
Abstract
Purpose
Various aberrations in the fibroblast growth factor receptor genes FGFR1, FGFR2, and FGFR3 are found in different cancers, including breast cancer (BC). This study analyzed the impact of FGFR amplification on the BC prognosis.
Methods
The study included 894 BC patients. The amplification rates of FGFR1, FGFR2, and FGFR3 were evaluated on tissue microarrays using fluorescence in situ hybridization (FISH). Associations between these parameters and prognosis were analyzed using multivariate Cox regression analyses.
Results
FGFR1 FISH was assessable in 503 samples, FGFR2 FISH in 447, and FGFR3 FISH in 562. The FGFR1 amplification rate was 6.6% (n = 33). Increased FGFR2 copy numbers were seen in 0.9% (n = 4); only one patient had FGFR3 amplification (0.2%). Most patients with FGFR1 amplification had luminal B-like tumors (69.7%, n = 23); only 32.6% (n = 153) of patients without FGFR1 amplification had luminal B-like BC. Other patient and tumor characteristics appeared similar between these two groups. Observed outcome differences between BC patients with and without FGFR1 amplification did not achieve statistical significance; however, there was a trend toward poorer distant metastasis-free survival in BC patients with FGFR1 amplification (HR = 2.08; 95% CI 0.98 to 4.39, P = 0.05).
Conclusion
FGFR1 amplification occurs most frequently in patients with luminal B-like BC. The study showed a nonsignificant correlation with the prognosis, probably due to the small sample size. Further research is therefore needed to address the role of FGFR1 amplifications in early BC patients. FGFR2 and FGFR3 amplifications are rare in patients with primary BC.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Breast cancer (BC) is the most common malignant tumor in women [17]. Treatment decisions in breast cancer patients are based on tumor predictive markers [estrogen receptor (ER), progesterone receptor (PR), human epidermal growth factor receptor 2 (HER2)], some of which are also prognostic markers (ER, PR, HER2, Ki-67).
The prognostic and predictive values of many different biomarkers in relation to breast cancer have been evaluated in recent years. The discovery of HER2 amplification/overexpression as a therapeutic target and the development of the first anti-HER2 agent, trastuzumab, were pioneering advances [53]. In the era of personalized medicine, more and more genetic aberrations in potentially targetable oncogenic driver genes, such as copy number variations of CCND1 and PIK3CA mutations, are now being investigated [9, 40].
Another promising biomarker in breast cancer is the fibroblast growth factor receptor 1 gene (FGFR1, chromosomal region: 8p11.2-p12). It belongs to a family of receptor tyrosine kinases, activated by fibroblast growth factors, that influence the downstream MAPK, PI3K-AKT-mTOR, and STAT pathways. Stimulation of FGFR1 physiologically leads to proliferation, survival, migration, and angiogenesis [14, 59]. Amplification of FGFR1 is found in several types of cancer (e.g., nonsmall cell lung carcinoma, head and neck tumors, breast cancer, ovarian cancer, bladder cancer, and rhabdomyosarcoma) [10, 11, 22, 38, 52], with a frequency of up to 10% in breast cancer [61]. Chromosomal aberration has been found to be associated with FGFR1 overexpression, luminal B subtype (16–27%), negative PR expression, and high Ki-67 protein expression [61]. In addition, breast cancer cell lines with FGFR1 amplification harbor endocrine resistance that can be reversed by RNA silencing, and FGFR1-amplified breast cancers have been reported to be associated with a poorer prognosis [13].
In addition to FGFR1, fibroblast growth factor receptor 2 (FGFR2 gene, chromosome 10) and fibroblast growth factor receptor 3 (FGFR3 gene, chromosome 4) belong to the same family of receptor tyrosine kinases and are linked to breast cancer susceptibility. Single-nucleotide polymorphisms (SNPs) in FGFR2 locus 10q26 have been reported to have the strongest association with breast cancer risk in genome-wide association studies [12, 15, 28, 35,36,37]. FGFR2-amplified breast cancer was found with frequencies of up to 4.4% [7]. Wein et al. [63] reported the case of a patient with FGFR2-amplified metastatic hormone receptor-positive breast cancer, who benefited from therapy with the mTOR inhibitor everolimus and exemestane. The authors also carried out an analysis of the METABRIC (Molecular Taxonomy of Breast Cancer) dataset [9] and found a 1.8% rate of FGFR2 amplification in breast cancer, associated with a poorer prognosis and resistance to endocrine therapy [63]. FGFR3 has also been linked to an influence on endocrine resistance [57] and the risk of breast cancer (e.g., via SNPs) [1], but the amplification rate has been reported to be less than 1% [26].
Since fibroblast growth factor receptor (FGFR) alterations are found in a variety of cancers [64], several FGFR inhibitors—both pan-FGFR and also selective FGFR inhibitors—have been developed and tested in clinical trials [30, 62]. The results of these studies will show whether cancer patients are able to benefit from this targeted therapy. However, assuming that there is an association between FGFR1 amplification and prognostically unfavorable luminal B breast cancer, it may be hypothesized that FGFR inhibitors may improve the prognosis, particularly in patients who are suffering from highly proliferative, hormone receptor-positive breast cancer with FGFR1 amplification.
The aim of this study was to investigate the amplification rates of FGFR1, FGFR2, and FGFR3 in patients with breast cancer and their impact on prognosis.
Materials and methods
Patient cohort
The Bavarian Breast Cancer Cases and Control (BBCC) study, described in detail elsewhere [16], was a case–control study that initially included 1538 women with breast cancer, who received various treatments in accordance with University Breast Center guidelines at the University Breast Center for Franconia, which is part of the University Hospital Erlangen (Bavaria, Germany). Tumor samples were collected from 1997 to 2007 [65, 66]. Approval for the study was obtained from the local ethics committee at the University of Erlangen (ref. numbers 2700 and 297_17 Bc). The study was conducted in concordance to “Reporting recommendations for tumor marker prognostic studies (REMARK)” [34].
Collection of clinical and histopathological data
Clinical and follow-up data were obtained from the patients’ records. Data for histopathological parameters—TNM, grading, ER status, PR status, HER2 status, and proliferation rate measured with Ki-67 immunohistochemistry (IHC)—were obtained from the original pathology files. The detailed methods of assessing these parameters have been described elsewhere [16]. Molecular-like breast cancer subtypes were defined as follows:
-
Luminal A-like: ER-positive and/or PR-positive, in at least 10% of tumor cell nuclei (through December, 2009) or in at least 1% of tumor cell nuclei (since January, 2010); HER2-negative, Ki-67 < 14%
-
Luminal B-like (HER2-negative): ER-positive and/or PR-positive, HER2-negative, Ki-67 ≥ 14% [4];
-
HER2-enriched: HER2 + by either immunohistochemistry (IHC 3 +) [45] or fluorescence in situ hybridization (FISH) or both [43, 44, 46].
-
Basal-like or triple-negative: ER-negative, PR-negative, and HER2-negative.
Fluorescence in situ hybridization of FGFR1, FGFR2, and FGFR3
After tissue microarrays (TMAs) of formalin-fixed, paraffin-embedded tumor tissue had been built [16], fluorescence in situ hybridization (FISH) was performed in accordance with the manufacturer’s recommendations and in-house standards. The FISH probes used were ZytoLight SPEC FGFR1/CEN8 Dual Color Probe, ZytoLight SPEC FGFR2/CEN10 Dual Color Probe, and ZytoLight SPEC FGFR3/4p11 Dual Color Probe (all from ZytoVision GmbH, Bremerhaven, Germany). These each contained a green-labeled probe that targeted the FGFR gene locus (FGFR1, FGFR2, or FGFR3) and an orange-labeled probe that targeted the centromeric region of the particular chromosome (CEN8, CEN10, 4p11, respectively). For each TMA core, the green signal (FGFR gene locus) and orange signal (centromeric region) were counted in 20 tumor nuclei each, and the FGFR/CEN ratio was calculated. An FGFR/CEN ratio ≥ 2.0 was defined as amplification of each fibroblast growth factor receptor. To verify the validity of FISH staining positively (amplification) and negatively (no amplification), the following cell lines were used for validation: MDA MB-134, SUM-190, MFM-223, SNU-16, Kato III, HCC-70, MDA MB-361, BT-20, and MCF-7.
In order to rule out intratumoral heterogeneity of FGFR1 amplification, additional FGFR1 FISH analyses for a subgroup (n = 149) of the initial TMA cohort (TMA_1) were done. For the subcohort, FISH analyses were performed using two more TMAs (TMA_2 and TMA_3) that included each one further area of the tumor area unrelated to the tumor spot that was investigated initially in TMA_1. Results were shown with cross tabulations.
