
Abstract
Lipoprotein lipase (LPL) is a hydrolase that cleaves circulating triglycerides to release fatty acids to the surrounding tissues. The enzyme is synthesized in parenchymal cells and is transported to its site of action on the capillary endothelium by glycophosphatidylinositol (GPI)-anchored high-density lipoprotein-binding protein 1 (GPIHBP1). Inactivating mutations in LPL; in its cofactor, apolipoprotein (Apo) C2; or in GPIHBP1 cause severe hypertriglyceridemia. Here we describe an individual with complete deficiency of GPIHBP1. The proband was an Asian Indian boy who had severe chylomicronemia at 2 months of age. Array-based copy-number analysis of his genomic DNA revealed homozygosity for a 17.5-kb deletion that included GPIHBP1. A 44-year-old aunt with a history of hypertriglyceridemia and pancreatitis was also homozygous for the deletion. A bolus of intravenously administered heparin caused a rapid increase in circulating LPL and decreased plasma triglyceride levels in control individuals but not in two GPIHBP1-deficient patients. Thus, short-term treatment with heparin failed to attenuate the hypertriglyceridemia in patients with GPIHBP1 deficiency. The increasing resolution of copy number microarrays and their widespread adoption for routine cytogenetic analysis is likely to reveal a greater role for submicroscopic deletions in Mendelian conditions. We describe the first neonate with complete GPIHBP1 deficiency due to homozygosity for a deletion of GPIHBP1.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Type I hyperlipoproteinemia is a rare, autosomal recessive disorder characterized by the accumulation of large triglyceride-rich lipoproteins in the circulation (chylomicronemia), which leads to the development of eruptive xanthomas, failure to thrive, and recurrent pancreatitis (Brunzell and Deeb 2001). The disorder is caused by mutations that disrupt the lipolytic processing of triglyceride-rich lipoproteins, including inactivating mutations in genes encoding lipoprotein lipase (LPL) (Havel and Gordon 1960), the enzyme that hydrolyzes circulating triglycerides; its cofactor, APOC2 (Breckenridge et al. 1978); a lipase-specific chaperone known as lipase maturation factor 1 (Peterfy et al. 2007); and the protein that anchors the enzyme to the endothelial surface, glycophosphatidylinositol (GPI)-anchored high-density lipoprotein-binding protein 1 (GPIHBP1) (Beigneux et al. 2007). GPIHBP1 was originally identified as a high-density lipoprotein (HDL)-binding protein (Ioka et al. 2003), but the subsequent finding that Gpihbp1 knockout mice have severe hypertriglyceridemia revealed an essential role for the protein in the action of LPL at capillary endothelial surfaces (Beigneux et al. 2007). LPL is synthesized primarily by adipocytes, skeletal myocytes, and cardiomyocytes and is secreted into the extracellular space where it binds proteoglycans (Brunzell and Deeb 2001). GPIHBP1 binds LPL in the capillary subendothelial spaces and translocates the enzyme to the luminal surfaces of endothelial cells (Davies et al. 2010). In mice lacking GPIHBP1, heparin-releasable LPL activity is reduced (Beigneux et al. 2007). GPIHBP1 appears to function as a platform for lipolysis of circulating triglycerides (Beigneux et al. 2007).
Human GPIHBP1 is located on the distal long arm of chromosome 8 and encodes a 184-amino-acid protein that contains three domains: N-terminal highly acidic, lymphocyte antigen (Ly6), and C-terminal to which the glycophosphatidylinositol is attached (Beigneux et al. 2009a). Mutations in the acidic and Ly6 domains interfere with the ability of GPIHBP1 to bind LPL and chylomicrons (Beigneux et al. 2009c; Gin et al. 2008). Several mutations in GPIHBP1 gene have been identified in patients with chylomicronemia (Table 1). Here, we describe the first reported patient with a complete absence of GPIHBP1 due to a deletion of GPIHBP1 and assess the therapeutic efficacy of heparin in this individual and in a second patient with hypertriglyceridemia who was homozygous for missense mutation c.203 G >A (p.Cys68Tyr) in GPIHBP1.
Materials and methods
Study participants
The study was approved by the Institutional Review Board of the University of Texas Southwestern Medical Center, USA. Written informed consent was obtained from all participants prior to collecting venous blood after a 12-h fast. Genomic DNA was extracted using the Gentra Puregene Blood Kit (Qiagen, Valencia, CA,USA) or the Easy-DNA kit (Invitrogen, Carlsbad, CA,USA) following the manufacturers’ protocol. Fasting plasma lipids and lipoproteins were measured by Quest Diagnostics.
Sequencing coding regions of candidate genes
The exons of LPL, APOC2, APOA5, and LMF1 were amplified from 50 ng of genomic DNA using the polymerase chain reaction (PCR) and oppositely oriented oligonucleotides that flank the exons of these genes (Supplementary Table 1). Amplification products were sequenced using dye-terminator chemistry and an ABI 3730xl DNA analyzer. Sequence variants were verified by manually inspecting chromatograms.
