
Abstract
KRAS mutations are the most frequent gain-of-function alterations in patients with lung adenocarcinoma (LADC) in the Western world. Although they have been identified decades ago, prior efforts to target KRAS signaling with single-agent therapeutic approaches such as farnesyl transferase inhibitors, prenylation inhibition, impairment of KRAS downstream signaling, and synthetic lethality screens have been unsuccessful. Moreover, the role of KRAS oncogene in LADC is still not fully understood, and its prognostic and predictive impact with regards to the standard of care therapy remains controversial. Of note, KRAS-related studies that included general non-small cell lung cancer (NSCLC) population instead of LADC patients should be very carefully evaluated. Recently, however, comprehensive genomic profiling and wide-spectrum analysis of other co-occurring genetic alterations have identified unique therapeutic vulnerabilities. Novel targeted agents such as the covalent KRAS G12C inhibitors or the recently proposed combinatory approaches are some examples which may allow a tailored treatment for LADC patients harboring KRAS mutations. This review summarizes the current knowledge about the therapeutic approaches of KRAS-mutated LADC and provides an update on the most recent advances in KRAS-targeted anti-cancer strategies, with a focus on potential clinical implications.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
Over the past 20 years, the formerly prevalent and widespread pessimism regarding the therapeutic approaches and prognosis of advanced-stage non-small lung cancer (NSCLC) has changed dramatically with the development of molecular profiling, targeted therapeutic agents, immune checkpoint inhibitors, and precision medicine [1]. These efforts have offered valuable insights into the mutational landscape of NSCLC, including the Kirsten rat sarcoma viral oncogene homolog (KRAS) mutation, which is the most common gain-of-function alteration, accounting for approximately 30% of lung adenocarcinomas (LADCs) in Western countries and about 10% of Asian LADCs [2, 3].
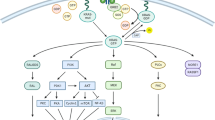
KRAS protein, encoded by the KRAS proto-oncogene, is a small guanine triphosphatase (GTPase) that serves as a binary switch in signal transduction for most receptor tyrosine kinases including EGFR, MET, or ALK, and plays a key role in regulating various cell functions [4, 5]. Oncogenic mutations of the KRAS gene mostly occur in exon 2 at codon 12, less frequently at codon 13 (3–5%) and rarely at exon 3 codon 61 (less than 1%) [5]. These alterations are missense mutations that impair the ability of KRAS to hydrolyze GTP, resulting in the constitutive activation of its effector pathways and thus cancer development and progression [6]. Because of its high frequency in LADC, several preclinical and clinical investigations have been conducted, seeking effective therapeutic approaches targeting KRAS mutation. Still, to date, no effective RAS inhibitors are currently used in routine clinical practice and the approaches for treating KRAS-mutant LADC mirror those for treating NSCLC that lacks a known driver mutation. In this review, we systematically analyze the clinically relevant aspects of KRAS-mutant NSCLC, mainly focusing on the clinicopathological relevance, therapeutic implications, and new treatment opportunities.
2 Clinical relevance of KRAS mutations in NSCLC
Molecular profiling of LADC patients shows that specific demographic and clinicopathological characteristics are associated with the presence of KRAS mutations. As national surveys indicate, KRAS mutations mostly occur in Caucasian or African-American patients and are far less frequent in Asian patients [2, 3, 5]. Based on the findings of a pooled analysis of resected NSCLC tumors, KRAS mutations tend to be more common among women and patients of younger age, although only the latter remained significant at the multivariate analysis (p = 0.044) [7, 8]. Notably, however, no histology- or race-specific analyses were performed in the above study with regards to the prevalence of KRAS mutations. Interestingly, smoking also leaves a molecular fingerprint on KRAS, since transition mutations (G12D) are more frequent in never smokers, whereas transversion mutations (G12C and G12V) are more often found among former or current smokers [9, 10]. In addition, smokers tend to have genetically more complex KRAS-mutant tumors, with higher mutational burden and higher frequency of major co-occurring mutations in TP53 or STK11, than those who have never smoked [10, 11]. The distribution of various KRAS mutational subtypes among patients with different smoking history is summarized in Fig. 1 [12].

KRAS mutational subtypes and smoking history in lung adenocarcinoma (LADC) [12]. In current (a) and former (b) smokers, KRAS G12C is the most common mutation, while KRAS G12D is the most frequent mutation among never smokers (c). Overall (d), the most frequently diagnosed KRAS mutational subtype in LADC patients is KRAS G12C, followed by KRAS G12V, KRAS G12D, and KRAS G12A
Recently, attention has also been drawn to the special histology and co-occurring mutations in KRAS-mutant lung cancer. Initial studies [13, 14] reported that although in a much lower percentage, KRAS mutations might be present not only in LADC but also in squamous cell lung cancer. However, recent analysis using up-to-date differential diagnostic criteria suggests that KRAS mutations do not occur in pure pulmonary squamous cell lung carcinomas, and in case detected, it is confined to LADC components in squamous cancer [15]. The other important issue is the clinical relevance of specific KRAS mutations and the presence of these mutations in combination with others. Variations in KRAS mutation subtypes have been associated with distinct biological behaviors that can lead to different clinical outcomes [16, 17]. For example, tumors with KRAS G12C mutations exhibited higher ERK1/2 phosphorylation than those with KRAS G12D [3, 18]. In support of this, a recent study using KRAS mutation-driven mouse models demonstrated higher efficacy of the MEK inhibitor selumetinib in KRAS G12C tumors compared with KRAS G12D tumors [18]. Accordingly, distinct KRAS mutations may lead to differential induction of signal transduction cascades and thus to specific drug sensitivity profiles [19]. As for co-occurring mutations, double mutants (KRAS and EGFR/ALK/BRAF) are rare in LADC, and KRAS mutations are typically present as a single-driver mutation [20,21,22]. However, KRAS mutations co-occur commonly with mutations in tumor suppressor genes including TP53, STK11 and KEAP1/NFE2L2, and a growing body of evidence suggests that these co-occurring mutations are associated with unique tumor characteristics and biological behaviors [23]. Taken together, the different amino acid substitutions in oncogenic KRAS and the presence of coexisting mutations highlight the need for genotype-specific analysis to identify clinically relevant subgroups of patients that may ultimately influence treatment decisions and prognosis [3].
3 The prognostic nature of KRAS mutations in LADC
The prognostic power of KRAS mutation alone in the general NSCLC population remains disputed. As in other malignancies, KRAS mutation was first reported to be a negative prognostic factor in NSCLC in the 1980s [24, 25]. However, although a considerable number of publications verified this finding [26,27,28,29,30], these studies were heterogeneous with regards to histology, tumor stage, and methodology. Slebos [25], Ohtaki [28], and Izar [29] investigated the prognosis in completely resected LADCs, while later studies were performed in stage IIIB–IV NSCLC patients [30,31,32]. The strongest proof of KRAS being a negative prognostic factor in NSCLC was reported by Mascaux et al. who conducted a meta-analysis of 53 studies and found that KRAS mutation correlated with a significantly worse prognosis (hazard ratio [HR] 1.40; p = 0.01; HR 1.5 for LADCs; p = 0.02) [33]. Contrary to this, in a study analyzing 998 LADCs, 318 of which harbored KRAS mutation, the authors concluded that KRAS mutation was not an individual prognostic factor [34]. One of the most comprehensive study, a meta-analysis of four individual trials of adjuvant chemotherapy in 1500 NSCLC patients (among them 300 KRAS-mutant cases), also reported that KRAS mutation had no prognostic value in this setting [8]. However, a more recent study that involved 1935 patients reported a clear advantage in overall survival (OS) for KRAS wild type patients, although the presence of mutation did not impact progression-free survival (PFS) [30]. Another recent pooled analysis of studies assessing the role of KRAS mutation in circulating tumor DNA also indicated poorer PFS and OS in KRAS-mutated genotypes [35].
The prevalence of KRAS mutations varies among different ethnic groups and ethnicity; therefore, it might also have an impact on prognosis [5]. A meta-analysis of 41 trials and 6939 patients concluded that KRAS mutation was a negative prognostic factor in NSCLC. Not surprisingly, these authors found that KRAS mutation only had a prognostic role in LADC (HR was 1.39; 95% CI 1.24–1.55). The authors also looked at ethnicity, comparing Asians and non-Asians, and found that the HR for Asians was much larger than that for non-Asians, implying that KRAS mutations have a worse prognosis in Asian patients [36]. These results were also backed up by a recent meta-analysis including over 9000 patients [37]. Of note, patients with EGFR mutant LADC have a better prognosis, and thus KRAS mutations’ prognostic power might be influenced by the inclusion/exclusion and the proportion of EGFR mutant cases in the study cohort.
Several studies suggest that due to the heterogeneity of KRAS mutations, specific mutational subtypes might have different effects on survival and treatment response. For example, in a mutation subtype-specific analysis of 505 stage III–IV LADC patients treated with chemotherapy, our group could not demonstrate prognostic or predictive potential of KRAS mutation. However, we found that G12V mutant patients had higher response rates and slightly longer PFS [31]. On the other hand, in two retrospective studies, the authors found a significantly shorter OS in patients with KRAS G12C mutation [38]. Garassino et al. further highlighted the role of subtype-specific KRAS mutation analysis when they conducted a preclinical study assessing the in vitro chemosensitivity of NSCLC cells. They found that G12V mutant tumor cells were more sensitive to cisplatin and, furthermore, that G12D mutation led to increased resistance to paclitaxel and sensitivity to sorafenib, while G12C mutation was associated with reduced response to cisplatin and increased sensitivity to paclitaxel and pemetrexed [19]. Villaruz et al. found a slightly increased OS in patients with G12C mutant tumors when compared with those with tumors harboring other KRAS mutation subtypes [34]. Table 1 summarizes a selection of the major studies about the prognostic relevance of KRAS status in early- and advanced-stage NSCLC.
4 Predictive role of KRAS mutations
4.1 Predictive value of KRAS mutations for response to chemotherapy
Despite the recent developments in NSCLC therapy, most patients with advanced-stage disease still receive platinum-based chemotherapy. Most studies do not suggest KRAS mutation as a predictive biomarker for response to chemotherapy. The predictive value of KRAS mutation in NSCLC was investigated in the metastatic setting in patients receiving definitive chemotherapy [39], in patients receiving adjuvant chemotherapy with radiation after surgery [40], and also in the phase III TRIBUTE trial where first-line carboplatin/paclitaxel plus erlotinib or placebo was compared in advanced-stage NSCLC [41]. In none of the above settings did KRAS prove to be a predictive factor for response rate, PFS, or OS.
More recently, results of the JBR10 trial, which studied the effects of postoperative vinorelbine or cisplatin in patients with resected stage IB or II NSCLC, were published. Remarkable benefit from chemotherapy was only reported in KRAS wild type patients; however, the difference did not prove to be statistically significant (p = 0.29) [42]. Neoadjuvant and perioperative chemotherapy sequences with carboplatin/paclitaxel or cisplatin/gemcitabine were compared in the phase III IFCT-0002 trial. KRAS-mutant tumors were shown to exhibit lower response to cytotoxic chemotherapy in univariate analysis, although KRAS mutation was not a significant predictor in multivariate analysis [43]. A recent retrospective analysis of patients with advanced-stage NSCLC also found that KRAS mutation is a predictor for poor OS when treated with cytotoxic chemotherapy [44]. Furthermore, it was shown that the co-existence of TP53 mutation predicts worse outcome [45]. Another aspect was shown in a study conducted in an Asian cohort that analyzed outcomes in patients receiving different chemotherapeutic regimens according to the KRAS mutation status. Significantly poorer PFSs and OSs were seen in patients with KRAS mutations when treated with pemetrexed or gemcitabine but not in those receiving taxanes [46]. Of note, a potential negative effect of KRAS codon 13 mutations was suggested by a clinical study [8] showing significantly shorter PFS and OS in patients with such mutations. As previously mentioned, in a preclinical study by Garassino et al., similar results were seen [19].
In summary, although KRAS mutations can be potentially considered as predictive biomarkers for chemotherapy in LADC, the exact type of mutation and the type of chemotherapy should also be taken into consideration.