Statistical analysis
Due to small numbers of FGFR2-amplified and FGFR3-amplified cases, statistical analysis was limited to FGFR1 gene status. Disease-free survival (DFS) was defined as the time from the date of diagnosis to the earliest date of disease progression (distant metastasis, local recurrence, death from any cause) or the date of censoring. Patients who were lost to follow-up before the maximum observation period of 10 years, or who were disease-free after the maximum observation time, were censored at the last date they were known to be disease-free or at the maximum observation time. Distant metastasis-free survival (DMFS), overall survival (OS), and local recurrence-free survival (LRFS) were defined similarly.
The primary objective was to study the impact of FGFR1 on DFS. For this purpose, a simple Cox regression analysis with FGFR1 amplification (yes/no) as predictor was performed in order to obtain an unadjusted hazard ratio (HR) with 95% confidence intervals (CI) and corresponding P values. Survival rates were estimated using the Kaplan–Meier product limit method. An adjusted HR for FGFR1 amplification was estimated using a multiple Cox regression model with FGFR1 amplification as predictor, along with well-known prognostic characteristics of DFS: age at diagnosis (continuous), body mass index (BMI, continuous), tumor stage (ordinal, T1 to T4), tumor grade (ordinal, 1 to 3), ER status (positive versus negative), PR status (positive versus negative), HER2 status (positive versus negative), and Ki-67 (continuous, 0–100%). Lymph-node stage was incorporated into the model as a stratification factor (N0, N +) rather than a predictor, as the proportional hazards assumption was violated. Patients with missing information on FGFR1 gene status were excluded. Missing predictor values were imputed, and continuous predictors were used as natural cubic spline functions [50]. The proportional hazards assumptions were checked using the Grambsch–Therneau method [24].
Similar analyses were performed for the secondary objectives DMFS, OS, and LRFS. The association between immunohistochemical ER, PR, Ki-67 expression (0–100%), and FGFR1 amplification was also analyzed using summary statistics (median; interquartile range, IQR), box plots and Wilcoxon rank-sum tests. P values were not corrected for multiple testing.
All of the tests were two-sided, and a P value < 0.05 was regarded as statistically significant. Calculations were carried out using the R system for statistical computing (version 3.4.1; R Development Core Team, Vienna, Austria, 2017).
Results
Amplification of FGFR1 in invasive breast cancer
FGFR1 amplification rate in breast cancer
A total of 894 patients with breast cancer were initially included in the FGFR1 analysis. Patients with contralateral breast cancer, breast cancer with distant metastasis at diagnosis, missing FGFR1 data, and those without a positive observation time were excluded (n = 391; see Supplementary Table S1), resulting in a final sample size of 503 patients.
In this final cohort, amplification of the FGFR1 gene was observed in 6.6% (33 of 503).
Missing tissue cores were the most common reason for nonassessable cases. Cases with no signals, or barely visible signals, were then excluded.
In Fig. 1, one breast cancer case with FGFR1 amplification as well as one tumor without amplification but normal FGFR1 gene status are illustrated.

Illustration of FGFR1 fluorescence in situ hybridization (FISH). Tumor nuclei are marked using DAPI, the FGFR1 gene is depicted as green signal, the centromere (CEN8) is labeled with an orange signal (×1000, oil). One breast cancer case harbors FGFR1 amplification, whereas the other breast tumor shows normal gene copy number of FGFR1, respectively
Intratumoral homogeneity of FGFR1 amplification
When comparing different intratumoral areas that were not lying close to each other, we did not find intratumoral heterogeneity of FGFR1 amplification but perfect agreement regarding FGFR1 gene status (TMA_1 vs. TMA2: agreement in 64 of 64 cases; TMA_1 vs. TMA_3: agreement in 42 of 42 cases; see Supplementary Table S2).
However, it has to be noted that 3 of 64 cases (4.7%, TMA_1 vs. TMA2) and 2 of 42 cases (4.8%, TMA_1 vs. TMA_3), respectively, harbored slightly increased FGFR1 gene copy numbers with a FGFR1/CEN8 ratio each that was very close to the defined cut-off (≥ 2.0), but did not surpass 1.99 (data not shown).
Association of FGFR1 gene status and clinical and pathological parameters
The mean age of the patients with FGFR1-amplified breast cancer was 60 years, and they had a mean BMI of 27 kg/m2. More than half of these patients had pT1 tumors (54.5%) and 36.4% had positive lymph-node stages.
Most breast cancer patients with FGFR1 amplification showed moderate differentiation (G2, 69.7%) and a luminal subtype with positive hormone receptor status and predominantly HER2- negative status (ER+, 90.9%; PR+, 78.8%; HER2+, 9.1%). The mean proliferation rate assessed using Ki-67 expression amounted to 24.8%. Table 1 lists the characteristics of the patients and tumors relative to FGFR1 amplification.
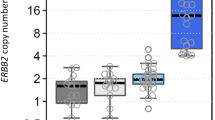
Patients with breast cancer who had normal FGFR1 copy numbers showed lower expression rate measurements (IHC) for the estrogen receptor (median, 70%; IQR, 10% to 80%) than patients with FGFR1 amplification (median, 80%; IQR, 60% to 90%; Fig. 2a). Such associations were not seen for the progesterone receptor (Fig. 2b) or Ki-67 IHC (Fig. 2c).

a Distribution of estrogen receptor (ER) expression relative to FGFR1 amplification status (P < 0.01). b Distribution of progesterone receptor (PR) expression relative to FGFR1 amplification status (P = 0.16). c Distribution of Ki-67 expression relative to FGFR1 amplification status (P = 0.11)
There were no cases of increased copy numbers in more than one of the FGFR genes (i.e., FGFR1 and FGFR2 and/or FGFR3 amplification).
Survival rates in FGFR1-amplified breast cancer.
Disease-free survival
The median follow-up period for the primary study aim of DFS was 10.0 years for patients both with and without FGFR1 amplification. No significant differences were observed between breast cancer patients with and without FGFR1 amplification in relation to DFS. The unadjusted HR was 1.60 (95% CI 0.88 to 2.89) and the adjusted HR was 1.25 (95% CI 0.67 to 2.32). The 5-year and 10-year survival rates are shown in Table 2.
Distant metastasis-free survival, overall survival, and local recurrence-free survival
Breast cancer patients with FGFR1 amplification had a poorer DMFS than patients without amplification (P = 0.04, unadjusted analysis); however, this difference in DMFS outcome did not achieve significance in the adjusted analysis (P = 0.05). No significant impact of FGFR1 amplification on the other secondary outcomes, OS and LRFS, was observed. Survival rates and HRs are presented in Tables 2 and 3. Kaplan–Meier curves are shown in Fig. 3.

a Kaplan–Meier curves for disease-free survival (DFS) relative to FGFR1 amplification status with log-rank test P value. b Kaplan–Meier curves for overall survival (OS) relative to FGFR1 amplification status with log-rank test P value. c Kaplan–Meier curves for distant metastasis-free survival (DMFS) relative to FGFR1 amplification status with log-rank test P value. d Kaplan–Meier curves for local recurrence-free survival (LRFS) relative to FGFR1 amplification status with log-rank test P value
FGFR2 and FGFR3 amplification in breast cancer
The evaluation of FGFR2 and FGFR3 gene status only revealed a very low frequency of copy number aberrations for each gene in the FISH analysis. FGFR2 was amplified in 0.9% (four of 447 cases assessable using FGFR2 FISH); only one case harbored FGFR3 amplification (0.2%, one of 562 cases assessable using FGFR3 FISH). Due to the low numbers of amplified cases, FGFR2 and FGFR3 gene status was excluded from further survival analysis.
Discussion
This study investigated the amplification rates of the fibroblast growth factor receptor genes FGFR1, FGFR2, and FGFR3 in patients with breast cancer. In view of the very small numbers of FGFR2-amplified and FGFR3-amplified cases, the analyses were on outcomes in patients with FGFR1 amplification. Amplification of FGFR1 was seen in 6.6% of assessable BC cases. Earlier studies have reported higher amplification rates of FGFR1 and the corresponding chromosomal region 8p11-12 (8.7–13.2%) [7, 13, 19, 54]. The fact that the frequency of FGFR1 amplification in the present study was lower might be due to different methods of evaluating gene status (e.g., multiplex ligation-dependent probe amplification), a different composition of the cohort (e.g., varying distribution of intrinsic subtypes), and the fact that the FGFR1 gene was not always included in the amplification unit in the earlier studies. The present study did not investigate variations in gene copy numbers for other genes included in the previously described 8p11.2-p12 amplicon [9].