Genotyping and copy number variation (CNV) detection
Array-based single nucleotide polymorphism (SNP) genotyping was performed on genomic DNA from proband 1 using the human 1-M Beadchip (Illumina, San Diego, CA, USA) according to the manufacturer’s Infinium II Assay protocol. Briefly, 750 ng of genomic DNA was amplified, enzymatically fragmented, and precipitated with isopropanol, as described (Illumina’s Infinium II Assay). The fragmented genomic DNA was hybridized with an Illumina Human 1-M beadchip that contained 50-mer probes attached to a silicon-based array that interrogates 1,072,820 SNP. A single-base extension reaction was performed to label the annealed DNA in an allele-specific manner. The hybridized array was scanned using the Illumina BeadArray Reader to image the laser-excited fluorophores for each probe. Data was analyzed using the Beadstudio Genotyping Module (v3.3.7). Individual probe log ratios [logR = log2 (sample intensity/expected reference intensity)] were plotted by their genomic positions using the Genome Browser in the Illumina Genome Viewer (v3.2.9). A smoothing line was generated using a 200-kb moving average window to enhance visualization of the copy number gains and losses in the Genome Viewer.
Probe-intensity data was analyzed using Partek Genomic Suite software (Partek, St. Louis, MO, USA). Briefly, copy number was estimated from intensity values relative to baseline copy number calculated from array results of seven unrelated individuals with other genetic disorders. Genomic segmentation algorithms were used to identify genomic regions that differed significantly in signal intensity. Genotype data was imported into Partek Genomic Suite software and analyzed using a Hidden Markov Model (Churchill 1989) to identify regions of homozygosity. For comparison, we examined genotype data from an offspring of a second-cousin mating and from six European Americans who were offspring of nonconsanguinious matings. Size distributions of the regions of homozygosity are represented as a quantile plot.
GPIHBP1 deletion mapping and pedigree analysis
To define the deletion breakpoints, oppositely oriented oligonucleotides complementary to sequences flanking the putative deletion were used to amplify the deletion junction. The primers used to amplify the junction were as follows: forward, 5′-GCATCAGGTATGAAGAGTTTGG-3′ and reverse, 5′-CAGGGTCTGTGTGGAAGAGG-3′.
Heparin infusion
Fasting blood samples were collected from each patient from a heparin lock. A total of 100 U/kg heparin was infused intravenously (APP Pharmaceuticals, Schaumburg, IL, USA). Venous blood samples were then collected in citrate ethylenediaminetetraacetate (EDTA) tubes 5, 10, 15, 30, 60, and 90 min after the heparin infusion. Blood samples were maintained at 4°C, and plasma was isolated by centrifugation within 30 min and stored at −80°C. Triglyceride levels were measured using the Vitros chemistry system (Ortho-Clinical Diagnostics, Rochester, NY, USA) following the manufacturer’s protocol. LPL and hepatic lipase (HL) mass were measured using a direct sandwich enzyme-linked immunosorbent assay (ELISA), as previously described (Bensadoun 1996; Peterson et al. 1992).
Results
Infant presents with chylomicronemia
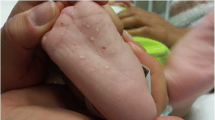
Proband 1 was a breast-fed male infant of Asian Indian ancestry. The patient was the offspring of unrelated parents with normal plasma triglyceride levels who both came from a small town in Gujarat state in India. The patient presented at 2 months of age with anemia, lethargy, and failure to thrive. Sepsis was initially suspected and ruled out. His plasma triglyceride level exceeded the limits of the assay (>25,000 mg/dl), and his cholesterol level was 1,056 mg/dl. Tests of liver and thyroid function were normal, as was the urinalysis. Magnetic resonance imaging of his head revealed an increase in lipid content in the central venous sinus on T1-weighted images, as reported previously (Koral et al. 2010).
The patient was placed on a low-fat diet and given fluids intravenously. After 24 h, his plasma triglyceride level had fallen to 19,960 mg/dl, with a plasma cholesterol level of 940 mg/dl. He was switched from Tolorex formula (27 kcal/oz, 1.3% fat) to Portagen (Mead Johnson), a milk-protein-based dietary formula that contains 40% fat (with 87% from medium-chain triglycerides). He was discharged after 12 days, with plasma triglyceride and cholesterol levels of 1,615 mg/dl and 397 mg/dl, respectively.
At 16-months of age, the proband’s height was 73 cm and he weighed 7.9 kg., which corresponds to the second percentile for age and sex. He is on a fat-restricted diet comprising Portagen, soy oil, and multivitamins. At 25-months of age, his plasma triglyceride level was 905 mg/dl, with total cholesterol of 146 mg/dl, HDL-cholesterol (C) of 16 mg/dl, and an low-density lipoprotein (LDL)-C level of 44 mg/dl.
No mutations identified in the coding regions of LPL, APOA5, APOC2, or LMF1
The coding regions of LPL, APOA5, APOC2, and LMF1 were sequenced; no nonsynonymous mutations were identified that were not also present in dbSNP (Supplementary Table 2).