4.2 Predictive value of KRAS mutations for response to targeted therapy
One of the major debates over the predictive role of KRAS-mutant status of NSCLC patients takes place in the field of EGFR-targeted therapies [5]. Most published data, including a meta-analysis of 22 studies, suggest that KRAS mutational status is a significant negative predictor for EGFR tyrosine kinase inhibitors (TKIs) [41, 47,48,49]. Accordingly, KRAS-mutated patients treated with EGFR TKIs have a trend for worse objective response rates (ORR), PFS, and OS compared with patients without KRAS mutation [41, 48, 49]. However, despite the convincing results, controversies still exist and not all studies have reached the same conclusions [46, 50]. A possible explanation for these discrepancies in the literature might be that the response to EGFR TKI is greatly influenced not only by the presence or absence of KRAS mutations but also by the involved KRAS codons and the type of amino acid substitutions [5, 51]. In support of this, a recent study showed poorer treatment efficacy in the case of G12C and G12V KRAS mutations but promising response rates in G12D and G12S KRAS-mutant NSCLC patients treated with EGFR TKIs [52]. All in all, patients with KRAS-mutant NSCLC generally have a poor response to EGFR inhibitors; however, due to the heterogeneity of various KRAS mutations, KRAS mutational analysis cannot be recommended as a tool to select NSCLC patients for EGFR TKI therapy.
4.3 Predictive value of KRAS mutations for response to anti-vascular therapy
Although the RAS pathway has been shown to affect VEGF expression, very few studies investigated the influence of KRAS mutation on the efficacy of anti-angiogenic therapy [53]. Only two groups reported that G12V, G12A [54], and G12D [55] KRAS mutations are associated with poor outcome in patients with colorectal cancer (CRC) receiving bevacizumab. As for NSCLC, a phase II trial evaluated the addition of neoadjuvant bevacizumab to chemotherapy and found that no patients with KRAS mutation (0 out of 10) demonstrated pathological response to neoadjuvant bevacizumab and chemotherapy, while 35% of KRAS wild type patients had significant response [56]. Furthermore, in a recent single-center retrospective study from our group, KRAS mutation, and especially G12D mutation, was shown to be a predictor of significantly worse PFS and OS in advanced-stage NSCLC patients treated with bevacizumab plus platinum-based chemotherapy [57]. Table 2 summarizes the available data on the predictive value of KRAS mutations for therapeutic response in NSCLC.
4.4 Predictive value of KRAS mutations for response to immune checkpoint inhibition therapy
Programmed cell death protein 1 (PD-1) expression has been shown to be in close connection with KRAS status, and KRAS mutations were described as possible biomarkers for immune checkpoint inhibitors [58]. Also, a clinical benefit was reported to PD-1 inhibitors in KRAS-mutant patients [59]. The elevated expression of programmed cell death ligand 1 (PD-L1) has been demonstrated in KRAS-mutant cells, and it was also shown that ERK activation mediates the upregulation of PD-L1 by KRAS mutations [60]. On the contrary, Reiniger et al. did not find significant relations between PD-L1 expression and KRAS status in LADC [61]. It was also reported that pembrolizumab (a PD-1 inhibitor) or an ERK inhibitor might prevent CD3+ T cells becoming apoptotic by recovering tumor immunity thus preventing immune escape [62]. In another study, however, Gettinger et al. found increased response and survival with nivolumab monotherapy and driver mutations of EGFR or KRAS did not show significant effect on survival or treatment response [63]. Altogether, further clinical experience is needed to determine whether KRAS mutation is a useful predictive factor for immunotherapy in NSCLC. Table 2 includes three trials where the predictive role of the KRAS mutational status for immune checkpoint inhibition therapy was studied.
5 KRAS as a therapeutic target in NSCLC
5.1 Pitfalls of KRAS mutation targeting in NSCLC
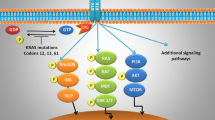
Because of its high mutation frequency in NSCLC, KRAS is an appealing target. However, the development of targeted therapies for KRAS-mutant lung cancers has long been marked by frustration [64,65,66]. For decades, KRAS was considered undruggable due to its exceptionally high affinity to GTP/GDP, to the absence of known allosteric binding sites, and to the presence of extensive post-transcriptional modifications [3, 7, 67]. KRAS protein shows high resistance against small-molecule modulation, since it is a small protein with a relatively smooth surface without clear binding pockets (besides its GTP/GDP binding pocket) [68]. Under physiological conditions in vivo, GTP almost exclusively occupies all potential binding sites with extremely high affinity. The development of KRAS inhibitors that achieve adequate blood concentration enough to displace GTP is, therefore, an almost improbable task [68, 69]. In addition, the binding of small-molecule inhibitors is also influenced by the interactions of KRAS with other proteins that make the surface of the KRAS protein shallow [68]. Importantly, indirect targeting of the molecules within the KRAS signaling pathway also proved to be almost ineffective due to the complexity and biological heterogeneity of KRAS mutations in NSCLC [68, 70]. All in all, despite enormous efforts, to date, almost all identified compounds that could effectively and directly target mutant KRAS have failed. However, with new technologies in drug development and novel mechanistic insights into RAS biology, new targeted therapeutic agents are under development with promising preclinical activity (Fig. 2).

A chronicle of KRAS mutation in lung cancer. Major biological discoveries and key clinical trials. During its more than 30-year history, our knowledge of KRAS mutation in lung cancer has progressed through a series of phases. Although the relationship between RAS genes and lung cancer was described in 1984, the first clinical trials investigating the efficacy of indirect KRAS inhibitors were carried out only in the early 2000s. Since then, large numbers of both direct and indirect KRAS inhibitors have been developed and tested. However, until recently, efforts to target the RAS family proteins were mostly ineffective in the clinics. At the same time, in the past years, a worldwide awakening of interest led to rapid translational progress and to the discovery of novel direct covalent KRAS G12C-inhibitors, some of which have been tested in clinical trials. The renewed enthusiasm and biological and clinical progress have changed the landscape of KRAS-mutated lung cancer and have led to the first serious discussions of whether RAS is indeed a druggable target. KRAS, Kirsten rat sarcoma viral oncogene homolog; MEK, MAPK/ERK kinase; mTOR, mammalian target of rapamycin; MET, MET proto-oncogene; Hsp90, heat shock protein 90; CDK4/6, cyclin-dependent kinases 4/6; FAK, focal adhesion kinase; OS, overall survival
5.2 Targeting KRAS membrane anchorage
RAS proteins require membrane associations to become biologically active [11, 68, 71]. The membrane anchorage of KRAS is dependent on posttranslational modification of the CAAX motif by farnesyltransferases. Initial preclinical studies with farnesyltransferase inhibitors (FTIs) demonstrated moderate success in blocking tumor cells both in vitro and in vivo. However, in the presence of FTIs, KRAS can be alternatively prenylated by geranylgeranyl-transferase-I, thus overcoming the effect of farnesyltransferase inhibition [6, 11, 72]. As expected, these results foreshadowed the disappointing clinical trials with FTIs that failed to improve outcomes in KRAS-mutant LADC patients [11, 73]. Still, some novel FTIs, when combined with other inhibitors such as geranylgeranyl-transferase inhibitors, showed potent anti-cancer activities in KRAS-driven pancreatic tumors. Nevertheless, the efficacy of these dual-functional therapeutic agents has not yet been investigated in LADC [74, 75]. Preclinical studies indicated that lung cancer cells might be sensitive to prenylation inhibition by bisphosphonates [76, 77]. Additionally, oral bisphosphonate use was associated with lower lung cancer risk among never smoker postmenopausal women in a large prospective study [78]. In isolated clinical cases, bisphosphonate therapy caused the regression of the primary lesion and its hepatic metastases in LADC [79]. Notably, however, a recent preclinical study demonstrated that the aminobisphosphonate compound zoledronic acid was ineffective in NSCLC cells harboring exon 2 codon 12 KRAS mutation, since this mutational subtype leads to prenylation-independent activation of KRAS. [80]. Nevertheless, the impact of the bisphosphonate treatment in KRAS-mutant LADC patients remains to be fully explored. Similarly, targeting other enzymes involved in the post-prenylation processing of RAS (e.g., the RAS converting CAAX endopeptidase 1 [Rce1] and isoprenylcysteine carboxyl methyltransferase [ICMT]) could as well inhibit the RAS-driven tumorigenesis [81]. In the past years, numerous Rce1 and ICMT inhibitors have been designed and investigated in several RAS-driven tumor entities. However, despite the encouraging results in vitro, the use of Rce1 and ICMT inhibitors could impact the normal function of other proteins as well in vivo, raising the questions about normal tissue toxicity and possible side effects of these inhibitors [82].
5.3 Targeting KRAS downstream signaling pathways
Another feasible approach to treat KRAS-mutated NSCLC might be to target the main signaling pathways controlled by the constitutively active mutant KRAS (i.e., the RAF-MEK-ERK or the PI3K/AKT/mTOR pathways). The inhibitors of these signaling pathways have been tested in different RAS-driven tumor types, and some of them showed promising activity in preclinical models [11]. The results of conducted trials in KRAS-mutant NSCLC in regards to downstream signaling pathway inhibition are summarized in Table 3. Notably, one of the most promising therapeutic agent was sorafenib, a multikinase inhibitor that showed promising results in preclinical settings and phase II clinical trials but only modest clinical activity in phase III trials with ORRs generally less than 10% and median PFS of approximately 3 months [11, 83,84,85]. Clinical outcomes for single-agent allosteric MEK inhibitors were also discouraging, since no clinical activity of selumetinib or trametinib was observed [86, 87]. As for other downstream inhibitors, the mTOR inhibitor ridaforolimus showed a moderate increase in PFS, but its clinical benefit was questionable with several side effects [88]. All in all, clinical trials investigating the efficacy of KRAS downstream inhibitors in monotherapy provided limited clinical benefit and substantial toxicity in most studies [11, 65] Yet, recent preclinical studies with patient-derived xenograft tumors highlighted the need for combination therapy in order to fully block KRAS signaling in lung cancer [89]. These results provide a strong therapeutic rationale to treat epithelial KRAS-mutant lung cancer with ERBB and MEK inhibitors, and mesenchymal-like KRAS-mutant lung cancer by combined therapy with FGFR and MEK inhibitors [3, 89]. To date, however, none of these findings have been translated into the clinics.
5.4 Synthetic lethal vulnerabilities in KRAS-mutant NSCLC
An alternative approach to direct targeting of KRAS-mutant cancer genes involves targeting co-dependent vulnerabilities or synthetic lethal partners that are preferentially essential for KRAS oncogenesis [90]. The therapeutic ablation of these secondary targets would hypothetically result in the selective death of KRAS-mutant but not KRAS wild type tumor cells [11, 91]. One of the therapeutic approaches inducing synthetic lethality included the proteasome inhibitor bortezomib [7]. However, in a small phase II clinical trial of 16 NSCLC patients with KRAS G12D mutation, bortezomib showed only a modest disease control rate of 40%, only 1 objective response (ORR 6%), and a PFS of 1 month [92]. The pharmacological inhibition of cyclin-dependent kinase (CDK) was as well of great clinical interest in the past years, and the selective CDK4/6 inhibitor abemaciclib showed indeed promising results both in phases I and III clinical trials. Accordingly, abemaciclib demonstrated significantly higher ORRs and PFS than erlotinib in pretreated patients with advanced-stage KRAS-mutant lung cancer patients, but no significant difference was observed in OS [93, 94]. The efficacy of other CDK4/6 inhibitors (including palbociclib in combination therapy with MEK inhibitors) is currently under investigation (NCT02022982 and NCT03170206). Finally, preclinical studies suggest that dual inhibition of discoidin domain receptor 1 (DDR1) and Notch pathways also hampers the growth of murine and human KRAS-mutant LADC; however, these results have not been yet validated in clinical trials [95].