In this study, BC patients with FGFR1 amplification showed a trend toward poorer outcomes, especially DMFS and LRFS. FGFR1 amplification was not an independent predictor of shorter DFS or OS. Thus, the study does not fully confirm the findings of an earlier report that FGFR1-amplified BC was associated with poorer OS in the overall cohort and that FGFR1 amplification was predictive of poor DFS, OS, and DMFS in ER-positive patients with BC [13]. Cuny et al. reported a shorter DFS in FGFR1-amplified BC in comparison with nonamplified carcinomas. Intriguingly, co-amplification of FGFR1 and the cyclin D1 gene (CCND1) showed even poorer DFS than increased FGFR1 copy numbers without CCND1 amplification [8]. It may be presumed that varying distributions of intrinsic subtypes contribute to these different findings. It should also be mentioned that the survival analyses in the present study were limited, as the cohort investigated included only 33 cases of FGFR1-amplified BC. In another study, FGFR1 amplification was not associated with relapse-free survival (RFS) or BC-specific survival. Instead, protein expression predicted shorter RFS in ER-positive/HER2-negative BC [56]. Further comprehensive studies are therefore needed in order to investigate the impact of FGFR1 amplification on survival in BC patients.
Estrogen receptor-positive BC [7] and luminal B BC [61] have been reported to show the highest frequency of FGFR1 amplifications. In one study, the amplification rates were 21.0% for luminal B, 12.7% for basal-like, 10.4% for luminal A, and 7.1% for HER2-positive invasive BC [29]. Interestingly, luminal A BC that expressed high levels of FGFR1 has been found to behave more aggressively, with a prognosis similar to that in luminal B tumors [51]. In the present study, most FGFR1-amplified tumors were ER-positive (90.9%), a finding that is consistent with the results published by Moelans et al. [39]. The present study showed that 69.7% of BCs with FGFR1 amplification harbored a luminal B phenotype, whereas the other intrinsic subtypes were only found at lower frequencies, up to 15.2%. However, due to the small numbers of FGFR1-amplified cases in the study, survival analysis of intrinsic subtypes relative to FGFR1 copy number status was not feasible.
Inhibition of FGFR1 has been regarded as a potential therapeutic target. On the assumption that the receptor tyrosine kinase (RTK) FGFR1 is the driver of the 8p11.2-p12 amplicon and represents a potential drug target in a variety of cancers, RTK-targeting small-molecule inhibitors against FGFR1 such as ponatinib, dovitinib, PD173074, and SU5402 were designed, and knockdown and preclinical pharmaceutical inhibition studies were carried out [2, 5, 10, 23, 31, 47, 62]. Multikinase inhibitors (TKIs), such as lucitanib (E-3810, NCT01283945), dovitinib (TKI258), nintedanib, and ponatinib [42], and selective FGFR inhibitors, such as AZD4547 [18] (NCT00979134, NCT01202591), BGJ398 (NCT01004224), LY2874455 (NCT01212107), and JNJ-42756493 (NCT01703481), have been tested in several phase I and II trials in cancers with FGFR1 aberrations [10]. In patients with advanced BC, the overall response rate was up to 50% and progression-free survival up to 10.9 months when the agents were combined with fulvestrant, but overall, the desired efficacy of FGFR inhibitors has not been achieved in (pre-)clinical studies [41, 42]. Administration of multi-TKIs was accompanied by asthenia, gastrointestinal symptoms, hypertension, and lymphopenia, whereas selective FGFR inhibitors led to hyperphosphatemia, gastrointestinal symptoms, nail toxicity, and stomatitis [2, 42]. Amplification of FGFR1 was seen in up to 43% of patients with invasive lobular cancer (ILC) of the breast, and it was associated with expression. FGFR1 inhibition has been found to reduce the viability of the BC cell line MDA-MB-134, which has similarities to ILC in relation to some copy number variations (including FGFR1) and protein expression [49]. In FGFR1-amplified ILC, ribosomal S6 kinase (RSK) inhibitors may be another potential drug target, as Xian et al. observed an effect of RSK on FGFR1-transformed cells [68]. In addition, simultaneous inhibition of FGFR1 and vascular endothelial growth factor receptor 1 (VEGFR1) may lead to anti-angiogenic effects in vivo [21]. ODM-203 acts as a selective dual blockade of FGFR and VEGFR, but may cause hyperphosphatemia and bilirubinemia [27]. There is evidence that high levels of FGFR1 expression are associated with resistance to anti-HER2 therapy in patients with HER2-positive BC [25]. It needs to be tested whether a combination of anti-HER2 therapy and FGFR inhibition might help to resolve this issue. In addition, FGFR inhibition may reverse resistance to endocrine therapy and anti-CDK4/6 therapy, and since 26.4% of FGFR1-amplified BC has been found to have PIK3CA alterations [26], it may be combined with inhibitors of the PI3K pathway. However, further investigation of the efficacy and safety of the inhibitors and combinations of these agents is needed [42].
FGFR1 amplification is associated with increased expression [33, 61]. However, it needs to be borne in mind that increased copy numbers may not always predict high levels of FGFR1 protein expression [48], so that inhibition might fail if treatment were to be selected on the basis of FGFR1 amplification. This may be related to the quality of the FISH assay and IHC assay used. Measurement of protein or mRNA expression may lead to better prediction of the response to FGFR inhibitors in head and neck cancer [20], but this issue has yet to be investigated in BC [42]. One limitation of our study is that, up to date, we did not analyze the association between FGFR1 amplification and protein expression of FGFR1. Further investigations have to show whether the copy number gain of the gene lead to increased expression of this growth factor receptor in our breast cancer cohort and whether FGFR1 overexpression might predict prognosis.
The 8p11-12 amplicon was significantly associated with DFS and distal recurrence [6]. However, it is still controversial whether FGFR1 itself is the driver oncogene, or whether another gene in the 8p11.2-p12 amplicon is responsible for the oncogenic potential [7, 13, 19]. FGFR1 amplification is apparently not always associated with FGFR1 overexpression, and the oncogenic amplicon 8p11.2-p12 is not always accompanied by FGFR1 amplification and sensitivity to FGFR inhibition [48, 55]. It therefore remains unclear whether FGFR1 is the appropriate drug target, or another gene in the amplicon, and this has led to ongoing discussion and investigation of this issue.
In addition to FGFR1 copy number aberrations, an FGFR1 SNP (rs17182023) has been investigated and was found to be associated with a reduced risk of BC and lower FGFR1 expression. By contrast, high levels of FGFR1 were associated with a poor outcome [67].
The FGFR2 locus has been shown to be one of the regions associated most strongly with the risk of BC in genome-wide association studies [12, 15, 35,36,37]. Rather than increased gene copy numbers, SNPs in the FGFR2 risk locus appear to be associated with the development of BC. Campbell et al. reported reduced FGFR2 expression and consequently—due to less influence of FGFR2 on the estrogen regulon—increased responsiveness to estrogen if one of three FGFR2 variants existed [3]. Copy number aberrations appear to be less important in BC. The FGFR2 gene was amplified in only 0.9% of the patients with BC in the present study, which is lower than the rate of 4.4% described in the literature [7]. This might be due to the composition of the cohort, since Turner et al. reported FGFR2 amplification in 4.0% of triple-negative BCs, but not in any other subtype [60]. In view of the poor prognosis for patients with TNBC without treatment and the current lack of an approved targeted therapy, FGFR inhibitors may be a treatment option in FGFR2-amplified/FGFR2-overexpressing TNBC. Initial preclinical data are promising [58], but this has yet to be confirmed in clinical studies.
The receptor tyrosine kinase FGFR3 may influence hormone receptor-positive BC that is resistant to tamoxifen [57]. However, amplification of FGFR3 is fairly exceptional in BC, as it was found in 0.2% of the present cohort and in 0.8% of BC cases investigated by Helsten et al. [26]. The SNP FGFR3_rs743682 was found to be associated with the risk of BC, but did not reach the same association level as the SNP FGFR2_ rs2981582 [1].
It is not only copy number variations and SNPs in the FGFR genes that can lead to high expression of the respective fibroblast growth factor receptor and up-regulated FGFR pathways. Mutations, rearrangements, post-transcriptional regulation, and isoform switching/alternative splicing, as well as stimulation via fibroblast growth factors from tumor or stromal cells, can also have an impact on the system [62]. For instance, the FGFR1 splice variant IIIb has been reported to inhibit cell growth [32]. These numerous changes should be borne in mind when FGFR-targeted therapy is being tested.
In conclusion, fibroblast growth factor receptor alterations (e.g., FGFR1 copy number variations and FGFR2 SNPs) influence the risk and prognosis in patients with breast cancer. Further investigations of the dysregulated FGFR pathways and the effects of FGFR inhibitors are needed. Due to the complexity of the FGFR1 amplicon itself, and because of the results that have been published so far, clarification of the driver gene of the FGFR1 amplicon 8p11.2-p12 is warranted in order to identify the potential target gene.