Array-based copy-number detection reveals homozygosity for GPIHBP1 deletion
The Illumina 1-M DNA BeadChip was used to screen for copy number variations (CNV) across the genome. Probe intensities were plotted as log ratios (logR) for each SNP and mapped to the genome using the Illumina Genome Viewer. A marked drop in the logR values was detected at the distal end of the long arm of chromosome 8 (Fig. 1). Probe intensity data was also analyzed using Partek Genomic Suite software (v6.4). Genomic segmentation algorithms were used to identify changes in DNA copy number in the patient and compared with seven unrelated individuals of European descent. A total of 12 probes at 8q24.3 had a mean copy number of 0.0062, suggesting with a high degree of confidence (p = 1.18e-6) that the patient was homozygous for a deletion.

Analysis of copy number variation (CNV) in GPIHBP1 in patient 1. Log ratios [logR = log2(sample intensity/expected reference intensity)] were calculated using the intensity measures for each probe (blue dot) within GenomeStudio (Illumina). Calculated log ratios were plotted genome wide and annotated with known genes using the Genome Browser within GenomeStudio. A smoothing line shown in red was calculated using a 200-kb sliding window. The region of reduced logR values represents the homozygous deletion, which includes the GPIHBP1 gene
Two probes within the predicted deletion, rs35660224 and rs11136270, had calculated logR values that were near the normal range. Proband 1 had the lowest raw intensity (R value) for these two probes (data not shown). Signal intensities of these probes in proband 1 were 52.7% and 36.3% of the signal intensity measured in seven unrelated individuals analyzed simultaneously. Thus, the signal detected for these probes likely represents background noise.
Based on microarray data, the deletion was predicted to be ∼18,400 bp in length and to contain the entire GPIHBP1 gene. The deletion breakpoints were sequenced and mapped to the National Center for Biotechnical Information (NCBI) reference genome (build 36.1). The deletion spanned 17,499 bp and included GPIHBP1 (Fig. 2a). The 5′ breakpoint was located in a short interspersed nuclear element (SINE) repeat. Although the 3′ breakpoint was not in a repetitive sequence, both breakpoints occurred in an identical 7-bp sequence (TGACCCT). The 7-bp motif was 1,124-bp upstream of exon 1 and 12,406-bp downstream of exon 4. Only a single copy of the 7-bp motif was present at the breakpoint of the deleted chromosome, making it difficult to determine the exact position of the DNA breakpoints (Fig. 2b).

Map of the deletion identified in patient 1. a Exons of GPIHBP1 are shown as black boxes. Repetitive sequences were identified and classified using RepeatMasker. Oligonucleotides flanking the deletion were used to amplify the intervening sequences. The 5′ deletion junction was located in a mammalian interspersed repeat (MIR) sequence, and the 3′ junction was in a noncoding sequence located 12,406-bp downstream of GPIHBP1. b A 7-bp sequence, TGACCCT, was present at each breakpoint of the normal allele and at the junction in the deleted allele. c Polymerase chain reaction (PCR) assays were performed to amplify the deletion and exon 1 of GPIHBP1. The amplified fragments were size-fractionated on a 1% agarose gel and the gel was stained with ethidium bromide. The oligonucliotide primers flanking the junctions generate an amplicon of 481 bp only when the deletion is present; primers are too far apart to amplify the ∼18-kb wild-type allele. Oligonucleotide primers flanking exon 1 amplify a 381-bp fragment. A negative control reaction in which no DNA was added to the reaction is shown
We developed a PCR assay to screen for the deletion in the family of proband 1 (Fig. 3a). When oligonucleotides flanking the deletion were used to amplify the intervening sequences, a PCR fragment of 481-bp was generated from the deleted allele (Fig. 2c); the wild-type allele did not generate a PCR fragment when the same oligonucleotides were used, as they flanked a ∼18-kb interval. To detect the wild-type allele, exon 1 of GPIHBP1 was amplified (Fig. 2c). As expected, both of the proband’s parents were heterozygous for the deletion, as were seven other family members (Fig. 3a). All family members heterozygous for the deletion had normal plasma triglyceride levels. A 44-year-old maternal aunt (III.9) was homozygous for the deletion (Fig. 3a). She had a history of hypertriglyceridemia and had pancreatitis at the age of 29 years. Her plasma triglyceride level was 981 mg/dl when she was not taking lipid-lowering medications.

Pedigrees of patient 1 (A) and patient 2 (B). a Family members of patient 1 were assayed for the presence of the deletion using the polymerase chain reaction (PCR) assay described in the legend of Fig. 2. Family members heterozygous and homozygous for the deletion are designated with half-filled and filled symbols, respectively. b The brother of patient 2 had a history of pancreatitis. The only individual available for analysis in this family was the proband. Ages and fasting plasma triglyceride levels are provided. DM diabetes mellitus, ND not determined
This mutation was not found in the Database of Genomic Variants. Two reports of copy-number gains of GPIBHP1 but no reports of copy-number losses were identified in the database (Kidd et al. 2008; Tuzun et al. 2005). The mutant GPIHBP1 alleles identified in proband 1 were presumably inherited from a common ancestor. To test this hypothesis, we determined the extent of homozygosity surrounding the deletion and distributions of the lengths of regions of homozygosity (ROH) in proband 1 as well as in six nonconsanguineous individuals of European descent and one individual whose parents were second cousins (Supplementary Fig. 1). Regions of homozygosity were identified from the Illumina genotypes using a Hidden Markov Model implemented within Partek Genomics Suite software. The homozygous deletion in proband 1 was located within a ∼6.1-Mb ROH. This ROH was the largest identified in the patient and nearly twice the length of the largest region in controls. Furthermore, proband 1 had nearly three times more large regions of homozygosity (>2 Mb) than did any of the controls. Taken together, these data indicate that the mutant GPIHBP1 alleles inherited by proband 1 were derived from a common ancestor due to recent inbreeding in the family.