5.5 Targeting direct regulators of KRAS activity
RAS protein is transformed into its active, GTP-bound state by interaction with guanine nucleotide exchange factors (GEFs) [96, 97]. The most-studied GEF for RAS is the protein Son of Sevenless (SOS) (for which two isoforms, SOS1 and SOS2, are known), which catalyzes the release of GDP and allows the binding of the more abundant GTP [96, 97]. Accordingly, the selective inhibition of SOS1 with small-molecule inhibitors such as the experimental BI 1701963 might allow KRAS blockade irrespective of KRAS mutation type [96, 98,99,100]. This highly specific SOS1 inhibitor reduces both KRAS-GTP levels and MAPK signaling in cellular and animal models [100, 101]. Furthermore, preclinical studies also suggest that BI 1701963 indeed blocks tumor growth both in G12 and G13 KRAS-mutant tumors, and the compound is selective for KRAS-mutant cell lines [100, 101]. The efficacy of the SOS1-binding pan-KRAS inhibitor BI 1701963 alone or in combination with the MEK inhibitor trametinib in patients with KRAS-mutated solid tumors is currently under investigation (NCT04111458).
5.6 Direct targeting of mutant KRAS
KRAS has been historically acknowledged a non-druggable target. However, according to the results of the latest preclinical findings, the landscape of G12C KRAS-mutated lung cancer might change. After the discovery of new allosteric regulatory pockets in GDP-RAS adjacent to the cysteine residue of KRAS G12C, compounds that target the guanine nucleotide-binding pocket (SML-8-73-1) or allele-specific inhibitors (ARS-853) have been reported [102,103,104]. Of note, the effects of both SML-8-73-1 and ARS-853 on mutant KRAS G12C are irreversible. SML-8-73-1 can covalently react with KRAS G12C, thus competing with GTP and GDP for active site binding in a cellular context even in the presence of a very high concentration of GTP [105]. Accordingly, by locking the KRAS-GDP state, these GDP-derived inhibitors can block the proliferative activity of the KRAS-mutant cells [103, 104]. Despite their preclinical inhibitory effects on KRAS G12C, follow-up studies also showed that the specificity of these inhibitors is somewhat low and may have off-target effects when used in the clinics [103,104,105]. ARS-853, on the other hand, does not compete with GTP for binding to KRAS, since it binds to a pocket nearby the nucleotide-binding pocket [106]. Hence, by making KRAS more preferential to accept GDP binding rather than GTP, it reduces the KRAS-GTP levels by more than 90% and increases the in vitro hydrolytic reaction and thus locking the KRAS in the GDP-bound state [103, 104, 106]. Accordingly, ARS-853 inactivates the RAS signaling by a trapping mechanism, by which KRAS G12C is trapped in the KRAS-GDP state [103, 104]. Importantly, similarly to SML-8-73-1 and SML-10-70-1, ARS-853 only binds to KRAS G12C and has no inhibitory effects on wild type KRAS and other types of mutant KRAS [68]. These findings were recently translated into mouse model studies where ARS-1620, a similar covalent compound with high potency and selectivity for KRAS G12C, induced durable tumor regression in different patient-derived tumor models [107]. Furthermore, recent studies also suggest a potential synergistic activity when ARS-853 is combined with receptor TKIs such as EGFR TKIs, indicating that covalent G12C-specific inhibitors might indeed be promising therapeutic agents used for the treatment of KRAS G12C-mutant NSCLC patients [103, 104, 108, 109]. To date, however, no clinical trials have been communicated with ARS-853 or ARS-1620 in KRAS-mutant NSCLC.
5.7 Novel direct covalent KRAS-G12C inhibitors: promising preclinical and clinical results
Recent discoveries of the aforementioned covalent KRAS G12C-specific inhibitors have led to the first serious discussions of whether RAS is indeed a druggable target. AMG 510 is a novel small molecule that covalently binds to the cysteine amino acid of KRAS G12C-mutant proteins, and thus, it locks KRAS in its inactive GDP-bound state irreversibly [110, 111]. In preclinical studies, treatment with AMG 510 induced the regression of KRAS G12C tumors and improved the efficacy of both chemotherapy and targeted agents [112]. Furthermore, AMG 510 therapy also resulted in a pro-inflammatory tumor microenvironment in immune-competent mice and produced durable responses alone and in combination with immune checkpoint inhibitors as well [112, 113]. As for its clinical benefit, in a recent phase I clinical trial in a small number of pretreated NSCLC patients (NCT03600883), a partial response was achieved in 54% and stable disease in 46% of the patients with a disease control rate of 100% [66, 111]. Importantly, the treatment was well-tolerated with the absence of dose-limiting toxicity and the occurrence of only a few drug-related side effects [66, 111]. A multicenter phase II clinical trial is currently ongoing [66]. MRTX 849 is another potent, mutation-selective, and orally available irreversible small-molecule inhibitor of KRAS G12C [114]. MRTX 894 also locks KRAS in an inactive GDP-bound state and blocks the KRAS-dependent signal transduction and cancer cell viability [3, 68]. In preclinical in vivo models, MRTX 894 treatment was associated with potent antitumor activity in different KRAS G12C-positive patient- and cell-derived tumors, with an overall response rate of 65% [114,115,116]. Meanwhile, with regards to its clinical efficacy, the first results of an ongoing phase I/II clinical trial (NCT 03785249) suggest promising clinical outcomes (especially in NSCLC patients) and favorable safety profile [68, 116]. Another potential direct KRAS G12C inhibitor might be the investigational, orally available JNJ-74699157 (ARS-3248), which is a new generation of the KRAS G12C inhibitor ARS-1620. A multicenter phase I clinical trial (NCT04006301) evaluating JNJ-74699157 started the enrollment in July 2019 and is currently ongoing [66, 68]. Further potential KRAS G12C inhibitors under development include the Eli Lilly drug LY3499446 (NCT04165031), the Pfizer drug tetrahydroquinazoline derivatives (US 2019/0248767A1), and the AstraZeneca drug tetracyclic compounds (WO 2019/110751 A1) [68].
5.8 Other therapeutic approaches to treat KRAS-mutant NSCLC
Besides the need to develop new, single-agent therapeutic compounds, the complexity of the RAS signaling pathway underscores the necessity for a variety of combination therapy as well. Consequently, combination screenings have been conducted using ARS-1620, AMG 510, and MRTX 849 to identify combinations that may enhance the therapeutic response [68, 109]. Accordingly, adding mTOR and IGF1R inhibitors to ARS-1620 greatly improves its effectiveness on KRAS G12C-mutant lung cancer cells in vitro and in mouse models [109]. Meanwhile, the combination of AMG 510 with multiple agents including different MEK inhibitors or the standard of care chemotherapeutic agent carboplatin resulted in the synergistic killing of tumor cells in vitro, thus providing rationale for this approach in the clinic [112]. As for the later-mentioned direct KRAS G12C inhibitor, combinations of MRTX 849 with agents including the HER family inhibitor afatinib, the CDK4/6 inhibitor palbociclib, the SHP2 inhibitor RMC-4550, and different mTOR pathway inhibitors demonstrated enhanced response and marked tumor regression in several cell-line panels and tumor models, including MRTX 849-refractory models as well [115]. Finally, since preclinical works support the hypothesis that KRAS mutations may be vulnerable to immune checkpoint inhibition, the evaluation of clinical response to combination therapy of direct and indirect KRAS inhibitors and immune checkpoint inhibitors is also justified [117].
As for other therapeutic agents, AZD4785 is a KRAS antisense oligonucleotide that targets the KRAS gene irrespective of its mutational status, thereby inhibiting the downstream effector pathways [118]. Despite the encouraging preclinical results showing significant antitumor activity and favorable safety profile in mice and monkeys bearing KRAS-mutant lung cancer, the first phase I clinical trial (NCT03101839) failed, possibly because AZD4785 targets both mutant and wild type KRAS protein [3, 118]. Accordingly, the development of AZD4785 was later discontinued. As RAS proteins are highly immunogenic, another potential therapeutic approach might be the adoptive transfer of genetically engineered tumor antigen-specific T cells into patients with KRAS-mutant tumors [119]. Pharmacological studies are still in a very early stage; however, the first results indeed show an in vitro efficacy of G12V-reactive CD4+ T cells against KRAS G12V-mutant NSCLC cells [66, 120].
6 Open questions and future challenges
While direct KRAS G12C inhibitors have shown promising results in some solid tumors including LADC, the development of new potential therapeutic strategies for the treatment of KRAS-mutated lung cancer is a work in progress, and many questions remain.
1. Despite the promising results achieved with direct KRAS G12C inhibitors, approximately half of the G12C-mutant lung cancer patients show only a partial response to these therapeutic agents [121]. The mechanism of how cancer cells bypass inhibition to prevent maximal response to therapy is not yet fully understood. A possible explanation might be that some quiescent cells produce new KRAS G12C in response to suppressed mitogen-activated protein kinase output, which is maintained in its active, drug-insensitive state by the epidermal growth factor receptor and aurora kinase signaling [121]. Since the inhibitors bind only to the inactive conformation of KRAS, the cells with these adaptive changes bypass the effects of KRAS G12C inhibitors and resume to proliferate [121]. This adaptive process must be overcome if we are to achieve complete and durable responses in the clinic [121].
2. Distant organ metastases with unique microenvironmental features occur frequently in lung cancer. Some of these special microenvironments, including the blood-brain barrier in case of brain metastases, represent a potential challenge for targeted therapeutic agents to reach the tumor cells in appropriate concentrations. Thereby, it is still an open question which of the direct covalent KRAS inhibitors will be able to penetrate the blood-brain barrier or other metastatic site-specific barriers.
3. Whether direct inhibition of KRAS with these new compounds in monotherapy is sufficient also remains an open question. Accordingly, future studies evaluating the clinical efficacy and tolerability of direct covalent KRAS inhibitors in combination therapy with anti-EGFR therapies, immune checkpoint inhibitors, or upstream and downstream RAS signaling inhibitors are needed.
4. Although KRAS G12C inhibitors are putting KRAS’s non-druggability reputation to the test, only 35 to 45% of all KRAS-mutant LADC patients harbor this variant [12]. Therefore, selective inhibitors or broader-acting pan-KRAS agents are needed for patients with non-G12C KRAS mutations. In non-clinical studies, the novel SOS1 inhibitors demonstrated increased antitumor activity irrespective of KRAS mutation type, yet these findings were not yet translated into the clinics.
7 Conclusions
To summarize, although KRAS mutations represent one of the most common oncogenic driver mutations in lung cancer, KRAS has been historically acknowledged a non-druggable target. Indeed, to date, no effective RAS inhibitors are used in routine clinical practice. Furthermore, the predictive role of KRAS mutation in patients receiving chemo-, targeted, anti-vascular, or immunotherapy needs to be clarified. Nevertheless, recent data on the novel direct covalent KRAS G12C inhibitors AMG 510 and MRTX 849 appear to be promising both in preclinical and clinical settings. Other therapeutic approaches such as combinatory therapy with targeted agents, immune checkpoint inhibitors, KRAS downstream inhibitors, or the newly developed direct covalent inhibitors are also encouraging but require further clinical testing. At the same time, mechanisms of adaptive resistance that limits the therapeutic potential of conformation-specific KRAS G12C inhibition might represent a possible future challenge that must be overcome for durable responses. All in all, despite the historical lack of progress, the emergence of new promising agents might change the therapeutic landscape of KRAS-mutant LADC. Yet, many questions remain and the clinical relevance of KRAS gene mutations warrants further investigations.
Data availability
Not applicable.
References
Herbst, R. S., Morgensztern, D., & Boshoff, C. (2018). The biology and management of non-small cell lung cancer. Nature, 553(7689), 446–454. https://doi.org/10.1038/nature25183.
Dearden, S., Stevens, J., Wu, Y. L., & Blowers, D. (2013). Mutation incidence and coincidence in non small-cell lung cancer: meta-analyses by ethnicity and histology (mutMap). Annals of Oncology, 24(9), 2371–2376. https://doi.org/10.1093/annonc/mdt205.
Yang, H., Liang, S.-Q., Schmid, R. A., & Peng, R.-W. (2019). New horizons in KRAS-mutant lung cancer: dawn after darkness. Frontiers in Oncology, 9, 953–953. https://doi.org/10.3389/fonc.2019.00953.