Abbreviations
- BBCC:
-
Bavarian Breast Cancer Cases and Control (study)
- BC:
-
Breast cancer
- BMI:
-
Body mass index
- CI:
-
Confidence interval(s)
- DFS:
-
Disease-free survival
- DMFS:
-
Distant metastasis-free survival
- ER:
-
Estrogen receptor
- FGFR:
-
Fibroblast growth factor receptor
- FISH:
-
Fluorescence in situ hybridization
- HER2:
-
Human epidermal growth factor receptor 2
- HR:
-
Hazard ratio
- IHC:
-
Immunohistochemistry
- ILC:
-
Invasive lobular cancer
- IQR:
-
Interquartile range
- LRFS:
-
Local recurrence-free survival
- OS:
-
Overall survival
- PR:
-
Progesterone receptor
- RFS:
-
Relapse-free survival
- RSK:
-
Ribosomal S6 kinase
- RTK:
-
Receptor tyrosine kinase
- TKI:
-
Multikinase inhibitor
- TMA:
-
Tissue microarray
- VEGFR1:
-
Vascular endothelial growth factor receptor 1
References
Agarwal D, Pineda S, Michailidou K, Herranz J, Pita G, Moreno LT, Alonso MR, Dennis J, Wang Q, Bolla MK, Meyer KB, Menendez-Rodriguez P, Hardisson D, Mendiola M, Gonzalez-Neira A, Lindblom A, Margolin S, Swerdlow A, Ashworth A, Orr N, Jones M, Matsuo K, Ito H, Iwata H, Kondo N, Hartman M, Hui M, Lim WY, Iau PT, Sawyer E, Tomlinson I, Kerin M, Miller N, Kang D, Choi J, Park SK, Noh D, Hopper JL, Schmidt DF, Makalic E, Southey MC, Teo SH, Yip CH, Sivanandan K, Tay W, Brauch H, Bruning T, Hamann U, Dunning AM, Shah M, Andrulis IL, Knight JA, Glendon G, Tchatchou S, Schmidt MK, Broeks A, Rosenberg EH, van’t Veer LJ, Fasching PA, Renner SP, Ekici AB, Beckmann MW, Shen C, Hsiung C, Yu J, Hou M, Blot W, Cai Q, Wu AH, Tseng C, Van Den Berg D, Stram DO, Cox A, Brock IW, Reed MW, Muir K, Lophatananon A, Stewart-Brown S, Siriwanarangsan P, Zheng W, Deming-Halverson S, Shrubsole MJ, Long J, Shu X, Lu W, Gao Y, Zhang B, Radice P, Peterlongo P, Manoukian S, Mariette F, Sangrajrang S, McKay J, Couch FJ, Toland AE, Yannoukakos D, Fletcher O, Johnson N, dos Santos SI, Peto J et al (2014) FGF receptor genes and breast cancer susceptibility: results from the Breast Cancer Association Consortium. Br J Cancer 110:1088–1100. https://doi.org/10.1038/bjc.2013.769
Andre F, Bachelot T, Campone M, Dalenc F, Perez-Garcia JM, Hurvitz SA, Turner N, Rugo H, Smith JW, Deudon S, Shi M, Zhang Y, Kay A, Porta DG, Yovine A, Baselga J (2013) Targeting FGFR with dovitinib (TKI258): preclinical and clinical data in breast cancer. Clin Cancer Res 19:3693–3702. https://doi.org/10.1158/1078-0432.CCR-13-0190
Campbell TM, Castro MAA, de Santiago I, Fletcher MNC, Halim S, Prathalingam R, Ponder BAJ, Meyer KB (2016) FGFR2 risk SNPs confer breast cancer risk by augmenting oestrogen responsiveness. Carcinogenesis 37:741–750. https://doi.org/10.1093/carcin/bgw065
Cheang MC, Chia SK, Voduc D, Gao D, Leung S, Snider J, Watson M, Davies S, Bernard PS, Parker JS, Perou CM, Ellis MJ, Nielsen TO (2009) Ki67 index, HER2 status, and prognosis of patients with luminal B breast cancer. J Natl Cancer Inst 101:736–750. https://doi.org/10.1093/jnci/djp082
Cheng W, Wang M, Tian X, Zhang X (2017) An overview of the binding models of FGFR tyrosine kinases in complex with small molecule inhibitors. Eur J Med Chem 126:476–490. https://doi.org/10.1016/j.ejmech.2016.11.052
Chin K, DeVries S, Fridlyand J, Spellman PT, Roydasgupta R, Kuo WL, Lapuk A, Neve RM, Qian Z, Ryder T, Chen F, Feiler H, Tokuyasu T, Kingsley C, Dairkee S, Meng Z, Chew K, Pinkel D, Jain A, Ljung BM, Esserman L, Albertson DG, Waldman FM, Gray JW (2006) Genomic and transcriptional aberrations linked to breast cancer pathophysiologies. Cancer Cell 10:529–541. https://doi.org/10.1016/j.ccr.2006.10.009
Courjal F, Cuny M, Simony-Lafontaine J, Louason G, Speiser P, Zeillinger R, Rodriguez C, Theillet C (1997) Mapping of DNA amplifications at 15 chromosomal localizations in 1875 breast tumors: definition of phenotypic groups. Cancer Res 57:4360–4367
Cuny M, Kramar A, Courjal F, Johannsdottir V, Iacopetta B, Fontaine H, Grenier J, Culine S, Theillet C (2000) Relating genotype and phenotype in breast cancer: an analysis of the prognostic significance of amplification at eight different genes or loci and of p53 mutations. Cancer Res 60:1077–1083
Curtis C, Shah SP, Chin SF, Turashvili G, Rueda OM, Dunning MJ, Speed D, Lynch AG, Samarajiwa S, Yuan Y, Graf S, Ha G, Haffari G, Bashashati A, Russell R, McKinney S, METABRIC Group, Langerod A, Green A, Provenzano E, Wishart G, Pinder S, Watson P, Markowetz F, Murphy L, Ellis I, Purushotham A, Borresen-Dale AL, Brenton JD, Tavare S, Caldas C, Aparicio S (2012) The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature 486:346–352. https://doi.org/10.1038/nature10983
Dienstmann R, Rodon J, Prat A, Perez-Garcia J, Adamo B, Felip E, Cortes J, Iafrate AJ, Nuciforo P, Tabernero J (2014) Genomic aberrations in the FGFR pathway: opportunities for targeted therapies in solid tumors. Ann Oncol 25:552–563. https://doi.org/10.1093/annonc/mdt419
Dutt A, Ramos AH, Hammerman PS, Mermel C, Cho J, Sharifnia T, Chande A, Tanaka KE, Stransky N, Greulich H, Gray NS, Meyerson M (2011) Inhibitor-sensitive FGFR1 amplification in human non-small cell lung cancer. PLoS ONE 6:e20351. https://doi.org/10.1371/journal.pone.0020351
Easton DF, Pooley KA, Dunning AM, Pharoah PD, Thompson D, Ballinger DG, Struewing JP, Morrison J, Field H, Luben R, Wareham N, Ahmed S, Healey CS, Bowman R, Meyer KB, Haiman CA, Kolonel LK, Henderson BE, Le Marchand L, Brennan P, Sangrajrang S, Gaborieau V, Odefrey F, Shen CY, Wu PE, Wang HC, Eccles D, Evans DG, Peto J, Fletcher O, Johnson N, Seal S, Stratton MR, Rahman N, Chenevix-Trench G, Bojesen SE, Nordestgaard BG, Axelsson CK, Garcia-Closas M, Brinton L, Chanock S, Lissowska J, Peplonska B, Nevanlinna H, Fagerholm R, Eerola H, Kang D, Yoo KY, Noh DY, Ahn SH, Hunter DJ, Hankinson SE, Cox DG, Hall P, Wedren S, Liu J, Low YL, Bogdanova N, Schurmann P, Dork T, Tollenaar RA, Jacobi CE, Devilee P, Klijn JG, Sigurdson AJ, Doody MM, Alexander BH, Zhang J, Cox A, Brock IW, MacPherson G, Reed MW, Couch FJ, Goode EL, Olson JE, Meijers-Heijboer H, van den Ouweland A, Uitterlinden A, Rivadeneira F, Milne RL, Ribas G, Gonzalez-Neira A, Benitez J, Hopper JL, McCredie M, Southey M, Giles GG, Schroen C, Justenhoven C, Brauch H, Hamann U, Ko YD, Spurdle AB, Beesley J, Chen X, Mannermaa A, Kosma VM, Kataja V, Hartikainen J, Day NE et al (2007) Genome-wide association study identifies novel breast cancer susceptibility loci. Nature 447:1087–1093. https://doi.org/10.1038/nature05887
Elbauomy Elsheikh S, Green AR, Lambros MB, Turner NC, Grainge MJ, Powe D, Ellis IO, Reis-Filho JS (2007) FGFR1 amplification in breast carcinomas: a chromogenic in situ hybridisation analysis. Breast Cancer Res (BCR) 9:R23. https://doi.org/10.1186/bcr1665
Eswarakumar VP, Lax I, Schlessinger J (2005) Cellular signaling by fibroblast growth factor receptors. Cytokine Growth Factor Rev 16:139–149. https://doi.org/10.1016/j.cytogfr.2005.01.001