Identification of a second patient with mutations in GPIHBP1
DNA sequencing of patients from the Parkland Hospital Outpatient Lipid Clinic who had type I hyperlipidemia revealed GPIHBP1 mutations in a 36-year-old Hispanic woman (proband 2), who was the sixth child of nonconsanguineous parents from two different towns in El Salvador. The pedigree of her family is shown in Fig. 3b. The proband (II.7) had a history of intermittent abdominal pain from the age 11 years and was found to have hypertriglyceridemia during her first pregnancy at age 24 years. She was hospitalized several times for pancreatitis and placed on fish oil (2 g three times daily) and gemfibrozil (600 mg twice daily). She was counseled to follow a low-fat diet (∼12% fat) but continues to have intermittent abdominal pain requiring analgesia. She had no history of diabetes or eruptive xanthomas. An older brother (II.5) also has a history of acute recurrent pancreatitis but was not available for study (Fig. 3b).
At presentation, proband 2 weighed 63.5 kg and was 1.57 m tall [body mass index (BMI) = 25.8 kg/m2]. Physical examination revealed lipemia retinalis and several eruptive xanthomas on her elbows, but no hepatosplenomegaly. Her plasma cholesterol was 544 mg/dl, with a triglyceride level of 6,480 mg/dl. Liver function tests, fasting glucose, hemoglobin A1c (5.8%), and urinalysis were within normal limits. The coding regions of LPL, APOA5, APOC2, and GPIHBP1 were sequenced (Supplementary Table 2). The only nonsynonymous mutation identified was a homozygous substitution in GPIHBP1 (c.203 G > A), which causes cysteine at codon 68 to be replaced with tyrosine (C68Y). This mutation was recently identified in a 30-year-old Spanish woman with hypertriglyceridemia and pancreatitis (Coca-Prieto et al. 2011).
Intravenously administered bolus of heparin did not increase circulating levels of LPL in the two GPIHBP1-deficient patients
To determine whether heparin would mobilize LPL from peripheral tissues and attenuate the hypertriglyceridemia in the two GPIHBP1-deficient patients, as had previously been observed in Gpihbp1 −/− mice (Weinstein et al. 2008), we administered heparin as a bolus to both patients and monitored the effect on blood levels of LPL and triglycerides. A 44-year-old Caucasian man with a normal plasma triglyceride level, and the 34-year-old father of proband 1 (Fig. 3a, individual IV.4) were also studied as controls. Heparin infusion caused a rapid increase in immunodetectable LPL in both control individuals, but no increase in LPL mass was apparent in the samples from the GPIHBP1-deficient probands (Fig. 4a). The postheparin plasma levels of HL increased 32-fold in the unrelated control individuals and remained elevated during the 90-min experiment. In both probands and in the proband’s father, HL level increased after heparin administration then fell slightly to lower levels (Fig. 4b). Postheparin plasma triglyceride levels fell by ∼50% in both control individuals (Fig. 4c). The maximum decreases in plasma triglyceride levels were 10% in patient 1 and 20% in patient 2. (Fig. 4c). While this study was being performed, two other groups reported that a bolus of heparin failed to elicit appreciable increases of circulating LPL in hypertriglyceridemic patients with mutations in GPIHBP1 (Franssen et al. 2010; Olivecrona et al. 2010).

Plasma levels of lipoprotein lipase (LPL) (a), hepatic lipase (HL) (b), and triglycerides (c) after an intravenous bolus injection of heparin (100 U/kg). Blood was collected prior to (time 0) and at multiple time points after, heparin injection. Plasma was separated by centrifugation and used in each measurement. a LPL mass was measured by sandwich enzyme-linked immunosorbent assay (ELISA) (Peterson et al. 1992). b HL was measured using a sandwich ELISA assay (Bensadoun 1996). c Plasma triglycerides were measured at each time point and expressed as a proportion of the level at baseline (time 0)
Discussion
In this study, we identified and characterized the first patient with complete deficiency of GPIHBP1 due to homozygosity for a mutation that deletes the entire GPIHBP1 gene. The patient presented at 2 months of age with extremely severe hypertriglyceridemia (>25,000 mg/dl), comparable with that found in the worst cases of LPL deficiency. Thus, in contrast to mice, in which the hypertriglyceridemia associated with GPIHBP1 deficiency is modest prior to weaning and becomes progressively more severe with age (Beigneux et al. 2007), GPIHBP1 deficiency is fully manifest in human infants. Prolonged infusion of heparin has been shown to reduce plasma triglyceride levels in GPIHBP1-deficient individuals (Franssen et al. 2010), but infusion of a bolus of heparin into the two GPIHBP1-deficient patients in our study confirmed that acute heparin administration does not significantly lower plasma triglyceride levels. Our results do not rule out the possibility that higher doses or more prolonged administration of heparin might cause a reduction in plasma triglyceride level, as has been reported previously (Franssen et al. 2010), although chronic high-dose heparin treatment is accompanied by other significant risks.
