
Abstract
Despite advancements in cancer management, tumor relapse and metastasis are associated with poor outcomes in many cancers. Over the past decade, oncogene-driven carcinogenesis, dysregulated cellular signaling networks, dynamic changes in the tissue microenvironment, epithelial-mesenchymal transitions, protein expression within regulatory pathways, and their part in tumor progression are described in several studies. However, the complexity of metabolic enzyme expression is considerably under evaluated. Alterations in cellular metabolism determine the individual phenotype and behavior of cells, which is a well-recognized hallmark of cancer progression, especially in the adaptation mechanisms underlying therapy resistance. In metabolic symbiosis, cells compete, communicate, and even feed each other, supervised by tumor cells. Metabolic reprogramming forms a unique fingerprint for each tumor tissue, depending on the cellular content and genetic, epigenetic, and microenvironmental alterations of the developing cancer. Based on its sensing and effector functions, the mechanistic target of rapamycin (mTOR) kinase is considered the master regulator of metabolic adaptation. Moreover, mTOR kinase hyperactivity is associated with poor prognosis in various tumor types. In situ metabolic phenotyping in recent studies highlights the importance of metabolic plasticity, mTOR hyperactivity, and their role in tumor progression. In this review, we update recent developments in metabolic phenotyping of the cancer ecosystem, metabolic symbiosis, and plasticity which could provide new research directions in tumor biology. In addition, we suggest pathomorphological and analytical studies relating to metabolic alterations, mTOR activity, and their associations which are necessary to improve understanding of tumor heterogeneity and expand the therapeutic management of cancer.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
Metabolic alterations and bioenergetic adaptation mechanisms are essential components of the metabolic ecosystem and play a key role in cancer progression. In the past two decades, considerable advancements have been achieved in this field of research. Warburg and Minami were the first to describe altered tumor metabolism in 1923 [1]. Approximately one century later, metabolic symbiosis of tumor tissues, including alterations within the tumor microenvironment, were highlighted among the main hallmarks of cancer [2]. The term “Warburg effect” was coined in the 1970s, referring to rapid glucose uptake and high-rate lactate secretion into the tumor microenvironment (increased acidification). This fundamental feature of the anabolic processes is necessary for cancer progression and tumor cell proliferation, where the conversion of glucose to lactate provides a large pool of glycolytic metabolites and fuels the pentose phosphate and other macromolecule synthetic pathways. Glucose utilization is influenced by the interactions of cancer cells and their surrounding microenvironment, which further impacts tumor evolution (progression, adaptation mechanisms, therapy responses, and metastasis). The heterogeneity and the hierarchy of tumor tissues determine the complex ecosystem [3]; moreover, metabolic heterogeneity and metabolic symbiosis influence the homeostasis of the whole organ.
Proliferating and inflammatory cells invade the microenvironment causing continual destruction of non-tumorous cells, contributing to the development of cancer within actively participating stromal elements. Tumor cells reorganize the cellular milieu (fibroblasts, immune, mesenchymal, and other non-tumorous cells, etc.), coordinating the evolution of tumor tissue [4]. The extracellular matrix (ECM) provides nutrients and building blocks for the proliferating tumor cells. Metabolic “waste” such as metabolites and activating cellular elements accumulate and play a role in the paracrine regulatory functions of surrounding cells. This activity results in ECM remodeling via signaling mechanisms in all cells or related matrix elements [5, 6]. Considering tissue heterogeneity, parts of the cell population may vary between well/moderately/poorly differentiated morphology. Furthermore, the mutation and protein expression patterns of tumor tissue must also be considered by pathologists and clinicians during differential diagnosis. Consequently, these factors influence the therapeutic management of cancer. However, the prognostic capacity of the initial primary biopsy is limited and does not represent the real complexity and alterations of relapsed or metastatic tissues (following therapeutic intervention). Genetic and epigenetic regulation and protein expression of the developing tumor can be altered by several factors, e.g., inflammation and hypoxia (inefficient angiogenesis). Therefore, it is essential to examine cancer on a cellular, phenotypic, and genomic level, to greater characterize these features and understand their clinical relevance. There are many histopathological reviews about tumor mapping and stromal and immune cells within their ECM structure. In vivo molecular imaging with positron emission tomography (PET) and magnetic resonance imaging (MRI) allow in situ morphological analysis and emphasize the significance of metabolic and cellular heterogeneity. These results suggest that multicellular symbiosis has complex, competitive advantages in metabolic tumor ecosystems supporting the high adaptation potential (plasticity) for malignant cells during tumor growth, resistance, and metastasis. Cancer can be considered as a dynamic ecosystem where tumor cells cooperate among each other and host neighboring non-tumorous cells within their microenvironment. Understanding these interactions at tissue level modifies and develops the therapeutic management of cancer, with the application of targeted and immune checkpoint inhibitors combined with conventional therapies. Promising ongoing and upcoming investigations expand the availability of actionable targets and targeted therapies against cancer.