Fachal L, Aschard H, Beesley J, Barnes DR, Allen J, Kar S, Pooley KA, Dennis J, Michailidou K, Turman C, Soucy P, Lemacon A, Lush M, Tyrer JP, Ghoussaini M, Marjaneh MM, Jiang X, Agata S, Aittomaki K, Alonso MR, Andrulis IL, Anton-Culver H, Antonenkova NN, Arason A, Arndt V, Aronson KJ, Arun BK, Auber B, Auer PL, Azzollini J, Balmana J, Barkardottir RB, Barrowdale D, Beeghly-Fadiel A, Benitez J, Bermisheva M, Bialkowska K, Blanco AM, Blomqvist C, Blot W, Bogdanova NV, Bojesen SE, Bolla MK, Bonanni B, Borg A, Bosse K, Brauch H, Brenner H, Briceno I, Brock IW, Brooks-Wilson A, Bruning T, Burwinkel B, Buys SS, Cai Q, Caldes T, Caligo MA, Camp NJ, Campbell I, Canzian F, Carroll JS, Carter BD, Castelao JE, Chiquette J, Christiansen H, Chung WK, Claes KBM, Clarke CL, Collaborators GS, Collaborators E, Collee JM, Cornelissen S, Couch FJ, Cox A, Cross SS, Cybulski C, Czene K, Daly MB, de la Hoya M, Devilee P, Diez O, Ding YC, Dite GS, Domchek SM, Dork T, Dos-Santos-Silva I, Droit A, Dubois S, Dumont M, Duran M, Durcan L, Dwek M, Eccles DM, Engel C, Eriksson M, Evans DG, Fasching PA, Fletcher O, Floris G, Flyger H et al (2020) Fine-mapping of 150 breast cancer risk regions identifies 191 likely target genes. Nat Genet. https://doi.org/10.1038/s41588-019-0537-1
Fasching PA, Weihbrecht S, Haeberle L, Gasparyan A, Villalobos IE, Ma Y, Ekici AB, Wachter DL, Hartmann A, Beckmann MW, Slamon DJ, Press MF (2014) HER2 and TOP2A amplification in a hospital-based cohort of breast cancer patients: associations with patient and tumor characteristics. Breast Cancer Res Treat 145:193–203. https://doi.org/10.1007/s10549-014-2922-x
Ferlay J, Soerjomataram I, Dikshit R, Eser S, Mathers C, Rebelo M, Parkin DM, Forman D, Bray F (2015) Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer 136:E359–386. https://doi.org/10.1002/ijc.29210
Gavine PR, Mooney L, Kilgour E, Thomas AP, Al-Kadhimi K, Beck S, Rooney C, Coleman T, Baker D, Mellor MJ, Brooks AN, Klinowska T (2012) AZD4547: an orally bioavailable, potent, and selective inhibitor of the fibroblast growth factor receptor tyrosine kinase family. Cancer Res 72:2045–2056. https://doi.org/10.1158/0008-5472.can-11-3034
Gelsi-Boyer V, Orsetti B, Cervera N, Finetti P, Sircoulomb F, Rouge C, Lasorsa L, Letessier A, Ginestier C, Monville F, Esteyries S, Adelaide J, Esterni B, Henry C, Ethier SP, Bibeau F, Mozziconacci MJ, Charafe-Jauffret E, Jacquemier J, Bertucci F, Birnbaum D, Theillet C, Chaffanet M (2005) Comprehensive profiling of 8p11-12 amplification in breast cancer. Mol Cancer Res (MCR) 3:655–667. https://doi.org/10.1158/1541-7786.MCR-05-0128
Goke F, Franzen A, Hinz TK, Marek LA, Yoon P, Sharma R, Bode M, von Maessenhausen A, Lankat-Buttgereit B, Goke A, Golletz C, Kirsten R, Boehm D, Vogel W, Kleczko EK, Eagles JR, Hirsch FR, Van Bremen T, Bootz F, Schroeck A, Kim J, Tan AC, Jimeno A, Heasley LE, Perner S (2015) FGFR1 expression levels predict BGJ398 sensitivity of FGFR1-dependent head and neck squamous cell cancers. Clin Cancer Res 21:4356–4364. https://doi.org/10.1158/1078-0432.CCR-14-3357
Golfmann K, Meder L, Koker M, Volz C, Borchmann S, Tharun L, Dietlein F, Malchers F, Florin A, Buttner R, Rosen N, Rodrik-Outmezguine V, Hallek M, Ullrich RT (2018) Synergistic anti-angiogenic treatment effects by dual FGFR1 and VEGFR1 inhibition in FGFR1-amplified breast cancer. Oncogene 37:5682–5693. https://doi.org/10.1038/s41388-018-0380-3
Gorringe KL, Jacobs S, Thompson ER, Sridhar A, Qiu W, Choong DY, Campbell IG (2007) High-resolution single nucleotide polymorphism array analysis of epithelial ovarian cancer reveals numerous microdeletions and amplifications. Clin Cancer Res 13:4731–4739. https://doi.org/10.1158/1078-0432.CCR-07-0502
Gozgit JM, Wong MJ, Moran L, Wardwell S, Mohemmad QK, Narasimhan NI, Shakespeare WC, Wang F, Clackson T, Rivera VM (2012) Ponatinib (AP24534), a multitargeted Pan-FGFR inhibitor with activity in multiple FGFR-amplified or mutated cancer models. Mol Cancer Ther 11:690–699. https://doi.org/10.1158/1535-7163.mct-11-0450
Grambsch PM, Therneau TM (1994) Proportional hazards tests and diagnostics based on weighted residuals. Biometrika 81:515–526
Hanker AB, Garrett JT, Estrada MV, Moore PD, Ericsson PG, Koch JP, Langley E, Singh S, Kim PS, Frampton GM, Sanford E, Owens P, Becker J, Groseclose MR, Castellino S, Joensuu H, Huober J, Brase JC, Majjaj S, Brohee S, Venet D, Brown D, Baselga J, Piccart M, Sotiriou C, Arteaga CL (2017) HER2-overexpressing breast cancers amplify FGFR signaling upon acquisition of resistance to dual therapeutic blockade of HER2. Clin Cancer Res 23:4323–4334. https://doi.org/10.1158/1078-0432.CCR-16-2287
Helsten T, Elkin S, Arthur E, Tomson BN, Carter J, Kurzrock R (2016) The FGFR landscape in cancer: analysis of 4,853 tumors by next-generation sequencing. Clin Cancer Res 22:259–267. https://doi.org/10.1158/1078-0432.ccr-14-3212
Holmstrom TH, Moilanen AM, Ikonen T, Bjorkman ML, Linnanen T, Wohlfahrt G, Karlsson S, Oksala R, Korjamo T, Samajdar S, Rajagopalan S, Chelur S, Narayanan K, Ramachandra RK, Mani J, Nair R, Gowda N, Anthony T, Dhodheri S, Mukherjee S, Ujjinamatada RK, Srinivas N, Ramachandra M, Kallio PJ (2019) ODM-203, a selective inhibitor of FGFR and VEGFR, shows strong antitumor activity, and induces antitumor immunity. Mol Cancer Ther 18:28–38. https://doi.org/10.1158/1535-7163.MCT-18-0204
Hunter DJ, Kraft P, Jacobs KB, Cox DG, Yeager M, Hankinson SE, Wacholder S, Wang Z, Welch R, Hutchinson A, Wang J, Yu K, Chatterjee N, Orr N, Willett WC, Colditz GA, Ziegler RG, Berg CD, Buys SS, McCarty CA, Feigelson HS, Calle EE, Thun MJ, Hayes RB, Tucker M, Gerhard DS, Fraumeni JF Jr, Hoover RN, Thomas G, Chanock SJ (2007) A genome-wide association study identifies alleles in FGFR2 associated with risk of sporadic postmenopausal breast cancer. Nat Genet 39:870–874. https://doi.org/10.1038/ng2075
Jang M, Kim E, Choi Y, Lee H, Kim Y, Kim J, Kang E, Kim SW, Kim I, Park S (2012) FGFR1 is amplified during the progression of in situ to invasive breast carcinoma. Breast Cancer Res 14:R115. https://doi.org/10.1186/bcr3239
Katoh M (2016) FGFR inhibitors: Effects on cancer cells, tumor microenvironment and whole-body homeostasis (Review). Int J Mol Med 38:3–15. https://doi.org/10.3892/ijmm.2016.2620
Liang G, Liu Z, Wu J, Cai Y, Li X (2012) Anticancer molecules targeting fibroblast growth factor receptors. Trends Pharmacol Sci 33:531–541. https://doi.org/10.1016/j.tips.2012.07.001
Liu Z, Neiss N, Zhou S, Henne-Bruns D, Korc M, Bachem M, Kornmann M (2007) Identification of a fibroblast growth factor receptor 1 splice variant that inhibits pancreatic cancer cell growth. Cancer Res 67:2712–2719. https://doi.org/10.1158/0008-5472.CAN-06-3843