Development has proceeded normally in patient 1 in our study, and sequelae unrelated to hypertriglyceridemia have not been noted in this patient or in other GPIHBP1-deficient patients. Similarly, abnormalities reported to date in mice lacking GPIHBP1 are all attributable to defects in LPL function (Beigneux et al. 2007). Young and colleagues found that GPIHBP1 is readily identifiable in many mammalian species, including monotremes, but not in fish, amphibians, or birds (Beigneux et al. 2009a). The recent appearance of GPIHBP1 in vertebrate evolution is also consistent with a highly specific function for the protein. These findings, together with the observation that distribution of GPIHBP1 expression across tissues in general parallels that of LPL (Beigneux et al. 2009a) suggests that GPIHBP1 does not have significant biological activity independent of its role in LPL action.
Defining the extent of the physiological roles of GPIHBP1 has important therapeutic implications. In humans, the phenotype of GPIHBP1 deficiency appears indistinguishable from that of LPL deficiency, whereas in mice, the hypertriglyceridemia is milder at birth in the Gpihbp −/− mice than in the Lpl −/− mice (Beigneux et al. 2007). If the protein functions solely to facilitate hydrolysis of circulating triglycerides in humans, then GPIHBP1 deficiency should be fully treatable by lowering plasma triglyceride levels.
The release of LPL into the circulation by heparin is accompanied by a dramatic reduction in plasma triglyceride levels (Korn 1955). The finding that heparin administration also significantly reduces plasma triglyceride levels in Gpihbp1-deficient mice raised the possibility that heparin could be a potential therapy for GPIHBP1-deficient patients (Weinstein et al. 2008). A recent study found that plasma triglyceride levels could be markedly reduced by a 6-h heparin infusion (from 1,780 to 534 mg/dl) but that little reduction was seen at earlier time points (Franssen et al. 2010). When heparin was given to GPIHBP1-deficient patients as a bolus, little LPL was released into the circulation (Franssen et al. 2010; Olivecrona et al. 2010) and data from our study confirm that a single bolus of heparin does not significantly lower plasma triglyceride levels in these patients.
The deletion in patient 1 involved two 7-bp sequences that flank GPIHBP1 and are identical in sequence. The upstream breakpoint is located in a mammalian interspersed repeat (MIR), whereas the 3′ breakpoint is located 294 bp downstream of another MIR repeat. MIR repeat elements contain a tRNA-like domain and a “core” domain, followed by a variable long interspersed element (LINE)-like region (Gilbert and Labuda 1999). MIR repeats have been documented to be directly involved in, or adjacent to, breakpoints of deletions that cause a variety of other diseases, including polycystic kidney disease (Bergmann et al. 2005), phenylketonuria (Kozak et al. 2006), maturity-onset diabetes of the young (MODY) (Ellard et al. 2007), and congenital afibrinogenemia (Spena et al. 2004). It is possible that the deletion observed in patient 1 occurred as a result of MIR-mediated nonhomologous end joining (Shaw and Lupski 2005; Toffolatti et al. 2002).
The parents of patient 1 were not known to be related but were both from a small town within Gujarat state in northwestern India. The high level of endogamy within Indian subpopulations has resulted in distinct genetic diversity between ethnicities (Indian Genome Variation Consortium 2008) and a high prevalence of founder mutations within particular communities (Colah et al. 2010; Zaidi et al. 2009). The region of homozygosity surrounding the GPIHBP1 deletion in patient 1 extended 6.1 Mb, which is greater than expected when compared with individuals of European ancestry. The presence of longer-than-expected homozygous genomic segments in patient 1 may reflect the local genetic substructure. The multilineal inheritance of the deletion in this family is consistent with this view. To more fully address this question would require analysis and comparison of additional individuals from this region of India.
We also identified an individual with chylomicronemia who was homozygous for a C68Y mutation in GPIHBP1 that was previously identified in a Spanish woman with chylomicronemia (Coca-Prieto et al. 2011). This mutation is one of six in GPIHBP1 that have been identified in patients with chylomicronemia (Table 1). A seventh mutation, G56R, is likely not to be a causative mutation for chylomicronemia, as reported (Gin et al. 2007; Wang and Hegele 2007). All mutations, excluding the gene deletion described in this study, are missense mutations involving residues in the Ly6 domain of the protein. The Ly6 domain contains ten highly conserved cysteine residues that are predicted to form a three-fingered structure, based on structural studies of other proteins with Ly6 domains (Huang et al. 2007; Kjaergaard et al. 2008). Substitution of alanine for any of these cysteines, including C68, does not interfere with trafficking of GPIHBP1 in cultured cells but abolishes LPL binding in a cell-free assay (Beigneux et al. 2009c). Olivicrona et al. (2010) identified three siblings who were compound heterozygotes for mutations in two different cysteine residues (C65S and C68G), and both of these mutations were also associated with failure to bind LPL.