In the middle of the last century, it was stated that different cell subpopulations exist in “monoclonal cancer” [7,8,9]. Fidler confirmed tumor heterogeneity (intracellular heterogeneity) using B16 melanoma cell lines as an in vivo experimental model in 1978 [10]. Accordingly, it was demonstrated that only a few highly metastatic cells were present in the original B16 tumor population. This was verified with the use of an adapted classical fluctuation origin test, by Luria and Delbrück. Considerable variations in the number and sites of pulmonary metastases were detected using different cell “clones.” In the 1980s, Miller highlighted the phrase intratumoral heterogeneity in Cancer and Metastasis Reviews [11] and finally, Miller and Heppner coined the terms tumor heterogeneity and intratumoral heterogeneity in subsequent publications [12, 13]. Currently, several types of tissue heterogeneities can be distinguished, e.g., cellular, environmental, genomic, epigenomic, phenotypic, and metabolic heterogeneity. All of these have influencing effects on tissue structure, resulting in the development of tissue heterogeneity, diversity, and plasticity of cancer (Fig. 1). This complex network (ecosystem) serves as the basis for developing tumors [3].

Main participants of tumor heterogeneity and the complex cellular milieu in tumor tissue. a Many different factors influence tumor development in correlation with the hallmarks of cancer. In the last century, terminology relating to tumor tissue heterogeneity was introduced. Each type of heterogeneity has been extensively studied; based on these, the figure describes the associated areas of cancer research. The summarized “heterogeneity section” consists of subcategories including cellular elements, genetic diversity, immune, microenvironmental, and metabolic alterations of the tumor (these main elements and their contributors are indicated). The shown heterogeneities in the sector contribute to the individual phenotypes, which constitute the complex ecosystem (with tumor and non-tumorous cells in tissue structure). Stress and starving conditions influence tissue heterogeneity and cellular proliferation, differentiation, and survival mechanisms. Unfortunately, evolutionary mechanisms may select highly aggressive cell populations in the developing ecosystem, contributing to disease relapse or metastasis in patients. (APCs, antigen-presenting cells; NKs, natural killer cells; EMT, epithelial-mesenchymal transition; MET, mesenchymal-epithelial transition; CSC, cancer stem cell; CAFs, cancer-associated fibroblasts; ECM, extracellular matrix). b Metabolic complexity of the tumor tissue in correlation with vascularization. The altered concentrations of glucose-lactate, oxygen-ROS (reactive oxygen species), H+ (pH)-metabolic waste, and amino acids (essential and nonessential amino acids — EAAs/NEAAs); and their altering gradients are labeled with blue/red triangles. The labeling of different cellular elements and other matrix components are also indicated
Environmental adaptation, metabolic activity, and rewiring are critical for maintaining the survival and growth of the cancer cell population. Furthermore, these processes have central regulatory roles in tumor progression and therapeutic failure. Heterogeneous metabolic activity in tumors is characterized based on morphological and pathological examinations, tissue distributions of different tracers via functional imaging analysis, and the staining patterns of several metabolic transporters and enzyme expressions. Metabolic heterogeneity and its consequences (metabolic plasticity) also affect tumor cell survival and growth in vivo. Metabolic plasticity occurs when the “swamp occupies the healthy organized tissue” and induces metabolic collapse and the final death of the host organism. In this way, the metabolic plasticity of the tumor tissue ensures survival of the tumor population at all costs. Therefore, better characterization of the metabolic phenotypes and a deeper understanding of the metabolic dependencies of tumor and stromal elements and their network could improve the therapeutic management of cancer using novel or already available antimetabolic drugs. Finally, mapping metabolic heterogeneity (at tissue level) is necessary via analytical and pathomorphological examinations. This study aims to contribute to this growing area of research by exploring the available publications and summarizing the upcoming developments in metabolic heterogeneity and their implications in a variety of cancers. Recent developments in multicellular model systems and morphological and imaging studies have improved the understanding of the complexity of tissue heterogeneity from a metabolism perspective. Based on these findings, a multidisciplinary approach could improve personalized therapies and patient outcomes.