Luo J, Liu S, Leung S, Gru AA, Tao Y, Hoog J, Ho J, Davies SR, Allred DC, Salavaggione AL, Snider J, Mardis ER, Nielsen TO, Ellis MJ (2017) An mRNA gene expression-based signature to identify FGFR1-amplified estrogen receptor-positive breast tumors. J Mol Diag (JMD) 19:147–161. https://doi.org/10.1016/j.jmoldx.2016.09.007
McShane LM, Altman DG, Sauerbrei W, Taube SE, Gion M, Clark GM (2006) REporting recommendations for tumor MARKer prognostic studies (REMARK). Breast Cancer Res Treat 100:229–235. https://doi.org/10.1007/s10549-006-9242-8
Michailidou K, Beesley J, Lindstrom S, Canisius S, Dennis J, Lush MJ, Maranian MJ, Bolla MK, Wang Q, Shah M, Perkins BJ, Czene K, Eriksson M, Darabi H, Brand JS, Bojesen SE, Nordestgaard BG, Flyger H, Nielsen SF, Rahman N, Turnbull C, Fletcher O, Peto J, Gibson L, dos-Santos-Silva I, Chang-Claude J, Flesch-Janys D, Rudolph A, Eilber U, Behrens S, Nevanlinna H, Muranen TA, Aittomaki K, Blomqvist C, Khan S, Aaltonen K, Ahsan H, Kibriya MG, Whittemore AS, John EM, Malone KE, Gammon MD, Santella RM, Ursin G, Makalic E, Schmidt DF, Casey G, Hunter DJ, Gapstur SM, Gaudet MM, Diver WR, Haiman CA, Schumacher F, Henderson BE, Le Marchand L, Berg CD, Chanock SJ, Figueroa J, Hoover RN, Lambrechts D, Neven P, Wildiers H, van Limbergen E, Schmidt MK, Broeks A, Verhoef S, Cornelissen S, Couch FJ, Olson JE, Hallberg E, Vachon C, Waisfisz Q, Meijers-Heijboer H, Adank MA, van der Luijt RB, Li J, Liu J, Humphreys K, Kang D, Choi JY, Park SK, Yoo KY, Matsuo K, Ito H, Iwata H, Tajima K, Guenel P, Truong T, Mulot C, Sanchez M, Burwinkel B, Marme F, Surowy H, Sohn C, Wu AH, Tseng CC, Van Den Berg D, Stram DO, Gonzalez-Neira A, Benitez J et al (2015) Genome-wide association analysis of more than 120,000 individuals identifies 15 new susceptibility loci for breast cancer. Nat Genet 47:373–380. https://doi.org/10.1038/ng.3242
Michailidou K, Hall P, Gonzalez-Neira A, Ghoussaini M, Dennis J, Milne RL, Schmidt MK, Chang-Claude J, Bojesen SE, Bolla MK, Wang Q, Dicks E, Lee A, Turnbull C, Rahman N, Fletcher O, Peto J, Gibson L, Dos Santos Silva I, Nevanlinna H, Muranen TA, Aittomaki K, Blomqvist C, Czene K, Irwanto A, Liu J, Waisfisz Q, Meijers-Heijboer H, Adank M, van der Luijt RB, Hein R, Dahmen N, Beckman L, Meindl A, Schmutzler RK, Muller-Myhsok B, Lichtner P, Hopper JL, Southey MC, Makalic E, Schmidt DF, Uitterlinden AG, Hofman A, Hunter DJ, Chanock SJ, Vincent D, Bacot F, Tessier DC, Canisius S, Wessels LF, Haiman CA, Shah M, Luben R, Brown J, Luccarini C, Schoof N, Humphreys K, Li J, Nordestgaard BG, Nielsen SF, Flyger H, Couch FJ, Wang X, Vachon C, Stevens KN, Lambrechts D, Moisse M, Paridaens R, Christiaens MR, Rudolph A, Nickels S, Flesch-Janys D, Johnson N, Aitken Z, Aaltonen K, Heikkinen T, Broeks A, Veer LJ, van der Schoot CE, Guenel P, Truong T, Laurent-Puig P, Menegaux F, Marme F, Schneeweiss A, Sohn C, Burwinkel B, Zamora MP, Perez JI, Pita G, Alonso MR, Cox A, Brock IW, Cross SS, Reed MW, Sawyer EJ, Tomlinson I, Kerin MJ, Miller N, Henderson BE et al (2013) Large-scale genotyping identifies 41 new loci associated with breast cancer risk. Nat Genet 45:353–361, 361e351–352. https://doi.org/10.1038/ng.2563
Michailidou K, Lindstrom S, Dennis J, Beesley J, Hui S, Kar S, Lemacon A, Soucy P, Glubb D, Rostamianfar A, Bolla MK, Wang Q, Tyrer J, Dicks E, Lee A, Wang Z, Allen J, Keeman R, Eilber U, French JD, Qing Chen X, Fachal L, McCue K, McCart Reed AE, Ghoussaini M, Carroll JS, Jiang X, Finucane H, Adams M, Adank MA, Ahsan H, Aittomaki K, Anton-Culver H, Antonenkova NN, Arndt V, Aronson KJ, Arun B, Auer PL, Bacot F, Barrdahl M, Baynes C, Beckmann MW, Behrens S, Benitez J, Bermisheva M, Bernstein L, Blomqvist C, Bogdanova NV, Bojesen SE, Bonanni B, Borresen-Dale AL, Brand JS, Brauch H, Brennan P, Brenner H, Brinton L, Broberg P, Brock IW, Broeks A, Brooks-Wilson A, Brucker SY, Bruning T, Burwinkel B, Butterbach K, Cai Q, Cai H, Caldes T, Canzian F, Carracedo A, Carter BD, Castelao JE, Chan TL, David Cheng TY, Seng Chia K, Choi JY, Christiansen H, Clarke CL, Collaborators N, Collee M, Conroy DM, Cordina-Duverger E, Cornelissen S, Cox DG, Cox A, Cross SS, Cunningham JM, Czene K, Daly MB, Devilee P, Doheny KF, Dork T, Dos-Santos-Silva I, Dumont M, Durcan L, Dwek M, Eccles DM, Ekici AB, Eliassen AH, Ellberg C, Elvira M et al (2017) Association analysis identifies 65 new breast cancer risk loci. Nature 551:92–94. https://doi.org/10.1038/nature24284
Missiaglia E, Selfe J, Hamdi M, Williamson D, Schaaf G, Fang C, Koster J, Summersgill B, Messahel B, Versteeg R, Pritchard-Jones K, Kool M, Shipley J (2009) Genomic imbalances in rhabdomyosarcoma cell lines affect expression of genes frequently altered in primary tumors: an approach to identify candidate genes involved in tumor development. Genes Chromosomes Cancer 48:455–467. https://doi.org/10.1002/gcc.20655
Moelans CB, de Weger RA, Monsuur HN, Vijzelaar R, van Diest PJ (2010) Molecular profiling of invasive breast cancer by multiplex ligation-dependent probe amplification-based copy number analysis of tumor suppressor and oncogenes. Mod Pathol 23:1029–1039. https://doi.org/10.1038/modpathol.2010.84
Pang B, Cheng S, Sun SP, An C, Liu ZY, Feng X, Liu GJ (2014) Prognostic role of PIK3CA mutations and their association with hormone receptor expression in breast cancer: a meta-analysis. Scientific reports 4:6255. https://doi.org/10.1038/srep06255
Pearson A, Smyth E, Babina IS, Herrera-Abreu MT, Tarazona N, Peckitt C, Kilgour E, Smith NR, Geh C, Rooney C, Cutts R, Campbell J, Ning J, Fenwick K, Swain A, Brown G, Chua S, Thomas A, Johnston SRD, Ajaz M, Sumpter K, Gillbanks A, Watkins D, Chau I, Popat S, Cunningham D, Turner NC (2016) High-level clonal FGFR amplification and response to FGFR inhibition in a translational clinical trial. Cancer Discov 6:838–851. https://doi.org/10.1158/2159-8290.CD-15-1246
Perez-Garcia J, Muñoz-Couselo E, Soberino J, Racca F, Cortes J (2018) Targeting FGFR pathway in breast cancer. Breast 37:126–133. https://doi.org/10.1016/j.breast.2017.10.014
Press MF, Sauter G, Buyse M, Fourmanoir H, Quinaux E, Tsao-Wei DD, Eiermann W, Robert N, Pienkowski T, Crown J, Martin M, Valero V, Mackey JR, Bee V, Ma Y, Villalobos I, Campeau A, Mirlacher M, Lindsay M-A, Slamon DJ (2016) HER2 gene amplification testing by fluorescent in situ hybridization (FISH): comparison of the ASCO-College of American Pathologists Guidelines With FISH Scores Used for Enrollment in Breast Cancer International Research Group Clinical Trials. J Clin Oncol 34:3518–3528. https://doi.org/10.1200/jco.2016.66.6693