Fernandez-Medarde, A., & Santos, E. (2011). Ras in cancer and developmental diseases. Genes & Cancer, 2(3), 344–358. https://doi.org/10.1177/1947601911411084.
Timar, J. (2014). The clinical relevance of KRAS gene mutation in non-small-cell lung cancer. Current Opinion in Oncology, 26(2), 138–144. https://doi.org/10.1097/cco.0000000000000051.
O'Bryan, J. P. (2019). Pharmacological targeting of RAS: recent success with direct inhibitors. Pharmacological Research, 139, 503–511. https://doi.org/10.1016/j.phrs.2018.10.021.
Matikas, A., Mistriotis, D., Georgoulias, V., & Kotsakis, A. (2017). Targeting KRAS mutated non-small cell lung cancer: a history of failures and a future of hope for a diverse entity. Critical Reviews in Oncology/Hematology, 110, 1–12. https://doi.org/10.1016/j.critrevonc.2016.12.005.
Shepherd, F. A., Domerg, C., Hainaut, P., Janne, P. A., Pignon, J. P., Graziano, S., et al. (2013). Pooled analysis of the prognostic and predictive effects of KRAS mutation status and KRAS mutation subtype in early-stage resected non-small-cell lung cancer in four trials of adjuvant chemotherapy. Journal of Clinical Oncology, 31(17), 2173–2181. https://doi.org/10.1200/jco.2012.48.1390.
Riely, G. J., Kris, M. G., Rosenbaum, D., Marks, J., Li, A., Chitale, D. A., Nafa, K., Riedel, E. R., Hsu, M., Pao, W., Miller, V. A., & Ladanyi, M. (2008). Frequency and distinctive spectrum of KRAS mutations in never smokers with lung adenocarcinoma. Clinical Cancer Research, 14(18), 5731–5734. https://doi.org/10.1158/1078-0432.Ccr-08-0646.
Redig, A. J., Chambers, E. S., Lydon, C. A., Dahlberg, S. E., Alden, R. S., & Janne, P. A. (2016). Genomic complexity in KRAS mutant non-small cell lung cancer (NSCLC) from never/light-smokers v smokers. Journal of Clinical Oncology, 34(15_suppl), 9087–9087. https://doi.org/10.1200/JCO.2016.34.15_suppl.9087.
Ferrer, I., Zugazagoitia, J., Herbertz, S., John, W., Paz-Ares, L., & Schmid-Bindert, G. (2018). KRAS-mutant non-small cell lung cancer: from biology to therapy. Lung Cancer, 124, 53–64. https://doi.org/10.1016/j.lungcan.2018.07.013.
Dogan, S., Shen, R., Ang, D. C., Johnson, M. L., D'Angelo, S. P., Paik, P. K., Brzostowski, E. B., Riely, G. J., Kris, M. G., Zakowski, M. F., & Ladanyi, M. (2012). Molecular epidemiology of EGFR and KRAS mutations in 3,026 lung adenocarcinomas: higher susceptibility of women to smoking-related KRAS-mutant cancers. Clinical Cancer Research: An Official Journal of the American Association for Cancer Research, 18(22), 6169–6177. https://doi.org/10.1158/1078-0432.CCR-11-3265.
Le Calvez, F., Mukeria, A., Hunt, J. D., Kelm, O., Hung, R. J., Taniere, P., et al. (2005). TP53 and KRAS mutation load and types in lung cancers in relation to tobacco smoke: distinct patterns in never, former, and current smokers. Cancer Research, 65(12), 5076–5083. https://doi.org/10.1158/0008-5472.Can-05-0551.
Vachtenheim, J., Horakova, I., Novotna, H., Opaalka, P., & Roubkova, H. (1995). Mutations of K-ras oncogene and absence of H-ras mutations in squamous cell carcinomas of the lung. Clinical Cancer Research, 1(3), 359–365.
Rekhtman, N., Paik, P. K., Arcila, M. E., Tafe, L. J., Oxnard, G. R., Moreira, A. L., Travis, W. D., Zakowski, M. F., Kris, M. G., & Ladanyi, M. (2012). Clarifying the spectrum of driver oncogene mutations in biomarker-verified squamous carcinoma of lung: lack of EGFR/KRAS and presence of PIK3CA/AKT1 mutations. Clinical Cancer Research, 18(4), 1167–1176. https://doi.org/10.1158/1078-0432.Ccr-11-2109.
Renaud, S., Seitlinger, J., Falcoz, P.-E., Schaeffer, M., Voegeli, A.-C., Legrain, M., Beau-Faller, M., & Massard, G. (2016). Specific KRAS amino acid substitutions and EGFR mutations predict site-specific recurrence and metastasis following non-small-cell lung cancer surgery. British Journal of Cancer, 115(3), 346–353. https://doi.org/10.1038/bjc.2016.182.
Wiesweg, M., Kasper, S., Worm, K., Herold, T., Reis, H., Sara, L., Metzenmacher, M., Abendroth, A., Darwiche, K., Aigner, C., Wedemeyer, H. H., Helfritz, F. A., Stuschke, M., Schumacher, B., Markus, P., Paul, A., Rahmann, S., Schmid, K. W., & Schuler, M. (2019). Impact of RAS mutation subtype on clinical outcome-a cross-entity comparison of patients with advanced non-small cell lung cancer and colorectal cancer. Oncogene, 38(16), 2953–2966. https://doi.org/10.1038/s41388-018-0634-0.
Li, S., Liu, S., Deng, J., Akbay, E. A., Hai, J., Ambrogio, C., Zhang, L., Zhou, F., Jenkins, R. W., Adeegbe, D. O., Gao, P., Wang, X., Paweletz, C. P., Herter-Sprie, G. S., Chen, T., Gutiérrez-Quiceno, L., Zhang, Y., Merlino, A. A., Quinn, M. M., Zeng, Y., Yu, X., Liu, Y., Fan, L., Aguirre, A. J., Barbie, D. A., Yi, X., & Wong, K. K. (2018). Assessing therapeutic efficacy of MEK inhibition in a KRAS(G12C)-driven mouse model of lung cancer. Clinical Cancer Research, 24(19), 4854–4864. https://doi.org/10.1158/1078-0432.Ccr-17-3438.
Garassino, M. C., Marabese, M., Rusconi, P., Rulli, E., Martelli, O., Farina, G., Scanni, A., & Broggini, M. (2011). Different types of K-Ras mutations could affect drug sensitivity and tumour behaviour in non-small-cell lung cancer. Annals of Oncology, 22(1), 235–237. https://doi.org/10.1093/annonc/mdq680.
Imielinski, M., Berger, A. H., Hammerman, P. S., Hernandez, B., Pugh, T. J., Hodis, E., et al. (2012). Mapping the hallmarks of lung adenocarcinoma with massively parallel sequencing. Cell, 150(6), 1107–1120. https://doi.org/10.1016/j.cell.2012.08.029.
Lee, B., Lee, T., Lee, S. H., Choi, Y. L., & Han, J. (2016). Clinicopathologic characteristics of EGFR, KRAS, and ALK alterations in 6,595 lung cancers. Oncotarget, 7(17), 23874–23884. https://doi.org/10.18632/oncotarget.8074.
Li, S., Li, L., Zhu, Y., Huang, C., Qin, Y., Liu, H., Ren-Heidenreich, L., Shi, B., Ren, H., Chu, X., Kang, J., Wang, W., Xu, J., Tang, K., Yang, H., Zheng, Y., He, J., Yu, G., & Liang, N. (2014). Coexistence of EGFR with KRAS, or BRAF, or PIK3CA somatic mutations in lung cancer: a comprehensive mutation profiling from 5125 Chinese cohorts. British Journal of Cancer, 110(11), 2812–2820. https://doi.org/10.1038/bjc.2014.210.
Arbour, K. C., Jordan, E., Kim, H. R., Dienstag, J., Yu, H. A., Sanchez-Vega, F., Lito, P., Berger, M., Solit, D. B., Hellmann, M., Kris, M. G., Rudin, C. M., Ni, A., Arcila, M., Ladanyi, M., & Riely, G. J. (2018). Effects of co-occurring genomic alterations on outcomes in patients with KRAS-mutant non-small cell lung cancer. Clinical Cancer Research, 24(2), 334–340. https://doi.org/10.1158/1078-0432.Ccr-17-1841.
Rodenhuis, S., van de Wetering, M. L., Mooi, W. J., Evers, S. G., van Zandwijk, N., & Bos, J. L. (1987). Mutational activation of the K-ras oncogene. A possible pathogenetic factor in adenocarcinoma of the lung. The New England Journal of Medicine, 317(15), 929–935. https://doi.org/10.1056/nejm198710083171504.
Slebos, R. J., Kibbelaar, R. E., Dalesio, O., Kooistra, A., Stam, J., Meijer, C. J., et al. (1990). K-ras oncogene activation as a prognostic marker in adenocarcinoma of the lung. The New England Journal of Medicine, 323(9), 561–565. https://doi.org/10.1056/nejm199008303230902.
Kern, J. A., Slebos, R. J., Top, B., Rodenhuis, S., Lager, D., Robinson, R. A., Weiner, D., & Schwartz, D. A. (1994). C-erbB-2 expression and codon 12 K-ras mutations both predict shortened survival for patients with pulmonary adenocarcinomas. The Journal of Clinical Investigation, 93(2), 516–520. https://doi.org/10.1172/jci117001.
Mitsudomi, T., Steinberg, S. M., Oie, H. K., Mulshine, J. L., Phelps, R., Viallet, J., Pass, H., Minna, J. D., & Gazdar, A. F. (1991). ras gene mutations in non-small cell lung cancers are associated with shortened survival irrespective of treatment intent. Cancer Research, 51(18), 4999–5002.
Ohtaki, Y., Shimizu, K., Kakegawa, S., Nagashima, T., Nakano, T., Atsumi, J., et al. (2014). Postrecurrence survival of surgically resected pulmonary adenocarcinoma patients according to EGFR and KRAS mutation status. Molecular and clinical oncology, 2(2), 187–196. https://doi.org/10.3892/mco.2013.237.
Izar, B., Zhou, H., Heist, R. S., Azzoli, C. G., Muzikansky, A., Scribner, E. E., et al. (2014). The prognostic impact of KRAS, its codon and amino acid specific mutations, on survival in resected stage I lung adenocarcinoma. Journal of Thoracic Oncology, 9(9), 1363–1369. https://doi.org/10.1097/jto.0000000000000266.
Guan, J. L., Zhong, W. Z., An, S. J., Yang, J. J., Su, J., Chen, Z. H., Yan, H. H., Chen, Z. Y., Huang, Z. M., Zhang, X. C., Nie, Q., & Wu, Y. L. (2013). KRAS mutation in patients with lung cancer: a predictor for poor prognosis but not for EGFR-TKIs or chemotherapy. Annals of Surgical Oncology, 20(4), 1381–1388. https://doi.org/10.1245/s10434-012-2754-z.
Cserepes, M., Ostoros, G., Lohinai, Z., Raso, E., Barbai, T., Timar, J., Rozsas, A., Moldvay, J., Kovalszky, I., Fabian, K., Gyulai, M., Ghanim, B., Laszlo, V., Klikovits, T., Hoda, M. A., Grusch, M., Berger, W., Klepetko, W., Hegedus, B., & Dome, B. (2014). Subtype-specific KRAS mutations in advanced lung adenocarcinoma: a retrospective study of patients treated with platinum-based chemotherapy. European Journal of Cancer, 50(10), 1819–1828. https://doi.org/10.1016/j.ejca.2014.04.001.
Ihle, N. T., Byers, L. A., Kim, E. S., Saintigny, P., Lee, J. J., Blumenschein, G. R., Tsao, A., Liu, S., Larsen, J. E., Wang, J., Diao, L., Coombes, K. R., Chen, L., Zhang, S., Abdelmelek, M. F., Tang, X., Papadimitrakopoulou, V., Minna, J. D., Lippman, S. M., Hong, W. K., Herbst, R. S., Wistuba, I. I., Heymach, J. V., & Powis, G. (2012). Effect of KRAS oncogene substitutions on protein behavior: implications for signaling and clinical outcome. Journal of the National Cancer Institute, 104(3), 228–239. https://doi.org/10.1093/jnci/djr523.