The deletion of GPIHBP1 described in this study was identified using microarray technology that provides efficient high-resolution screening of copy number variation across the entire human genome. Historically, copy number variations have primarily been associated with sporadic congenital syndromic disorders, but submicroscopic variations play an important role in several Mendelian disorders. Some 5% of neurofibromatosis cases are caused by deletions of the entire NF1 gene (Williams et al. 2009), whereas ∼10–15% of familial hypercholesterolemia (Hobbs et al. 1992) and 65% of Duchenne muscular dystrophy (Prior and Bridgeman 2005) are due to gene deletions. Analysis of several fully sequenced human genomes indicates that the number of submicroscopic deletions is far greater than was previously recognized (Mills et al. 2011). Thus, whereas the substantial majority of single gene disorders are attributable to single nucleotide mutations, the increasing resolution of copy number microarrays and their widespread adoption for routine cytogenetic analysis is likely to reveal a greater role for submicroscopic deletions in Mendelian conditions as well as in sporadic conditions and complex traits.
References
Beigneux AP, Davies BS, Gin P, Weinstein MM, Farber E, Qiao X, Peale F, Bunting S, Walzem RL, Wong JS, Blaner WS, Ding ZM, Melford K, Wongsiriroj N, Shu X, de Sauvage F, Ryan RO, Fong LG, Bensadoun A, Young SG (2007) Glycosylphosphatidylinositol-anchored high-density lipoprotein-binding protein 1 plays a critical role in the lipolytic processing of chylomicrons. Cell Metab 5:279–291
Beigneux AP, Davies BS, Bensadoun A, Fong LG, Young SG (2009a) GPIHBP1, a GPI-anchored protein required for the lipolytic processing of triglyceride-rich lipoproteins. J Lipid Res 50(Suppl):S57–S62
Beigneux AP, Franssen R, Bensadoun A, Gin P, Melford K, Peter J, Walzem RL, Weinstein MM, Davies BS, Kuivenhoven JA, Kastelein JJ, Fong LG, Dallinga-Thie GM, Young SG (2009b) Chylomicronemia with a mutant GPIHBP1 (Q115P) that cannot bind lipoprotein lipase. Arterioscler Thromb Vasc Biol 29:956–962
Beigneux AP, Gin P, Davies BS, Weinstein MM, Bensadoun A, Fong LG, Young SG (2009c) Highly conserved cysteines within the Ly6 domain of GPIHBP1 are crucial for the binding of lipoprotein lipase. J Biol Chem 284:30240–30247
Bensadoun A (1996) Sandwich immunoassay for measurement of human hepatic lipase. Methods Enzymol 263:333–338
Bergmann C, Kupper F, Schmitt CP, Vester U, Neuhaus TJ, Senderek J, Zerres K (2005) Multi-exon deletions of the PKHD1 gene cause autosomal recessive polycystic kidney disease (ARPKD). J Med Genet 42:e63
Breckenridge WC, Little JA, Steiner G, Chow A, Poapst M (1978) Hypertriglyceridemia associated with deficiency of apolipoprotein C-II. N Engl J Med 298:1265–1273
Brunzell J, Deeb S (2001) Lipoprotein Lipase, apolipoprotein CII and Hepatic Lipase Deficiencies. In: Scriver C, Beaudet A, Sly W, Valle D (eds) The Metabolic and Molecular Bases of Inherited Disease. McGraw-Hill, New York, pp 2789–2816
Charriere S, Peretti N, Bernard S, et al (2011) Gpihbp1 c89f neomutation and hydrophobic c-terminal domain g175r mutation in two pedigrees with severe hyperchylomicronemia. J Clin Endocrinol Metab
Churchill GA (1989) Stochastic models for heterogeneous DNA sequences. Bull Math Biol 51:79–94
Coca-Prieto I, Kroupa O, Gonzalez-Santos P, Magne J, Olivecrona G, Ehrenborg E, Valdivielso P (2011) Childhood-onset chylomicronaemia with reduced plasma lipoprotein lipase activity and mass: identification of a novel GPIHBP1 mutation. J Intern Med
Colah R, Gorakshakar A, Phanasgaonkar S, D’Souza E, Nadkarni A, Surve R, Sawant P, Master D, Patel R, Ghosh K, Mohanty D (2010) Epidemiology of beta-thalassaemia in Western India: mapping the frequencies and mutations in sub-regions of Maharashtra and Gujarat. Br J Haematol 149:739–747
Davies BS, Beigneux AP, Barnes RH 2nd, Tu Y, Gin P, Weinstein MM, Nobumori C, Nyren R, Goldberg I, Olivecrona G, Bensadoun A, Young SG, Fong LG (2010) GPIHBP1 is responsible for the entry of lipoprotein lipase into capillaries. Cell Metab 12:42–52
Ellard S, Thomas K, Edghill EL, Owens M, Ambye L, Cropper J, Little J, Strachan M, Stride A, Ersoy B, Eiberg H, Pedersen O, Shepherd MH, Hansen T, Harries LW, Hattersley AT (2007) Partial and whole gene deletion mutations of the GCK and HNF1A genes in maturity-onset diabetes of the young. Diabetologia 50:2313–2317