Press MF, Seoane JA, Curtis C, Quinaux E, Guzman R, Sauter G, Eiermann W, Mackey JR, Robert N, Pienkowski T, Crown J, Martin M, Valero V, Bee V, Ma Y, Villalobos I, Slamon DJ (2019) Assessment of ERBB2/HER2 status in HER2-equivocal breast cancers by FISH and 2013/2014 ASCO-CAP guidelines. JAMA Oncol 5:366–375. https://doi.org/10.1001/jamaoncol.2018.6012
Press MF, Slamon DJ, Flom KJ, Park J, Zhou J-Y, Bernstein L (2002) Evaluation of HER-2/neu gene amplification and overexpression: comparison of frequently used assay methods in a molecularly characterized cohort of breast cancer specimens. J Clin Oncol 20:3095–3105. https://doi.org/10.1200/jco.2002.09.094
Press MF, Villalobos I, Santiago A, Guzman R, Cervantes M, Gasparyan A, Campeau A, Ma Y, Tsao-Wei DD, Groshen S (2016) Assessing the New American Society of Clinical Oncology/College of American Pathologists Guidelines for HER2 testing by fluorescence in situ hybridization: experience of an academic consultation practice. Arch Pathol Lab Med 140:1250–1258. https://doi.org/10.5858/arpa.2016-0009-OA
Raval SH, Singh RD, Joshi DV, Patel HB, Mody SK (2016) Recent developments in receptor tyrosine kinases targeted anticancer therapy. Vet World 9:80–90. https://doi.org/10.14202/vetworld.2016.80-90
Ray ME, Yang ZQ, Albertson D, Kleer CG, Washburn JG, Macoska JA, Ethier SP (2004) Genomic and expression analysis of the 8p11-12 amplicon in human breast cancer cell lines. Cancer Res 64:40–47
Reis-Filho JS, Simpson PT, Turner NC, Lambros MB, Jones C, Mackay A, Grigoriadis A, Sarrio D, Savage K, Dexter T, Iravani M, Fenwick K, Weber B, Hardisson D, Schmitt FC, Palacios J, Lakhani SR, Ashworth A (2006) FGFR1 emerges as a potential therapeutic target for lobular breast carcinomas. Clin Cancer Res 12:6652–6662. https://doi.org/10.1158/1078-0432.ccr-06-1164
Salmen J, Neugebauer J, Fasching PA, Haeberle L, Huober J, Wockel A, Rauh C, Schuetz F, Weissenbacher T, Kost B, Stickeler E, Klar M, Orlowska-Volk M, Windfuhr-Blum M, Heil J, Rom J, Sohn C, Fehm T, Mohrmann S, Loehberg CR, Hein A, Schulz-Wendtland R, Hartkopf AD, Brucker SY, Wallwiener D, Friese K, Hartmann A, Beckmann MW, Janni W, Rack B (2014) Pooled analysis of the prognostic relevance of progesterone receptor status in five German cohort studies. Breast Cancer Res Treat 148:143–151. https://doi.org/10.1007/s10549-014-3130-4
Shi YJ, Tsang JY, Ni YB, Chan SK, Chan KF, Tse GM (2016) FGFR1 is an adverse outcome indicator for luminal A breast cancers. Oncotarget 7:5063–5073. https://doi.org/10.18632/oncotarget.6563
Simon R, Richter J, Wagner U, Fijan A, Bruderer J, Schmid U, Ackermann D, Maurer R, Alund G, Knonagel H, Rist M, Wilber K, Anabitarte M, Hering F, Hardmeier T, Schonenberger A, Flury R, Jager P, Fehr JL, Schraml P, Moch H, Mihatsch MJ, Gasser T, Sauter G (2001) High-throughput tissue microarray analysis of 3p25 (RAF1) and 8p12 (FGFR1) copy number alterations in urinary bladder cancer. Cancer Res 61:4514–4519
Slamon DJ, Leyland-Jones B, Shak S, Fuchs H, Paton V, Bajamonde A, Fleming T, Eiermann W, Wolter J, Pegram M, Baselga J, Norton L (2001) Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. New Engl J Med 344:783–792. https://doi.org/10.1056/NEJM200103153441101
Stephens PJ, Tarpey PS, Davies H, Van Loo P, Greenman C, Wedge DC, Nik-Zainal S, Martin S, Varela I, Bignell GR, Yates LR, Papaemmanuil E, Beare D, Butler A, Cheverton A, Gamble J, Hinton J, Jia M, Jayakumar A, Jones D, Latimer C, Lau KW, McLaren S, McBride DJ, Menzies A, Mudie L, Raine K, Rad R, Chapman MS, Teague J, Easton D, Langerod A, Oslo Breast Cancer Consortium (OSBREAC), Lee MT, Shen CY, Tee BT, Huimin BW, Broeks A, Vargas AC, Turashvili G, Martens J, Fatima A, Miron P, Chin SF, Thomas G, Boyault S, Mariani O, Lakhani SR, van de Vijver M, van’t Veer L, Foekens J, Desmedt C, Sotiriou C, Tutt A, Caldas C, Reis-Filho JS, Aparicio SA, Salomon AV, Borresen-Dale AL, Richardson AL, Campbell PJ, Futreal PA, Stratton MR (2012) The landscape of cancer genes and mutational processes in breast cancer. Nature 486:400–404. https://doi.org/10.1038/nature11017
Theillet C, Adelaide J, Louason G, Bonnet-Dorion F, Jacquemier J, Adnane J, Longy M, Katsaros D, Sismondi P, Gaudray P et al (1993) FGFRI and PLAT genes and DNA amplification at 8p12 in breast and ovarian cancers. Genes Chromosomes Cancer 7:219–226
Tomiguchi M, Yamamoto Y, Yamamoto-Ibusuki M, Goto-Yamaguchi L, Fujiki Y, Fujiwara S, Sueta A, Hayashi M, Takeshita T, Inao T, Iwase H (2016) Fibroblast growth factor receptor-1 protein expression is associated with prognosis in estrogen receptor-positive/human epidermal growth factor receptor-2-negative primary breast cancer. Cancer Sci 107:491–498. https://doi.org/10.1111/cas.12897
Tomlinson DC, Knowles MA, Speirs V (2012) Mechanisms of FGFR3 actions in endocrine resistant breast cancer. Int J Cancer 130:2857–2866. https://doi.org/10.1002/ijc.26304
Tsimafeyeu I, Ludes-Meyers J, Stepanova E, Daeyaert F, Khochenkov D, Joose JB, Solomko E, Van Akene K, Peretolchina N, Yin W, Ryabaya O, Byakhov M, Tjulandin S (2016) Targeting FGFR2 with alofanib (RPT835) shows potent activity in tumour models. Eur J Cancer 61:20–28. https://doi.org/10.1016/j.ejca.2016.03.068
Turner N, Grose R (2010) Fibroblast growth factor signalling: from development to cancer. Nat Rev Cancer 10:116–129. https://doi.org/10.1038/nrc2780
Turner N, Lambros MB, Horlings HM, Pearson A, Sharpe R, Natrajan R, Geyer FC, van Kouwenhove M, Kreike B, Mackay A, Ashworth A, van de Vijver MJ, Reis-Filho JS (2010) Integrative molecular profiling of triple negative breast cancers identifies amplicon drivers and potential therapeutic targets. Oncogene 29:2013–2023. https://doi.org/10.1038/onc.2009.489
Turner N, Pearson A, Sharpe R, Lambros M, Geyer F, Lopez-Garcia MA, Natrajan R, Marchio C, Iorns E, Mackay A, Gillett C, Grigoriadis A, Tutt A, Reis-Filho JS, Ashworth A (2010) FGFR1 amplification drives endocrine therapy resistance and is a therapeutic target in breast cancer. Cancer Res 70:2085–2094. https://doi.org/10.1158/0008-5472.CAN-09-3746
Wang S, Ding Z (2017) Fibroblast growth factor receptors in breast cancer. Tumour Biol 39:1010428317698370. https://doi.org/10.1177/1010428317698370
Wein L, Savas P, Van Geelen C, Caramia F, Moodie K, Joshi S, Loi S (2017) FGFR2 amplification in metastatic hormone-positive breast cancer and response to an mTOR inhibitor. Ann Oncol 28:2025–2027. https://doi.org/10.1093/annonc/mdx194
Weiss J, Sos ML, Seidel D, Peifer M, Zander T, Heuckmann JM, Ullrich RT, Menon R, Maier S, Soltermann A, Moch H, Wagener P, Fischer F, Heynck S, Koker M, Schottle J, Leenders F, Gabler F, Dabow I, Querings S, Heukamp LC, Balke-Want H, Ansen S, Rauh D, Baessmann I, Altmuller J, Wainer Z, Conron M, Wright G, Russell P, Solomon B, Brambilla E, Brambilla C, Lorimier P, Sollberg S, Brustugun OT, Engel-Riedel W, Ludwig C, Petersen I, Sanger J, Clement J, Groen H, Timens W, Sietsma H, Thunnissen E, Smit E, Heideman D, Cappuzzo F, Ligorio C, Damiani S, Hallek M, Beroukhim R, Pao W, Klebl B, Baumann M, Buettner R, Ernestus K, Stoelben E, Wolf J, Nurnberg P, Perner S, Thomas RK (2010) Frequent and focal FGFR1 amplification associates with therapeutically tractable FGFR1 dependency in squamous cell lung cancer. Sci Transl Med 2:6293. https://doi.org/10.1126/scitranslmed.3001451