Mascaux, C., Iannino, N., Martin, B., Paesmans, M., Berghmans, T., Dusart, M., et al. (2005). The role of RAS oncogene in survival of patients with lung cancer: a systematic review of the literature with meta-analysis. British Journal of Cancer, 92(1), 131–139. https://doi.org/10.1038/sj.bjc.6602258.
Villaruz, L. C., Socinski, M. A., Cunningham, D. E., Chiosea, S. I., Burns, T. F., Siegfried, J. M., & Dacic, S. (2013). The prognostic and predictive value of KRAS oncogene substitutions in lung adenocarcinoma. Cancer, 119(12), 2268–2274. https://doi.org/10.1002/cncr.28039.
Fan, G., Zhang, K., Ding, J., & Li, J. (2017). Prognostic value of EGFR and KRAS in circulating tumor DNA in patients with advanced non-small cell lung cancer: a systematic review and meta-analysis. Oncotarget, 8(20), 33922–33932. https://doi.org/10.18632/oncotarget.15412.
Meng, D., Yuan, M., Li, X., Chen, L., Yang, J., Zhao, X., et al. (2013). Prognostic value of K-RAS mutations in patients with non-small cell lung cancer: a systematic review with meta-analysis. Lung Cancer, 81(1), 1–10. https://doi.org/10.1016/j.lungcan.2013.03.019.
Zhang, S.-M., Zhu, Q.-G., Ding, X.-X., Lin, S., Zhao, J., Guan, L., Li, T., He, B., & Zhang, H. Q. (2018). Prognostic value of EGFR and KRAS in resected non-small cell lung cancer: a systematic review and meta-analysis. Cancer Management and Research, 10, 3393–3404. https://doi.org/10.2147/CMAR.S167578.
Svaton, M., Fiala, O., Pesek, M., Bortlicek, Z., Minarik, M., Benesova, L., & Topolcan, O. (2016). The prognostic role of KRAS mutation in patients with advanced NSCLC treated with second- or third-line chemotherapy. Anticancer Research, 36(3), 1077–1082.
Rodenhuis, S., Boerrigter, L., Top, B., Slebos, R. J., Mooi, W. J., van't Veer, L., & van Zandwijk, N. (1997). Mutational activation of the K-ras oncogene and the effect of chemotherapy in advanced adenocarcinoma of the lung: a prospective study. Journal of Clinical Oncology, 15(1), 285–291. https://doi.org/10.1200/jco.1997.15.1.285.
Schiller, J. H., Adak, S., Feins, R. H., Keller, S. M., Fry, W. A., Livingston, R. B., Hammond, M. E. M., Wolf, B., Sabatini, L., Jett, J., Kohman, L., & Johnson, D. H. (2001). Lack of prognostic significance of p53 and K-ras mutations in primary resected non-small-cell lung cancer on E4592: a laboratory ancillary study on an Eastern Cooperative Oncology Group prospective randomized trial of postoperative adjuvant therapy. Journal of Clinical Oncology, 19(2), 448–457. https://doi.org/10.1200/jco.2001.19.2.448.
Eberhard, D. A., Johnson, B. E., Amler, L. C., Goddard, A. D., Heldens, S. L., Herbst, R. S., Ince, W. L., Jänne, P. A., Januario, T., Johnson, D. H., Klein, P., Miller, V. A., Ostland, M. A., Ramies, D. A., Sebisanovic, D., Stinson, J. A., Zhang, Y. R., Seshagiri, S., & Hillan, K. J. (2005). Mutations in the epidermal growth factor receptor and in KRAS are predictive and prognostic indicators in patients with non-small-cell lung cancer treated with chemotherapy alone and in combination with erlotinib. Journal of Clinical Oncology, 23(25), 5900–5909. https://doi.org/10.1200/jco.2005.02.857.
Tsao, M. S., Aviel-Ronen, S., Ding, K., Lau, D., Liu, N., Sakurada, A., Whitehead, M., Zhu, C. Q., Livingston, R., Johnson, D. H., Rigas, J., Seymour, L., Winton, T., & Shepherd, F. A. (2007). Prognostic and predictive importance of p53 and RAS for adjuvant chemotherapy in non small-cell lung cancer. Journal of Clinical Oncology, 25(33), 5240–5247. https://doi.org/10.1200/jco.2007.12.6953.
Zalcman, G., Beau-Faller, M., Creveuil, C., de Fraipont, F., Mounawar, M., Richard, N., et al. (2008). Use of Ras effector RASSF1A promoter gene methylation and chromosome 9p loss of heterozygosity (LOH) to predict progression-free survival (PFS) in perioperative chemotherapy (CT) phase III trial IFCT-0002 in resectable non-small cell lung cancer. Journal of Clinical Oncology, 26(15_suppl), 7500–7500. https://doi.org/10.1200/jco.2008.26.15_suppl.7500.
Hames, M. L., Chen, H., Iams, W., Aston, J., Lovly, C. M., & Horn, L. (2016). Correlation between KRAS mutation status and response to chemotherapy in patients with advanced non-small cell lung cancer. Lung Cancer, 92, 29–34. https://doi.org/10.1016/j.lungcan.2015.11.004.
Shepherd, F. A., Lacas, B., Le Teuff, G., Hainaut, P., Janne, P. A., Pignon, J. P., et al. (2017). Pooled analysis of the prognostic and predictive effects of TP53 comutation status combined with KRAS or EGFR mutation in early-stage resected non-small-cell lung cancer in four trials of adjuvant chemotherapy. Journal of Clinical Oncology, 35(18), 2018–2027. https://doi.org/10.1200/jco.2016.71.2893.
Sun, J. M., Hwang, D. W., Ahn, J. S., Ahn, M. J., & Park, K. (2013). Prognostic and predictive value of KRAS mutations in advanced non-small cell lung cancer. PLoS One, 8(5), e64816. https://doi.org/10.1371/journal.pone.0064816.
Massarelli, E., Varella-Garcia, M., Tang, X., Xavier, A. C., Ozburn, N. C., Liu, D. D., Bekele, B. N., Herbst, R. S., & Wistuba, I. I. (2007). KRAS mutation is an important predictor of resistance to therapy with epidermal growth factor receptor tyrosine kinase inhibitors in non-small-cell lung cancer. Clinical Cancer Research, 13(10), 2890–2896. https://doi.org/10.1158/1078-0432.Ccr-06-3043.
Linardou, H., Dahabreh, I. J., Kanaloupiti, D., Siannis, F., Bafaloukos, D., Kosmidis, P., Papadimitriou, C. A., & Murray, S. (2008). Assessment of somatic k-RAS mutations as a mechanism associated with resistance to EGFR-targeted agents: a systematic review and meta-analysis of studies in advanced non-small-cell lung cancer and metastatic colorectal cancer. The Lancet Oncology, 9(10), 962–972. https://doi.org/10.1016/s1470-2045(08)70206-7.
Mao, C., Qiu, L. X., Liao, R. Y., Du, F. B., Ding, H., Yang, W. C., et al. (2010). KRAS mutations and resistance to EGFR-TKIs treatment in patients with non-small cell lung cancer: a meta-analysis of 22 studies. Lung Cancer, 69(3), 272–278. https://doi.org/10.1016/j.lungcan.2009.11.020.
Garassino, M. C., Martelli, O., Broggini, M., Farina, G., Veronese, S., Rulli, E., et al. (2013). Erlotinib versus docetaxel as second-line treatment of patients with advanced non-small-cell lung cancer and wild-type EGFR tumours (TAILOR): a randomised controlled trial. The Lancet Oncology, 14(10), 981–988. https://doi.org/10.1016/s1470-2045(13)70310-3.
Metro, G., Chiari, R., Duranti, S., Siggillino, A., Fischer, M. J., Giannarelli, D., Ludovini, V., Bennati, C., Marcomigni, L., Baldi, A., Giansanti, M., Minotti, V., & Crinò, L. (2012). Impact of specific mutant KRAS on clinical outcome of EGFR-TKI-treated advanced non-small cell lung cancer patients with an EGFR wild type genotype. Lung Cancer, 78(1), 81–86. https://doi.org/10.1016/j.lungcan.2012.06.005.
Zer, A., Ding, K., Lee, S. M., Goss, G. D., Seymour, L., Ellis, P. M., Hackshaw, A., Bradbury, P. A., Han, L., O’Callaghan, C. J., Tsao, M. S., & Shepherd, F. A. (2016). Pooled analysis of the prognostic and predictive value of KRAS mutation status and mutation subtype in patients with non-small cell lung cancer treated with epidermal growth factor receptor tyrosine kinase inhibitors. Journal of Thoracic Oncology, 11(3), 312–323. https://doi.org/10.1016/j.jtho.2015.11.010.
Chin, L., Tam, A., Pomerantz, J., Wong, M., Holash, J., Bardeesy, N., Shen, Q., O'Hagan, R., Pantginis, J., Zhou, H., Horner II, J. W., Cordon-Cardo, C., Yancopoulos, G. D., & DePinho#, R. A. (1999). Essential role for oncogenic Ras in tumour maintenance. Nature, 400(6743), 468–472. https://doi.org/10.1038/22788.
Fiala, O., Buchler, T., Mohelnikova-Duchonova, B., Melichar, B., Matejka, V. M., Holubec, L., Kulhankova, J., Bortlicek, Z., Bartouskova, M., Liska, V., Topolcan, O., Sedivcova, M., & Finek, J. (2016). G12V and G12A KRAS mutations are associated with poor outcome in patients with metastatic colorectal cancer treated with bevacizumab. Tumour Biology, 37(5), 6823–6830. https://doi.org/10.1007/s13277-015-4523-7.
Bruera, G., Cannita, K., Tessitore, A., Russo, A., Alesse, E., Ficorella, C., & Ricevuto, E. (2015). The prevalent KRAS exon 2 c.35 G>A mutation in metastatic colorectal cancer patients: a biomarker of worse prognosis and potential benefit of bevacizumab-containing intensive regimens? Critical Reviews in Oncology/Hematology, 93(3), 190–202. https://doi.org/10.1016/j.critrevonc.2014.10.004.
Chaft, J. E., Rusch, V., Ginsberg, M. S., Paik, P. K., Finley, D. J., Kris, M. G., Price, K. A. R., Azzoli, C. G., Fury, M. G., Riely, G. J., Krug, L. M., Downey, R. J., Bains, M. S., Sima, C. S., Rizk, N., Travis, W. D., & Rizvi, N. A. (2013). Phase II trial of neoadjuvant bevacizumab plus chemotherapy and adjuvant bevacizumab in patients with resectable nonsquamous non-small-cell lung cancers. Journal of Thoracic Oncology, 8(8), 1084–1090. https://doi.org/10.1097/JTO.0b013e31829923ec.
Ghimessy, A. K., Gellert, A., Schlegl, E., Hegedus, B., Raso, E., Barbai, T., et al. (2019). KRAS mutations predict response and outcome in advanced lung adenocarcinoma patients receiving first-line bevacizumab and platinum-based chemotherapy. Cancers (Basel), 11(10). https://doi.org/10.3390/cancers11101514.
D'Incecco, A., Andreozzi, M., Ludovini, V., Rossi, E., Capodanno, A., Landi, L., et al. (2015). PD-1 and PD-L1 expression in molecularly selected non-small-cell lung cancer patients. British Journal of Cancer, 112(1), 95–102. https://doi.org/10.1038/bjc.2014.555.
Dong, Z. Y., Zhong, W. Z., Zhang, X. C., Su, J., Xie, Z., Liu, S. Y., Tu, H. Y., Chen, H. J., Sun, Y. L., Zhou, Q., Yang, J. J., Yang, X. N., Lin, J. X., Yan, H. H., Zhai, H. R., Yan, L. X., Liao, R. Q., Wu, S. P., & Wu, Y. L. (2017). Potential predictive value of TP53 and KRAS mutation status for response to PD-1 blockade immunotherapy in lung adenocarcinoma. Clinical Cancer Research, 23(12), 3012–3024. https://doi.org/10.1158/1078-0432.Ccr-16-2554.