2 Metabolic consequences of frequent genetic mutations
RAS, p53, and MYC are common driver mutations (mutated oncogene/tumor suppressor and amplified genes) that cause regulatory alterations, affecting the cancer metabolism directly and indirectly [14]. MYC regulates metabolic enzymes, which orchestrate metabolic homeostasis, and plays a critical role in tumorigenesis, proliferation, and cell growth. This transcription factor also regulates several metabolic enzymes and transporters (e.g., phosphofructokinase 1 — PFK1, hexokinase — HK, lactate dehydrogenase A— LDHA, glutaminase — GLS, ATP citrate lyase — ACLY, amino acid transporter solute carrier family 1 member 2 — SLC1A5, glucose transporter 1 — GLUT1, monocarboxylate transporter 4 — MCT4). Additionally, MYC is involved in glucose/glutamine sensing and mTOR-dependent Akt functions, which regulate the bioenergetic balance of tumor cell growth and survival [15]. Several studies underline the importance of oncogenic Ras-mediated metabolic shifts in tumor progression, which could influence and enhance the MYC-regulated cellular events via activation of the Raf/Mek/Erk pathways. Moreover, RAS promotes anabolic mechanisms involved in fueling mitochondrial ATP generation. Increased autophagy is observed in RAS-driven tumor cells. Additionally, extracellular albumin and lipid consumption can be increased through macropinocytosis [16, 17]. These mechanisms help to utilize both extra- and intracellular resources and recycle many different metabolites via anabolic shifts to generate new cells in the growing tumor population. These RAS-driven alterations, non-oxidative pentose phosphate pathways (PPP), glutamine/glutathione metabolism–mediated maintaining of redox homeostasis, and reactive oxygen species (ROS) detoxification are important in cancer progression [18]. In addition, p53 regulates cellular metabolism by multiple mechanisms and is often referred to as the guardian of the genome and cellular integrity. It can be activated in several cellular stress situations, including starving conditions, e.g., energy and nutrient depletion. In correlation with these, AMPK activates p53 either directly or through other mechanisms [19,20,21].
Consequently, glycolysis is reduced, and mitochondrial respiration is increased directly by TP53-induced glycolysis and apoptosis regulator (TIGAR). In turn, this reduces the activity of Akt/mTOR, NF-κB, PPP, glucose transporters, and the expression of glycolytic enzymes (PFKs; pyruvate dehydrogenase kinase 2 — PDH2) and increases PDH activation [22, 23]. In parallel, mitochondrial oxidative phosphorylation is positively regulated by p53 in cells which facilitate pyruvate–acetyl-CoA conversion and mitochondrial glutaminolysis by GLS2, promoting mitochondrial oxidative processes and enhancing glutathione production, thus maintaining redox homeostasis [24]. Furthermore, it was described that p53 can directly bind and depress sterol regulatory element-binding transcription factor 1 (SREBP1), inhibit lipid synthesis, and enhance fatty acid (FA) oxidation in cells [23]. Moreover, mutant p53 could have additional roles in metabolic reprogramming with active participation. It was reported that these mutant proteins could bind and activate transcription factors, including SREBP1/2 and ETS2, which upregulate both lipid and nucleotide biosynthesis in malignant cells [25]. These results highlight TP53 mutation and its part in the loss of unfavorable regulatory functions. However, they could also have a direct gain-of-function effect within the metabolic regulatory network and mutation hotspot in a cell type–dependent manner. Beyond the most frequently mutated driver genes, several other mutations also have metabolic consequences during tumorigenesis. Dynamic tissue remodeling, signaling alterations due to oncogenic mutations, and their metabolic products have been widely investigated and mapped.