Wöckel A, Festl J, Stüber T, Brust K, Krockenberger M, Heuschmann PU, Jírů-Hillmann S, Albert U-S, Budach W, Follmann M, Janni W, Kopp I, Kreienberg R, Kühn T, Langer T, Nothacker M, Scharl A, Schreer I, Link H, Engel J, Fehm T, Weis J, Welt A, Steckelberg A, Feyer P, König K, Hahne A, Baumgartner T, Kreipe HH, Knoefel WT, Denkinger M, Brucker S, Lüftner D, Kubisch C, Gerlach C, Lebeau A, Siedentopf F, Petersen C, Bartsch HH, Schulz-Wendtland R, Hahn M, Hanf V, Müller-Schimpfle M, Henscher U, Roncarati R, Katalinic A, Heitmann C, Honegger C, Paradies K, Bjelic-Radisic V, Degenhardt F, Wenz F, Rick O, Hölzel D, Zaiss M, Kemper G, Budach V, Denkert C, Gerber B, Tesch H, Hirsmüller S, Sinn H-P, Dunst J, Münstedt K, Bick U, Fallenberg E, Tholen R, Hung R, Baumann F, Beckmann MW, Blohmer J, Fasching P, Lux MP, Harbeck N, Hadji P, Hauner H, Heywang-Köbrunner S, Huober J, Hübner J, Jackisch C, Loibl S, Lück H-J, von Minckwitz G, Möbus V, Müller V, Nöthlings U, Schmidt M, Schmutzler R, Schneeweiss A, Schütz F, Stickeler E, Thomssen C, Untch M, Wesselmann S, Bücker A, Buck A, Stangl S (2018) Interdisciplinary screening, diagnosis, therapy and follow-up of breast cancer. Guideline of the DGGG and the DKG (S3-Level, AWMF Registry Number 032/045OL, December 2017)—Part 2 with recommendations for the therapy of primary, recurrent and advanced breast cancer. Geburtshilfe Frauenheilkd 78:1056–1088. https://doi.org/10.1055/a-0646-4630
Wöckel A, Festl J, Stüber T, Brust K, Stangl S, Heuschmann PU, Albert U-S, Budach W, Follmann M, Janni W, Kopp I, Kreienberg R, Kühn T, Langer T, Nothacker M, Scharl A, Schreer I, Link H, Engel J, Fehm T, Weis J, Welt A, Steckelberg A, Feyer P, König K, Hahne A, Kreipe HH, Knoefel WT, Denkinger M, Brucker S, Lüftner D, Kubisch C, Gerlach C, Lebeau A, Siedentopf F, Petersen C, Bartsch HH, Schulz-Wendtland R, Hahn M, Hanf V, Müller-Schimpfle M, Henscher U, Roncarati R, Katalinic A, Heitmann C, Honegger C, Paradies K, Bjelic-Radisic V, Degenhardt F, Wenz F, Rick O, Hölzel D, Zaiss M, Kemper G, Budach V, Denkert C, Gerber B, Tesch H, Hirsmüller S, Sinn H-P, Dunst J, Münstedt K, Bick U, Fallenberg E, Tholen R, Hung R, Baumann F, Beckmann MW, Blohmer J, Fasching PA, Lux MP, Harbeck N, Hadji P, Hauner H, Heywang-Köbrunner S, Huober J, Hübner J, Jackisch C, Loibl S, Lück H-J, von Minckwitz G, Möbus V, Müller V, Nöthlings U, Schmidt M, Schmutzler R, Schneeweiss A, Schütz F, Stickeler E, Thomssen C, Untch M, Wesselmann S, Bücker A, Krockenberger M (2018) Interdisciplinary screening, diagnosis, therapy and follow-up of breast cancer. guideline of the DGGG and the DKG (S3-Level, AWMF Registry Number 032/045OL, December 2017)—Part 1 with recommendations for the screening, diagnosis and therapy of breast cancer. Geburtshilfe Frauenheilkd 78:927–948. https://doi.org/10.1055/a-0646-4522
Wu J, Wang Y, Liu J, Chen Q, Pang D, Jiang Y (2018) Effects of FGFR1 gene polymorphisms on the risk of breast cancer and FGFR1 Protein expression. Cell Physiol Biochem 47:2569–2578. https://doi.org/10.1159/000491653
Xian W, Pappas L, Pandya D, Selfors LM, Derksen PW, de Bruin M, Gray NS, Jonkers J, Rosen JM, Brugge JS (2009) Fibroblast growth factor receptor 1-transformed mammary epithelial cells are dependent on RSK activity for growth and survival. Cancer Res 69:2244–2251. https://doi.org/10.1158/0008-5472.CAN-08-3398
Acknowledgements
We are grateful to A. Santiago, R. Guzman, and A. Gasparyan at USC for technical support, including creation of the tissue microarrays. Our thanks also go to Ivonne Villalobos (USC) for administrative support for this project. Special thanks are also due to all of the patients who participated in the study.
Funding
Research for the present study was supported by a grant from the Bavaria California Technology Center (www.bacatec.de). The FISH probes were sponsored by ZytoVision GmbH, Bremerhaven, Germany. Open Access funding provided by Projekt DEAL.
Author information
Authors and Affiliations
Contributions
All authors contributed to the study conception and design. Material preparation, data collection and analysis were performed by P.A. Fasching, R. Erber, M.F. Press, and L. Häberle. P. Gass was responsible for data management. The first draft of the manuscript was written by R. Erber and P.A. Fasching and all authors commented on previous versions of the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Conflicts of interest
P.A.F. has received honoraria from Roche, Pfizer, Novartis, and Celgene. His institution conducts research for Novartis, Cepheid, and BioNTech. H.A. has received honoraria from BMS, MSD, Roche, AstraZeneca, Boehringer Ingelheim, Abbvie, Jansen-Cilag, and Ipsen. R.E. has received honoraria from Roche, Eisai, Pfizer, and Novartis. The institution of H.A. and R.E. conducts research for AstraZeneca, Roche, Janssen-Cilag, NanoString Technologies, Novartis, Cepheid, and BioNTech. M.F.P. has received honoraria from Biocartis, Eli Lilly & Company, Zymeworks, Novartis, and Puma Biotechnology. His institution conducts research for Eli Lilly & Company, Zymeworks, Novartis, Cepheid, and Puma Biotechnology. P.G. received honoraria from Novartis, financial support for symposia from Novartis, Roche, and PharmaMar, and compensation for travel expenses from discovering hands® company. S.H. is founder, shareholder, and managing director of ZytoVision GmbH, Bremerhaven, Germany, the manufacturer of FISH probes used in this study. The other authors have no conflicts of interest to declare.
Ethical approval
All procedures performed in studies involving human participants were in accordance with the ethical standards of the local national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards. Approval for the study was obtained from the local ethics committee at the University of Erlangen (ref. numbers 2700 and 297_17 Bc). This article does not contain any studies with animals performed by any of the authors.
Informed consent
Informed consent was obtained from all individual participants included in the study.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Erber, R., Rübner, M., Davenport, S. et al. Impact of fibroblast growth factor receptor 1 (FGFR1) amplification on the prognosis of breast cancer patients. Breast Cancer Res Treat 184, 311–324 (2020). https://doi.org/10.1007/s10549-020-05865-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10549-020-05865-2