Lee, M.-H., Yanagawa, J., Li, R., Walser, T. C., Krysan, K., Wang, G., Goldman, J. W., Garon, E. B. G., Zeng, G., Sharma, S., Minna, J. D., Carbone, D., Dubinett, S. M., & Lee, J. M. (2015). Increased PD-L1 expression in KRAS mutated premalignant human bronchial epithelial cells is enhanced by LKB1 loss and mediated by ERK activation. Journal for Immunotherapy of Cancer, 3(Suppl 2), –P305. https://doi.org/10.1186/2051-1426-3-S2-P305.
Reiniger, L., Teglasi, V., Pipek, O., Rojko, L., Glasz, T., Vagvolgyi, A., et al. (2019). Tumor necrosis correlates with PD-L1 and PD-1 expression in lung adenocarcinoma. Acta Oncologica, 58(8), 1087–1094. https://doi.org/10.1080/0284186x.2019.1598575.
Chen, N., Fang, W., Lin, Z., Peng, P., Wang, J., Zhan, J., et al. (2017). KRAS mutation-induced upregulation of PD-L1 mediates immune escape in human lung adenocarcinoma. Cancer Immunology, Immunotherapy, 66(9), 1175–1187. https://doi.org/10.1007/s00262-017-2005-z.
Gettinger, S., Horn, L., Jackman, D., Spigel, D., Antonia, S., Hellmann, M., et al. (2018). Five-year follow-up of nivolumab in previously treated advanced non-small-cell lung cancer: results from the CA209-003 study. Journal of Clinical Oncology, 36(17), 1675–1684. https://doi.org/10.1200/jco.2017.77.0412.
Lindsay, C. R., Jamal-Hanjani, M., Forster, M., & Blackhall, F. (2018). KRAS: reasons for optimism in lung cancer. European Journal of Cancer, 99, 20–27. https://doi.org/10.1016/j.ejca.2018.05.001.
Roman, M., Baraibar, I., Lopez, I., Nadal, E., Rolfo, C., Vicent, S., et al. (2018). KRAS oncogene in non-small cell lung cancer: clinical perspectives on the treatment of an old target. Molecular Cancer, 17(1), 33. https://doi.org/10.1186/s12943-018-0789-x.
Friedlaender, A., Drilon, A., Weiss, G. J., Banna, G. L., & Addeo, A. (2020). KRAS as a druggable target in NSCLC: rising like a phoenix after decades of development failures. Cancer Treatment Reviews, 85, 101978. https://doi.org/10.1016/j.ctrv.2020.101978.
Gysin, S., Salt, M., Young, A., & McCormick, F. (2011). Therapeutic strategies for targeting ras proteins. Genes & Cancer, 2(3), 359–372. https://doi.org/10.1177/1947601911412376.
Nagasaka, M., Li, Y., Sukari, A., Ou, S.-H. I., Al-Hallak, M. N., & Azmi, A. S. (2020). KRAS G12C game of thrones, which direct KRAS inhibitor will claim the iron throne? Cancer Treatment Reviews, 84, 101974. https://doi.org/10.1016/j.ctrv.2020.101974.
Dang, C. V., Reddy, E. P., Shokat, K. M., & Soucek, L. (2017). Drugging the ‘undruggable’ cancer targets. Nature Reviews. Cancer, 17(8), 502–508. https://doi.org/10.1038/nrc.2017.36.
Lohinai, Z., Klikovits, T., Moldvay, J., Ostoros, G., Raso, E., Timar, J., Fabian, K., Kovalszky, I., Kenessey, I., Aigner, C., Renyi-Vamos, F., Klepetko, W., Dome, B., & Hegedus, B. (2017). KRAS-mutation incidence and prognostic value are metastatic site-specific in lung adenocarcinoma: poor prognosis in patients with KRAS mutation and bone metastasis. Scientific Reports, 7, 39721. https://doi.org/10.1038/srep39721.
Bagchi, S., Rathee, P., Jayaprakash, V., & Banerjee, S. (2018). Farnesyl transferase inhibitors as potential anticancer agents. Mini Reviews in Medicinal Chemistry, 18(19), 1611–1623. https://doi.org/10.2174/1389557518666180801110342.
End, D. W., Smets, G., Todd, A. V., Applegate, T. L., Fuery, C. J., Angibaud, P., Venet, M., Sanz, G., Poignet, H., Skrzat, S., Devine, A., Wouters, W., & Bowden, C. (2001). Characterization of the antitumor effects of the selective farnesyl protein transferase inhibitor R115777 in vivo and in vitro. Cancer Research, 61(1), 131–137.
Riely, G. J., Johnson, M. L., Medina, C., Rizvi, N. A., Miller, V. A., Kris, M. G., Pietanza, M. C., Azzoli, C. G., Krug, L. M., Pao, W., & Ginsberg, M. S. (2011). A phase II trial of Salirasib in patients with lung adenocarcinomas with KRAS mutations. Journal of Thoracic Oncology, 6(8), 1435–1437. https://doi.org/10.1097/JTO.0b013e318223c099.
Tanaka, A., Radwan, M. O., Hamasaki, A., Ejima, A., Obata, E., Koga, R., Tateishi, H., Okamoto, Y., Fujita, M., Nakao, M., Umezawa, K., Tamanoi, F., & Otsuka, M. (2017). A novel inhibitor of farnesyltransferase with a zinc site recognition moiety and a farnesyl group. Bioorganic & Medicinal Chemistry Letters, 27(16), 3862–3866. https://doi.org/10.1016/j.bmcl.2017.06.047.
Kazi, A., Xiang, S., Yang, H., Chen, L., Kennedy, P., Ayaz, M., Fletcher, S., Cummings, C., Lawrence, H. R., Beato, F., Kang, Y.'., Kim, M. P., Delitto, A., Underwood, P. W., Fleming, J. B., Trevino, J. G., Hamilton, A. D., & Sebti, S. M. (2019). Dual farnesyl and geranylgeranyl transferase inhibitor thwarts mutant KRAS-driven patient-derived pancreatic tumors. Clinical Cancer Research, 25(19), 5984–5996. https://doi.org/10.1158/1078-0432.Ccr-18-3399.
Xie, F., Li, P., Gong, J., Zhang, J., & Ma, J. (2015). The bisphosphonate zoledronic acid effectively targets lung cancer cells by inhibition of protein prenylation. Biochemical and Biophysical Research Communications, 467(4), 664–669. https://doi.org/10.1016/j.bbrc.2015.10.089.
Di Salvatore, M., Orlandi, A., Bagala, C., Quirino, M., Cassano, A., Astone, A., et al. (2011). Anti-tumour and anti-angiogenetic effects of zoledronic acid on human non-small-cell lung cancer cell line. Cell Proliferation, 44(2), 139–146. https://doi.org/10.1111/j.1365-2184.2011.00745.x.
Tao, M. H., Chen, S., Freudenheim, J. L., Cauley, J. A., Johnson, K. C., Mai, X., Sarto, G. E., Wakelee, H., Boffetta, P., & Wactawski-Wende, J. (2018). Oral bisphosphonate use and lung cancer incidence among postmenopausal women. Annals of Oncology, 29(6), 1476–1485. https://doi.org/10.1093/annonc/mdy097.
Nagao, S., Hattori, N., Fujitaka, K., Iwamoto, H., Ohshimo, S., Kanehara, M., Ishikawa, N., Haruta, Y., Murai, H., & Kohno, N. (2011). Regression of a primary pulmonary adenocarcinoma after zoledronic acid monotherapy. Hiroshima Journal of Medical Sciences, 60(1), 7–9.
Kenessey, I., Kói, K., Horváth, O., Cserepes, M., Molnár, D., Izsák, V., et al. (2016). KRAS-mutation status dependent effect of zoledronic acid in human non-small cell cancer preclinical models. Oncotarget, 7(48), 79503–79514. https://doi.org/10.18632/oncotarget.12806.
Cox, A. D., Der, C. J., & Philips, M. R. (2015). Targeting RAS membrane association: back to the future for anti-RAS drug discovery? Clinical Cancer Research, 21(8), 1819–1827. https://doi.org/10.1158/1078-0432.Ccr-14-3214.
Cox, A. D., Fesik, S. W., Kimmelman, A. C., Luo, J., & Der, C. J. (2014). Drugging the undruggable RAS: mission possible? Nature Reviews. Drug Discovery, 13(11), 828–851. https://doi.org/10.1038/nrd4389.
Dingemans, A.-M. C., Mellema, W. W., Groen, H. J. M., van Wijk, A., Burgers, S. A., Kunst, P. W. A., et al. (2013). A Phase II study of sorafenib in patients with platinum-pretreated, advanced (Stage IIIb or IV) non–small cell lung cancer with a KRAS mutation. Clinical Cancer Research, 19(3), 743. https://doi.org/10.1158/1078-0432.CCR-12-1779.
Papadimitrakopoulou, V., Lee, J. J., Wistuba, I. I., Tsao, A. S., Fossella, F. V., Kalhor, N., Gupta, S., Byers, L. A., Izzo, J. G., Gettinger, S. N., Goldberg, S. B., Tang, X., Miller, V. A., Skoulidis, F., Gibbons, D. L., Shen, L., Wei, C., Diao, L., Peng, S. A., Wang, J., Tam, A. L., Coombes, K. R., Koo, J. S., Mauro, D. J., Rubin, E. H., Heymach, J. V., Hong, W. K., & Herbst, R. S. (2016). The BATTLE-2 study: a biomarker-integrated targeted therapy study in previously treated patients with advanced non-small-cell lung cancer. Journal of Clinical Oncology, 34(30), 3638–3647. https://doi.org/10.1200/jco.2015.66.0084.
Paz-Ares, L., Hirsh, V., Zhang, L., de Marinis, F., Yang, J. C., Wakelee, H. A., et al. (2015). Monotherapy Administration of sorafenib in patients with non-small cell lung cancer (mission) trial: a phase III, multicenter, placebo-controlled trial of sorafenib in patients with relapsed or refractory predominantly nonsquamous non-small-cell lung cancer after 2 or 3 previous treatment regimens. Journal of Thoracic Oncology, 10(12), 1745–1753. https://doi.org/10.1097/jto.0000000000000693.
Carter, C. A., Rajan, A., Keen, C., Szabo, E., Khozin, S., Thomas, A., Brzezniak, C., Guha, U., Doyle, L. A., Steinberg, S. M., Xi, L., Raffeld, M., Tomita, Y., Lee, M. J., Lee, S., Trepel, J. B., Reckamp, K. L., Koehler, S., Gitlitz, B., Salgia, R., Gandara, D., Vokes, E., & Giaccone, G. (2016). Selumetinib with and without erlotinib in KRAS mutant and KRAS wild-type advanced nonsmall-cell lung cancer. Annals of Oncology, 27(4), 693–699. https://doi.org/10.1093/annonc/mdw008.
Blumenschein Jr., G. R., Smit, E. F., Planchard, D., Kim, D. W., Cadranel, J., De Pas, T., et al. (2015). A randomized phase II study of the MEK1/MEK2 inhibitor trametinib (GSK1120212) compared with docetaxel in KRAS-mutant advanced non-small-cell lung cancer (NSCLC)dagger. Annals of Oncology, 26(5), 894–901. https://doi.org/10.1093/annonc/mdv072.
Riely, G. J., Brahmer, J. R., Planchard, D., Crinò, L., Doebele, R. C., Mas Lopez, L. A., et al. (2012). A randomized discontinuation phase II trial of ridaforolimus in non-small cell lung cancer (NSCLC) patients with KRAS mutations. Journal of Clinical Oncology, 30(15_suppl), 7531–7531. https://doi.org/10.1200/jco.2012.30.15_suppl.7531.
Manchado, E., Weissmueller, S., Morris, J. P. T., Chen, C. C., Wullenkord, R., Lujambio, A., et al. (2016). A combinatorial strategy for treating KRAS-mutant lung cancer. Nature, 534(7609), 647–651. https://doi.org/10.1038/nature18600.
