113
Selvaraj S, Dixon JR, Bansal V, Bing Ren B (2013) Whole-genome haplotype reconstruction using proximity-ligation and shotgun sequencing. Nat Biotechnol 31:1111–1118. doi:10.1038/nbt.2728
Song J, Irwin J, Dean C (2013) Remembering the prolonged cold of winter. Curr Biol 23:R807–R811. doi:10.1016/j.cub.2013.07.027
Swiezewski S, Crevillen P, Liu F, Ecker JR, Jerzmanowski A, Dean C (2007) Small RNA-mediated chromatin silencing directed to the 3′ region of the Arabidopsis gene encoding the developmental regulator, FLC. Proc Natl Acad Sci U S A 104:3633–3638
Swiezewski S, Liu F, Magusin A, Dean C (2009) Cold-induced silencing by long antisense transcripts of an Arabidopsis Polycomb target. Nature 462:799–802. doi:10.1038/nature08618
TCGA (The Cancer Genome Atlas Research Network), Weinstein JN, Collisson EA, Mills GB, Shaw KRB Ozenberger BA, Ellrott K, Shmulevich I, Sander C, Stuart JM (2013) The Cancer Genome Atlas Pan-Cancer analysis project. Nat Genet 45:1113–1118
van Oeveren J, de Ruiter M, Jesse T, van der Poel H, Tang J, Yalcin F, Janssen A, Volpin H, Stormo KE, Bogden R, van Eijk MJ, Prins M (2011) Sequence-based physical mapping of complex genomes by whole genome profiling. Genome Res 21:618–625
Waldmann TA (2003) Immunotherapy: past, present and future. Nat Med 9:269–277
Wang S, Debbie Wong D, Forrest K, Allen A, Chao S, Huang E, Maccaferri M, Salvi S, Milner SG, Cattivelli L, Mastrangelo AM, Whan A, Stephen S, Barker G, Wieseke R, Plieske J, IWGSC, Lillemo M, Mather D, Appels R, Dolferus R, Brown-Guedira G, Korol A, Akhunova AR, Feuillet C, Salse J, Morgante M, Pozniak C, Luo M-C, Dvorak J, Morell M, Dubcovsky J, Ganal M, Tuberosa R, Lawley C, Mikoulitch I, Cavanagh C, Edwards KJ, Hayden M, Akhunov E (2014) Characterization of polyploid wheat genomic diversity using a high-density 90,000 SNP array. Plant Biotechnol in press
Welch JS, Ley TJ, Link DC, Miller CA, Larson DE, Koboldt DC, Wartman LD, Lamprecht TL, Liu F, Xia J, Kandoth C, Fulton RS, McLellan MD, Dooling DJ, Wallis JW, Chen K, Harris CC, Schmidt HK, Kalicki-Veizer JM, Lu C, Zhang Q, Lin L, O’Laughlin MD, McMichael JF, Delehaunty KD, Fulton LA, Magrini VJ, McGrath SD, Demeter RT, Vickery TL, Hundal J, Cook LL, Swift GW, Reed JP, Alldredge PA, Wylie TN, Walker JR, Watson MA, Heath SE, Shannon WD, Varghese N, Nagarajan R, Payton JE, Baty JD, Kulkarni S, Klco JM, Tomasson MH, Westervelt P, Walter MJ, Graubert TA, DiPersio JF, Ding L, Mardis ER, Wilson RK (2012) The origin and evolution of mutations in acute myeloid leukemia. Cell 150:264–278. doi:10.1016/j.cell.2012.06.023
Wicker T, Sabot F, Hua-Van A, Bennetzen JL, Capy P, Chalhoub B, Flavell A, Leroy P, Morgante M, Panaud O, Paux E, SanMiguel P, Schulman AH (2007) A unified classification system for eukaryotic transposable elements. Nat Rev Genet 8:973–982
Yang G, Zhang F, Hancock CN, Wessler SR (2007) Transposition of the rice miniature inverted repeat transposable element mPing in Arabidopsis thaliana. Proc Natl Acad Sci U S A 104:10962–10967
Yun L, Gu Y, Rajeevan H, Kidd KK (2014) Application of six IrisPlex SNPs and comparison of two eye color prediction systems in diverse Eurasia populations. Int J Legal Med. doi:10.1007/s00414-013-0953-1
Acknowledgments
The authors are grateful to Delphine Fleury for valuable inputs into the manuscript preparation.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article
Cite this article
Appels, R., Nystrom-Persson, J. & Keeble-Gagnere, G. Advances in genome studies in plants and animals. Funct Integr Genomics 14, 1–9 (2014). https://doi.org/10.1007/s10142-014-0364-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10142-014-0364-5