Franssen R, Young SG, Peelman F, Hertecant J, Sierts JA, Schimmel AW, Bensadoun A, Kastelein JJ, Fong LG, Dallinga-Thie GM, Beigneux AP (2010) Chylomicronemia with low postheparin lipoprotein lipase levels in the setting of GPIHBP1 defects. Circ Cardiovasc Genet 3:169–178
Gilbert N, Labuda D (1999) CORE-SINEs: eukaryotic short interspersed retroposing elements with common sequence motifs. Proc Natl Acad Sci U S A 96:2869–2874
Gin P, Beigneux AP, Davies B, Young MF, Ryan RO, Bensadoun A, Fong LG, Young SG (2007) Normal binding of lipoprotein lipase, chylomicrons, and apo-AV to GPIHBP1 containing a G56R amino acid substitution. Biochim Biophys Acta 1771:1464–1468
Gin P, Yin L, Davies BS, Weinstein MM, Ryan RO, Bensadoun A, Fong LG, Young SG, Beigneux AP (2008) The acidic domain of GPIHBP1 is important for the binding of lipoprotein lipase and chylomicrons. J Biol Chem 283:29554–29562
Havel RJ, Gordon RS Jr (1960) Idiopathic hyperlipemia: metabolic studies in an affected family. J Clin Invest 39:1777–1790
Hobbs HH, Brown MS, Goldstein JL (1992) Molecular genetics of the LDL receptor gene in familial hypercholesterolemia. Hum Mutat 1:445–466
Huang Y, Fedarovich A, Tomlinson S, Davies C (2007) Crystal structure of CD59: implications for molecular recognition of the complement proteins C8 and C9 in the membrane-attack complex. Acta Crystallogr D: Biol Crystallogr 63:714–721
Indian Genome Variation Consortium (2008) Genetic landscape of the people of India: a canvas for disease gene exploration. J Genet 87:3–20
Ioka RX, Kang MJ, Kamiyama S, Kim DH, Magoori K, Kamataki A, Ito Y, Takei YA, Sasaki M, Suzuki T, Sasano H, Takahashi S, Sakai J, Fujino T, Yamamoto TT (2003) Expression cloning and characterization of a novel glycosylphosphatidylinositol-anchored high density lipoprotein-binding protein, GPI-HBP1. J Biol Chem 278:7344–7349
Kidd JM, Cooper GM, Donahue WF, Hayden HS, Sampas N, Graves T, Hansen N, Teague B, Alkan C, Antonacci F, Haugen E, Zerr T, Yamada NA, Tsang P, Newman TL, Tuzun E, Cheng Z, Ebling HM, Tusneem N, David R, Gillett W, Phelps KA, Weaver M, Saranga D, Brand A, Tao W, Gustafson E, McKernan K, Chen L, Malig M, Smith JD, Korn JM, McCarroll SA, Altshuler DA, Peiffer DA, Dorschner M, Stamatoyannopoulos J, Schwartz D, Nickerson DA, Mullikin JC, Wilson RK, Bruhn L, Olson MV, Kaul R, Smith DR, Eichler EE (2008) Mapping and sequencing of structural variation from eight human genomes. Nature 453:56–64
Kjaergaard M, Hansen LV, Jacobsen B, Gardsvoll H, Ploug M (2008) Structure and ligand interactions of the urokinase receptor (uPAR). Front Biosci 13:5441–5461
Koral K, McMenamy J, Hauser N, Rollins N (2010) Hyperlipidemia resulting in abnormal density and signal intensity of blood in a neonate with lipoprotein lipase deficiency. AJNR Am J Neuroradiol 31:1999–2000
Korn ED (1955) Clearing factor, a heparin-activated lipoprotein lipase. I. Isolation and characterization of the enzyme from normal rat heart. J Biol Chem 215:1–14
Kozak L, Hrabincova E, Kintr J, Horky O, Zapletalova P, Blahakova I, Mejstrik P, Prochazkova D (2006) Identification and characterization of large deletions in the phenylalanine hydroxylase (PAH) gene by MLPA: evidence for both homologous and non-homologous mechanisms of rearrangement. Mol Genet Metab 89:300–309
Mills RE, Walter K, Stewart C, Handsaker RE, Chen K, Alkan C, Abyzov A, Yoon SC, Ye K, Cheetham RK, Chinwalla A, Conrad DF, Fu Y, Grubert F, Hajirasouliha I, Hormozdiari F, Iakoucheva LM, Iqbal Z, Kang S, Kidd JM, Konkel MK, Korn J, Khurana E, Kural D, Lam HY, Leng J, Li R, Li Y, Lin CY, Luo R, Mu XJ, Nemesh J, Peckham HE, Rausch T, Scally A, Shi X, Stromberg MP, Stutz AM, Urban AE, Walker JA, Wu J, Zhang Y, Zhang ZD, Batzer MA, Ding L, Marth GT, McVean G, Sebat J, Snyder M, Wang J, Eichler EE, Gerstein MB, Hurles ME, Lee C, McCarroll SA, Korbel JO (2011) Mapping copy number variation by population-scale genome sequencing. Nature 470:59–65
Olivecrona G, Ehrenborg E, Semb H, Makoveichuk E, Lindberg A, Hayden MR, Gin P, Davies BS, Weinstein MM, Fong LG, Beigneux AP, Young SG, Olivecrona T, Hernell O (2010) Mutation of conserved cysteines in the Ly6 domain of GPIHBP1 in familial chylomicronemia. J Lipid Res 51:1535–1545