Aguirre, A. J., & Hahn, W. C. (2018). Synthetic lethal vulnerabilities in KRAS-mutant cancers. Cold Spring Harbor Perspectives in Medicine, 8(8), a031518. https://doi.org/10.1101/cshperspect.a031518.
Downward, J. (2015). RAS synthetic lethal screens revisited: still seeking the elusive prize? Clinical Cancer Research: An Official Journal of the American Association for Cancer Research, 21(8), 1802–1809. https://doi.org/10.1158/1078-0432.CCR-14-2180.
Litvak, A. M., Drilon, A. E., Rekhtman, N., Pietanza, M. C., Chaft, J. E., Woo, K., et al. (2015). Phase II trial of bortezomib in KRAS G12D mutant lung cancers. Journal of Clinical Oncology, 33(15_suppl), e19002. https://doi.org/10.1200/jco.2015.33.15_suppl.e19002.
Goldman, J. W., Shi, P., Reck, M., Paz-Ares, L., Koustenis, A., & Hurt, K. C. (2016). Treatment rationale and study design for the JUNIPER Study: a randomized phase III study of abemaciclib with best supportive care versus erlotinib with best supportive care in patients with stage IV non-small-cell lung cancer with a detectable KRAS mutation whose disease has progressed after platinum-based chemotherapy. Clinical Lung Cancer, 17(1), 80–84. https://doi.org/10.1016/j.cllc.2015.08.003.
Goldman, J. W., Mazieres, J., Barlesi, F., Koczywas, M., Dragnev, K. H., Göksel, T., et al. (2018). A randomized phase 3 study of abemaciclib versus erlotinib in previously treated patients with stage IV NSCLC with KRAS mutation: JUNIPER. Journal of Clinical Oncology, 36(15_suppl), 9025. https://doi.org/10.1200/JCO.2018.36.15_suppl.9025.
Ambrogio, C., Nadal, E., Villanueva, A., Gómez-López, G., Cash, T. P., Barbacid, M., & Santamaría, D. (2016). KRAS-driven lung adenocarcinoma: combined DDR1/Notch inhibition as an effective therapy. ESMO open, 1(5), e000076–e000076. https://doi.org/10.1136/esmoopen-2016-000076.
Hillig, R. C., Sautier, B., Schroeder, J., Moosmayer, D., Hilpmann, A., Stegmann, C. M., Werbeck, N. D., Briem, H., Boemer, U., Weiske, J., Badock, V., Mastouri, J., Petersen, K., Siemeister, G., Kahmann, J. D., Wegener, D., Böhnke, N., Eis, K., Graham, K., Wortmann, L., von Nussbaum, F., & Bader, B. (2019). Discovery of potent SOS1 inhibitors that block RAS activation via disruption of the RAS-SOS1 interaction. Proceedings of the National Academy of Sciences of the United States of America, 116(7), 2551–2560. https://doi.org/10.1073/pnas.1812963116.
Winter, J. J., Anderson, M., Blades, K., Brassington, C., Breeze, A. L., Chresta, C., et al. (2015). Small molecule binding sites on the Ras:SOS complex can be exploited for inhibition of Ras activation. Journal of Medicinal Chemistry, 58(5), 2265–2274. https://doi.org/10.1021/jm501660t.
Sun, Q., Burke, J. P., Phan, J., Burns, M. C., Olejniczak, E. T., Waterson, A. G., et al. (2012). Discovery of small molecules that bind to K-Ras and inhibit Sos-mediated activation. Angewandte Chemie (International Ed. in English), 51(25), 6140–6143. https://doi.org/10.1002/anie.201201358.
Maurer, T., Garrenton, L. S., Oh, A., Pitts, K., Anderson, D. J., Skelton, N. J., Fauber, B. P., Pan, B., Malek, S., Stokoe, D., Ludlam, M. J. C., Bowman, K. K., Wu, J., Giannetti, A. M., Starovasnik, M. A., Mellman, I., Jackson, P. K., Rudolph, J., Wang, W., & Fang, G. (2012). Small-molecule ligands bind to a distinct pocket in Ras and inhibit SOS-mediated nucleotide exchange activity. Proceedings of the National Academy of Sciences of the United States of America, 109(14), 5299–5304. https://doi.org/10.1073/pnas.1116510109.
Oncology, B. I. (2019). Boehringer Ingelheim advances first Pan-KRAS inhibitor BI 1701963 into clinical testing. https://www.boehringer-ingelheim.com/press-release/first-pan-kras-inhibitor-advances-clinical-testing.
Mullard, A. (2019). Cracking KRAS. Nature Reviews. Drug Discovery, 18(12), 887–891. https://doi.org/10.1038/d41573-019-00195-5.
Hunter, J. C., Gurbani, D., Ficarro, S. B., Carrasco, M. A., Lim, S. M., Choi, H. G., Xie, T., Marto, J. A., Chen, Z., Gray, N. S., & Westover, K. D. (2014). In situ selectivity profiling and crystal structure of SML-8-73-1, an active site inhibitor of oncogenic K-Ras G12C. Proceedings of the National Academy of Sciences of the United States of America, 111(24), 8895–8900. https://doi.org/10.1073/pnas.1404639111.
Lito, P., Solomon, M., Li, L. S., Hansen, R., & Rosen, N. (2016). Allele-specific inhibitors inactivate mutant KRAS G12C by a trapping mechanism. Science, 351(6273), 604–608. https://doi.org/10.1126/science.aad6204.
Patricelli, M. P., Janes, M. R., Li, L. S., Hansen, R., Peters, U., Kessler, L. V., Chen, Y., Kucharski, J. M., Feng, J., Ely, T., Chen, J. H., Firdaus, S. J., Babbar, A., Ren, P., & Liu, Y. (2016). Selective inhibition of oncogenic KRAS output with small molecules targeting the inactive state. Cancer Discovery, 6(3), 316–329. https://doi.org/10.1158/2159-8290.Cd-15-1105.
Lim, S. M., Westover, K. D., Ficarro, S. B., Harrison, R. A., Choi, H. G., Pacold, M. E., Carrasco, M., Hunter, J., Kim, N. D., Xie, T., Sim, T., Jänne, P. A., Meyerson, M., Marto, J. A., Engen, J. R., & Gray, N. S. (2014). Therapeutic targeting of oncogenic K-Ras by a covalent catalytic site inhibitor. Angewandte Chemie (International Ed. in English), 53(1), 199–204. https://doi.org/10.1002/anie.201307387.
Ostrem, J. M., Peters, U., Sos, M. L., Wells, J. A., & Shokat, K. M. (2013). K-Ras(G12C) inhibitors allosterically control GTP affinity and effector interactions. Nature, 503(7477), 548–551. https://doi.org/10.1038/nature12796.
Janes, M. R., Zhang, J., Li, L. S., Hansen, R., Peters, U., Guo, X., et al. (2018). Targeting KRAS mutant cancers with a covalent G12C-specific inhibitor. Cell, 172(3), 578-589.e517, doi:https://doi.org/10.1016/j.cell.2018.01.006.
Spencer-Smith, R., & O'Bryan, J. P. (2019). Direct inhibition of RAS: quest for the holy grail? Seminars in Cancer Biology, 54, 138–148. https://doi.org/10.1016/j.semcancer.2017.12.005.
Molina-Arcas, M., Moore, C., Rana, S., van Maldegem, F., Mugarza, E., Romero-Clavijo, P., Herbert, E., Horswell, S., Li, L. S., Janes, M. R., Hancock, D. C., & Downward, J. (2019). Development of combination therapies to maximize the impact of KRAS-G12C inhibitors in lung cancer. Science Translational Medicine, 11(510), eaaw7999. https://doi.org/10.1126/scitranslmed.aaw7999.
AMG 510 first to inhibit “undruggable” KRAS (2019). Cancer Discov, 9(8), 988-989, doi:https://doi.org/10.1158/2159-8290.Cd-nb2019-073.
Fakih, M., O'Neil, B., Price, T. J., Falchook, G. S., Desai, J., Kuo, J., et al. (2019). Phase 1 study evaluating the safety, tolerability, pharmacokinetics (PK), and efficacy of AMG 510, a novel small molecule KRASG12C inhibitor, in advanced solid tumors. Journal of Clinical Oncology, 37(15_suppl), 3003. https://doi.org/10.1200/JCO.2019.37.15_suppl.3003.
Canon, J., Rex, K., Saiki, A. Y., Mohr, C., Cooke, K., Bagal, D., et al. (2019). The clinical KRAS(G12C) inhibitor AMG 510 drives anti-tumour immunity. Nature, 575(7781), 217–223. https://doi.org/10.1038/s41586-019-1694-1.
Saiki, A. Y., Gaida, K., Rex, K., Achanta, P., Miguel, T. S., Koppada, N., et al. (2019). Abstract 4484: discovery and in vitro characterization of AMG 510–a potent and selective covalent small-molecule inhibitor of KRAS G12C. Cancer Research, 79(13 Supplement), 4484. https://doi.org/10.1158/1538-7445.AM2019-4484.
Papadopoulos, K. P., Ou, S.-H. I., Johnson, M. L., Christensen, J., Velastegui, K., Potvin, D., et al. (2019). A phase I/II multiple expansion cohort trial of MRTX849 in patients with advanced solid tumors with KRAS G12C mutation. Journal of Clinical Oncology, 37(15_suppl), TPS3161-TPS3161, doi:https://doi.org/10.1200/JCO.2019.37.15_suppl.TPS3161.
Christensen, J. G., Hallin, J., Engstrom, L. D., Hargis, L., Calinisan, A., Aranda, R., et al. (2019). The KRASG12C inhibitor, MRTX849, provides insight toward therapeutic susceptibility of KRAS mutant cancers in mouse models and patients. Cancer Discovery, CD-19-1167, doi:https://doi.org/10.1158/2159-8290.CD-19-1167.
Hallin, J., Engstrom, L. D., Hargis, L., Calinisan, A., Aranda, R., Briere, D. M., Sudhakar, N., Bowcut, V., Baer, B. R., Ballard, J. A., Burkard, M. R., Fell, J. B., Fischer, J. P., Vigers, G. P., Xue, Y., Gatto, S., Fernandez-Banet, J., Pavlicek, A., Velastagui, K., Chao, R. C., Barton, J., Pierobon, M., Baldelli, E., Patricoin III, E. F., Cassidy, D. P., Marx, M. A., Rybkin, I. I., Johnson, M. L., Ou, S. H. I., Lito, P., Papadopoulos, K. P., Jänne, P. A., Olson, P., & Christensen, J. G. (2020). The KRAS(G12C) inhibitor MRTX849 provides insight toward therapeutic susceptibility of KRAS-mutant cancers in mouse models and patients. Cancer Discovery, 10(1), 54–71. https://doi.org/10.1158/2159-8290.Cd-19-1167.
Adderley, H., Blackhall, F. H., & Lindsay, C. R. (2019). KRAS-mutant non-small cell lung cancer: converging small molecules and immune checkpoint inhibition. EBioMedicine, 41, 711–716. https://doi.org/10.1016/j.ebiom.2019.02.049.
Ross, S. J., Revenko, A. S., Hanson, L. L., Ellston, R., Staniszewska, A., Whalley, N., Pandey, S. K., Revill, M., Rooney, C., Buckett, L. K., Klein, S. K., Hudson, K., Monia, B. P., Zinda, M., Blakey, D. C., Lyne, P. D., & Macleod, A. R. (2017). Targeting KRAS-dependent tumors with AZD4785, a high-affinity therapeutic antisense oligonucleotide inhibitor of KRAS. Science Translational Medicine, 9(394), eaal5253. https://doi.org/10.1126/scitranslmed.aal5253.
Johnson, L. A., Morgan, R. A., Dudley, M. E., Cassard, L., Yang, J. C., Hughes, M. S., Kammula, U. S., Royal, R. E., Sherry, R. M., Wunderlich, J. R., Lee, C. C. R., Restifo, N. P., Schwarz, S. L., Cogdill, A. P., Bishop, R. J., Kim, H., Brewer, C. C., Rudy, S. F., VanWaes, C., Davis, J. L., Mathur, A., Ripley, R. T., Nathan, D. A., Laurencot, C. M., & Rosenberg, S. A. (2009). Gene therapy with human and mouse T-cell receptors mediates cancer regression and targets normal tissues expressing cognate antigen. Blood, 114(3), 535–546. https://doi.org/10.1182/blood-2009-03-211714.
Tran, E., Ahmadzadeh, M., Lu, Y. C., Gros, A., Turcotte, S., Robbins, P. F., et al. (2015). Immunogenicity of somatic mutations in human gastrointestinal cancers. Science, 350(6266), 1387–1390. https://doi.org/10.1126/science.aad1253.
Xue, J. Y., Zhao, Y., Aronowitz, J., Mai, T. T., Vides, A., Qeriqi, B., Kim, D., Li, C., de Stanchina, E., Mazutis, L., Risso, D., & Lito, P. (2020). Rapid non-uniform adaptation to conformation-specific KRAS(G12C) inhibition. Nature, 577(7790), 421–425. https://doi.org/10.1038/s41586-019-1884-x.
Khambata-Ford, S., Harbison, C. T., Hart, L. L., Awad, M., Xu, L. A., Horak, C. E., Dakhil, S., Hermann, R. C., Lynch, T. J., & Weber, M. R. (2010). Analysis of potential predictive markers of cetuximab benefit in BMS099, a phase III study of cetuximab and first-line taxane/carboplatin in advanced non-small-cell lung cancer. Journal of Clinical Oncology, 28(6), 918–927. https://doi.org/10.1200/jco.2009.25.2890.
Ludovini, V., Bianconi, F., Pistola, L., Chiari, R., Minotti, V., Colella, R., Giuffrida, D., Tofanetti, F. R., Siggillino, A., Flacco, A., Baldelli, E., Iacono, D., Mameli, M. G., Cavaliere, A., & Crinò, L. (2011). Phosphoinositide-3-kinase catalytic alpha and KRAS mutations are important predictors of resistance to therapy with epidermal growth factor receptor tyrosine kinase inhibitors in patients with advanced non-small cell lung cancer. Journal of Thoracic Oncology, 6(4), 707–715. https://doi.org/10.1097/JTO.0b013e31820a3a6b.
Fiala, O., Pesek, M., Finek, J., Benesova, L., Belsanova, B., & Minarik, M. (2013). The dominant role of G12C over other KRAS mutation types in the negative prediction of efficacy of epidermal growth factor receptor tyrosine kinase inhibitors in non-small cell lung cancer. Cancer Genetics, 206(1-2), 26–31. https://doi.org/10.1016/j.cancergen.2012.12.003.
Haura, E. B., Ricart, A. D., Larson, T. G., Stella, P. J., Bazhenova, L., Miller, V. A., Cohen, R. B., Eisenberg, P. D., Selaru, P., Wilner, K. D., & Gadgeel, S. M. (2010). A phase II study of PD-0325901, an oral MEK inhibitor, in previously treated patients with advanced non-small cell lung cancer. Clinical Cancer Research, 16(8), 2450–2457. https://doi.org/10.1158/1078-0432.Ccr-09-1920.
Smit, E. F., Dingemans, A. M., Thunnissen, F. B., Hochstenbach, M. M., van Suylen, R. J., & Postmus, P. E. (2010). Sorafenib in patients with advanced non-small cell lung cancer that harbor K-ras mutations: a brief report. Journal of Thoracic Oncology, 5(5), 719–720. https://doi.org/10.1097/JTO.0b013e3181d86ebf.
Hainsworth, J. D., Cebotaru, C. L., Kanarev, V., Ciuleanu, T. E., Damyanov, D., Stella, P., Ganchev, H., Pover, G., Morris, C., & Tzekova, V. (2010). A phase II, open-label, randomized study to assess the efficacy and safety of AZD6244 (ARRY-142886) versus pemetrexed in patients with non-small cell lung cancer who have failed one or two prior chemotherapeutic regimens. Journal of Thoracic Oncology, 5(10), 1630–1636. https://doi.org/10.1097/JTO.0b013e3181e8b3a3.
Martinez-Garcia, M., Banerji, U., Albanell, J., Bahleda, R., Dolly, S., Kraeber-Bodere, F., et al. (2012). First-in-human, phase I dose-escalation study of the safety, pharmacokinetics, and pharmacodynamics of RO5126766, a first-in-class dual MEK/RAF inhibitor in patients with solid tumors. Clinical Cancer Research, 18(17), 4806–4819. https://doi.org/10.1158/1078-0432.Ccr-12-0742.
Honda, K., Yamamoto, N., Nokihara, H., Tamura, Y., Asahina, H., Yamada, Y., Suzuki, S., Yamazaki, N., Ogita, Y., & Tamura, T. (2013). Phase I and pharmacokinetic/pharmacodynamic study of RO5126766, a first-in-class dual Raf/MEK inhibitor, in Japanese patients with advanced solid tumors. Cancer Chemotherapy and Pharmacology, 72(3), 577–584. https://doi.org/10.1007/s00280-013-2228-4.
Blumenschein Jr., G. R., Saintigny, P., Liu, S., Kim, E. S., Tsao, A. S., Herbst, R. S., et al. (2013). Comprehensive biomarker analysis and final efficacy results of sorafenib in the BATTLE trial. Clinical Cancer Research: An Official Journal of the American Association for Cancer Research, 19(24), 6967–6975. https://doi.org/10.1158/1078-0432.CCR-12-1818.
Janne, P. A., Shaw, A. T., Pereira, J. R., Jeannin, G., Vansteenkiste, J., Barrios, C., et al. (2013). Selumetinib plus docetaxel for KRAS-mutant advanced non-small-cell lung cancer: a randomised, multicentre, placebo-controlled, phase 2 study. The Lancet Oncology, 14(1), 38–47. https://doi.org/10.1016/s1470-2045(12)70489-8.
Chenard-Poirier, M., Kaiser, M., Boyd, K., Sriskandarajah, P., Constantinidou, A., Harris, S. J., et al. (2017). Results from the biomarker-driven basket trial of RO5126766 (CH5127566), a potent RAF/MEK inhibitor, in RAS- or RAF-mutated malignancies including multiple myeloma. Journal of Clinical Oncology, 35(15_suppl), 2506. https://doi.org/10.1200/JCO.2017.35.15_suppl.2506.
Janne, P. A., van den Heuvel, M. M., Barlesi, F., Cobo, M., Mazieres, J., Crino, L., et al. (2017). Selumetinib plus docetaxel compared with docetaxel alone and progression-free survival in patients with KRAS-mutant advanced non-small cell lung cancer: the SELECT-1 randomized clinical Trial. Jama, 317(18), 1844–1853. https://doi.org/10.1001/jama.2017.3438.
Gerber, D. E., Camidge, D. R., Morgensztern, D., Cetnar, J., Kelly, R. J., Ramalingam, S. S., Spigel, D. R., Jeong, W., Scaglioni, P. P., Zhang, S., Li, M., Weaver, D. T., Vaikus, L., Keegan, M., Horobin, J. C., & Burns, T. F. (2020). Phase 2 study of the focal adhesion kinase inhibitor defactinib (VS-6063) in previously treated advanced KRAS mutant non-small cell lung cancer. Lung Cancer, 139, 60–67. https://doi.org/10.1016/j.lungcan.2019.10.033.
Scolnick, E. M., Rands, E., Williams, D., & Parks, W. P. (1973). Studies on the nucleic acid sequences of Kirsten sarcoma virus: a model for formation of a mammalian RNA-containing sarcoma virus. Journal of Virology, 12(3), 458–463.
Malumbres, M., & Barbacid, M. (2003). RAS oncogenes: the first 30 years. Nature Reviews Cancer, 3(6), 459–465. https://doi.org/10.1038/nrc1097.
Santos, E., Martin-Zanca, D., Reddy, E. P., Pierotti, M. A., Della Porta, G., & Barbacid, M. (1984). Malignant activation of a K-ras oncogene in lung carcinoma but not in normal tissue of the same patient. Science, 223(4637), 661–664. https://doi.org/10.1126/science.6695174.
Adjei, A. A., Mauer, A., Bruzek, L., Marks, R. S., Hillman, S., Geyer, S., Hanson, L. J., Wright, J. J., Erlichman, C., Kaufmann, S. H., & Vokes, E. E. (2003). Phase II study of the farnesyl transferase inhibitor R115777 in patients with advanced non-small-cell lung cancer. Journal of Clinical Oncology, 21(9), 1760–1766. https://doi.org/10.1200/jco.2003.09.075.
Rinehart, J., Adjei, A. A., Lorusso, P. M., Waterhouse, D., Hecht, J. R., Natale, R. B., et al. (2004). Multicenter phase II study of the oral MEK inhibitor, CI-1040, in patients with advanced non-small-cell lung, breast, colon, and pancreatic cancer. Journal of Clinical Oncology, 22(22), 4456–4462. https://doi.org/10.1200/jco.2004.01.185.
Sequist, L. V., von Pawel, J., Garmey, E. G., Akerley, W. L., Brugger, W., Ferrari, D., Chen, Y., Costa, D. B., Gerber, D. E., Orlov, S., Ramlau, R., Arthur, S., Gorbachevsky, I., Schwartz, B., & Schiller, J. H. (2011). Randomized phase II study of erlotinib plus tivantinib versus erlotinib plus placebo in previously treated non-small-cell lung cancer. Journal of Clinical Oncology, 29(24), 3307–3315. https://doi.org/10.1200/jco.2010.34.0570.
Socinski, M. A., Goldman, J., El-Hariry, I., Koczywas, M., Vukovic, V., Horn, L., et al. (2013). A multicenter phase II study of ganetespib monotherapy in patients with genotypically defined advanced non-small cell lung cancer. Clinical Cancer Research, 19(11), 3068–3077. https://doi.org/10.1158/1078-0432.Ccr-12-3381.
Ramalingam, S., Goss, G., Rosell, R., Schmid-Bindert, G., Zaric, B., Andric, Z., Bondarenko, I., Komov, D., Ceric, T., Khuri, F., Samarzija, M., Felip, E., Ciuleanu, T., Hirsh, V., Wehler, T., Spicer, J., Salgia, R., Shapiro, G., Sheldon, E., Teofilovici, F., Vukovic, V., & Fennell, D. (2015). A randomized phase II study of ganetespib, a heat shock protein 90 inhibitor, in combination with docetaxel in second-line therapy of advanced non-small cell lung cancer (GALAXY-1). Annals of Oncology, 26(8), 1741–1748. https://doi.org/10.1093/annonc/mdv220.
Bradbury, P. A., Morris, D. G., Nicholas, G., Tu, D., Tehfe, M., Goffin, J. R., et al. (2018). Canadian Cancer Trials Group (CCTG) IND211: a randomized trial of pelareorep (Reolysin) in patients with previously treated advanced or metastatic non-small cell lung cancer receiving standard salvage therapy. Lung Cancer, 120, 142–148. https://doi.org/10.1016/j.lungcan.2018.03.005.
Kirsten, W. H., & Mayer, L. A. (1967). Morphologic responses to a murine erythroblastosis virus. Journal of the National Cancer Institute, 39(2), 311–335.
Funding
Open Access funding provided by Semmelweis University (SE). BD acknowledge funding from the Hungarian National Research, Development and Innovation Office (KH130356, NAP2-2017-1.2.1-NKP-0002, K129065, KNN121510). BD and VL were also supported by the Austrian Science Fund (FWF I3522, FWF I3977, and I4677). VL is a recipient of Janos Bolyai Research Scholarship of the Hungarian Academy of Sciences and the UNKP-19-4 New National Excellence Program of the Ministry for Innovation and Technology.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare that they have no competing interests.
Code availability
Not applicable.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Aron Ghimessy and Peter Radeczky are shared first authors.
Balazs Dome and Zsolt Megyesfalvi are shared last and corresponding authors.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ghimessy, A., Radeczky, P., Laszlo, V. et al. Current therapy of KRAS-mutant lung cancer. Cancer Metastasis Rev 39, 1159–1177 (2020). https://doi.org/10.1007/s10555-020-09903-9
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10555-020-09903-9