Peterfy M, Ben-Zeev O, Mao HZ, Weissglas-Volkov D, Aouizerat BE, Pullinger CR, Frost PH, Kane JP, Malloy MJ, Reue K, Pajukanta P, Doolittle MH (2007) Mutations in LMF1 cause combined lipase deficiency and severe hypertriglyceridemia. Nat Genet 39:1483–1487
Peterson J, Fujimoto WY, Brunzell JD (1992) Human lipoprotein lipase: relationship of activity, heparin affinity, and conformation as studied with monoclonal antibodies. J Lipid Res 33:1165–1170
Prior TW, Bridgeman SJ (2005) Experience and strategy for the molecular testing of Duchenne muscular dystrophy. J Mol Diagn 7:317–326
Shaw CJ, Lupski JR (2005) Non-recurrent 17p11.2 deletions are generated by homologous and non-homologous mechanisms. Hum Genet 116:1–7
Spena S, Duga S, Asselta R, Peyvandi F, Mahasandana C, Malcovati M, Tenchini ML (2004) Congenital afibrinogenaemia caused by uniparental isodisomy of chromosome 4 containing a novel 15-kb deletion involving fibrinogen Aalpha-chain gene. Eur J Hum Genet 12:891–898
Toffolatti L, Cardazzo B, Nobile C, Danieli GA, Gualandi F, Muntoni F, Abbs S, Zanetti P, Angelini C, Ferlini A, Fanin M, Patarnello T (2002) Investigating the mechanism of chromosomal deletion: characterization of 39 deletion breakpoints in introns 47 and 48 of the human dystrophin gene. Genomics 80:523–530
Tuzun E, Sharp AJ, Bailey JA, Kaul R, Morrison VA, Pertz LM, Haugen E, Hayden H, Albertson D, Pinkel D, Olson MV, Eichler EE (2005) Fine-scale structural variation of the human genome. Nat Genet 37:727–732
Wang J, Hegele RA (2007) Homozygous missense mutation (G56R) in glycosylphosphatidylinositol-anchored high-density lipoprotein-binding protein 1 (GPI-HBP1) in two siblings with fasting chylomicronemia (MIM 144650). Lipids Health Dis 6:23
Weinstein MM, Yin L, Beigneux AP, Davies BS, Gin P, Estrada K, Melford K, Bishop JR, Esko JD, Dallinga-Thie GM, Fong LG, Bensadoun A, Young SG (2008) Abnormal patterns of lipoprotein lipase release into the plasma in GPIHBP1-deficient mice. J Biol Chem 283:34511–34518
Williams VC, Lucas J, Babcock MA, Gutmann DH, Korf B, Maria BL (2009) Neurofibromatosis type 1 revisited. Pediatrics 123:124–133
Zaidi G, Sahu RP, Zhang L, George G, Bhavani N, Shah N, Bhatia V, Bhansali A, Jevalikar G, Jayakumar RV, Eisenbarth GS, Bhatia E (2009) Two novel AIRE mutations in autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy (APECED) among Indians. Clin Genet 76:441–448
Acknowledgments
We thank Christine Quinn for her assistance and Stephen Young for helpful discussions. We thank Dr. John Brunzell (University of Washington) for supplying us with the monoclonal antibodies 5F9 and 5D2 employed in the LPL ELISA.
Funding Support
JJR is supported by training grant 5TLIDK081181. This grant was supported by the National Institutes of Health (RL1HL092550, R01-DK54387, and PO1 HL20948).
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Communicated by: Ertan Mayatepek
Competing interest: None declared.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Supplementary Table 1
Oligonucleotides used to sequence candidate genes. (DOCX 11 kb)
Supplementary Table 2
Sequence variations in candidate genes in the proband (Patient 1). (DOCX 11 kb)
Supplementary Figure 1
Quantile (Q-Q) plot comparison of the size distribution of regions of homozygosity. Regions of homozygosity were identified from Illumina genotype data using Partek Genomics Suite software. After filtering regions overlapping chromosome centromeres, the size distributions of homozygous regions were compared with the corresponding distributions of zix unrelated Caucasians analyzed on the same array platform. Patient 1 (red diamonds) had longer regions of homozygosity, which are represented by a shift above the normal expectation (dashed line) in intervals of homozygosity > ∼1,200 kb. The GPIHBP1 gene deletion is located in a ∼6.1 Mb region of homozygosity (arrow). The distribution of regions of homozygosity in a sample from a second-cousin mating is plotted using blue triangles. (JPEG 49 kb)
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Rios, J.J., Shastry, S., Jasso, J. et al. Deletion of GPIHBP1 causing severe chylomicronemia. J Inherit Metab Dis 35, 531–540 (2012). https://doi.org/10.1007/s10545-011-9406-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10545-011-9406-5