During tumor progression, especially in metastatic processes, cells undergo an epithelial-mesenchymal transition (EMT); this process has unique demands and requires metabolic rewiring mechanisms. Mutation-dependent activation of Wnt/-catenin signaling is characteristic for mesenchymal transformation in epithelial cells, which lose their differentiated phenotype. For example, mutations in the adenomatous polyposis coli gene and altered Wnt signaling–related events are associated with familial adenomatous polyposis in colonic tumors [26]. Crosstalking between other signaling network kinases (as MAPK, MEK1, and LKB1) and oncogenic signaling pathways including Wnt, transforming growth factor-β (TGFβ), NOTCH, and JAK/STAT has several effects in tumor progression, especially in EMT. These signaling alterations induce the overexpression of SNAIL1-2/ZEB1-2/TWIST transcription factors (as a consequence of EMT signaling activation) and downregulate the expression of several glycolytic enzymes. Additionally, this promotes glutamine and asparagine metabolism. These genetic alterations, which result in Wnt/TGFβ/NOTCH signaling hyperactivity or other EMT-forcing events, alter the metabolic activity of cancer cells. The concomitant adaptation mechanisms initiate the anchorage-independent migration, the survival of cancer cells in particular conditions, and even the metastasis formation in the whole body [27]. In contrast with a proliferating tumor mass, metastatic cells have a “slow life,” which is characterized by their catabolism, resistance to cell death, generation of ATP from oxidative phosphorylation (OXPHOS), quenching ROS, and adaptation mechanisms using alternative energy sources such as autophagy rather than external sources [28,29,30].
It has been suggested that other relatively frequent mutations like EGFR and PI3KCA activations correlate with the in situ proliferation and metabolic alterations (e.g., in non-small cell lung cancer — NSCLCs and breast carcinomas). A positive correlation was found among PET-CT SUVmax, EGFR, glycolytic activity, and PI3KCA mutation status in NSCLCs and breast carcinomas [31, 32]. These results support the conclusion that not only TP53 and KRAS but also receptor tyrosine kinase pathway mutations are responsible for the metabolic switch to increased glycolysis and reduced OXPHOS [32,33,34]. Additionally, epigenetic alterations (e.g., hypermethylation) and PTEN suppression result in PDK1/MYC-dependent Akt/mTOR activation. These are commonly found in targeted receptor inhibitor-resistant carcinoma cells [35]. The loss of other negative regulators in these signaling pathways and their downstream consequences may be targetable with mTOR kinase inhibitors (e.g., in cancers harboring TSC1/2 and PTEN germline or somatic mutations).
Metabolic rewiring in cancer is associated with oncogenic alterations, which have been extensively examined in the past few years. The data summarized above demonstrates how the different oncogenic mutations and their metabolic consequences affect cancer metabolism. Finally, these advancements may improve understanding of oncogenic mutations and expand the therapeutic management with the addition of antimetabolic drugs to combined therapies.
3 Increased amount of oncometabolites
Germline or somatic mutations in metabolic genes can cause “inborn” errors in metabolism and increased cancer risk [36, 37]. Mutations and loss of function of certain metabolic enzymes (fumarate hydratase — FH and succinate dehydrogenase — SDH) in the tricarboxylic acid (TCA) cycle are described as precursors of rare inherited and renal cancers. In 2008, isocitrate dehydrogenase (IDH1/2) mutations and the accumulation of D-2-hydroxyglutarate (2HG) were discovered to have a tumorigenic role in gliomas [38]. “Oncometabolites” were distinguished from metabolic toxins and other small metabolites based on their promoting role in malignant transformation, tumor growth, and progression [39]. There are many observations regarding tumor cell–driven metabolic by-products in high intra- or extracellular concentrations as well as their role in propagating and promoting tumor growth. Due to their structural similarities, fumarate, succinate, and 2HG can competitively inhibit alpha-ketoglutarate (αKG)–dependent dioxygenases (increasing HIF1α, pseudohypoxia). These oncometabolites also competitively inhibit epigenetic regulatory proteins (histone lysine demethylases — KDMs, ten-eleven translocation — TET, and 5-methylcytosine hydroxylases) by influencing methylation and acetylation functions [39, 40]. Furthermore, these mutations and altered enzyme functions cause mitochondrial failures, resulting in increased ROS with several consequences (Fig. 2.).

The tumor-promoting effects of oncometabolites. a The classical oncometabolites (fumarate, succinate, D-2-hydroxyglutarate — 2HG) and their epigenetic and angiogenic effects are shown in the figure. The accumulation of these oncometabolites inhibit prolyl hydroxylases and stabilize HIF1α (causing hypoxia), and they have a direct regulatory role by competitively inhibiting dioxygenases, influencing promoter methylation — activation/inactivation of oncogenes and tumor suppressor genes, respectively. b Lactate (as an oncometabolite) causes an acidic microenvironment, which aids tumor progression. Tumor and stromal cells produce lactate in correlation with blood vessel distance and tumorigenic alterations (e.g., oncogene-driven pseudohypoxia). Lactate and decreased pH have many tumor promoter functions: inhibition of antitumor immune effector cells, induction of therapy resistance, and sourcing of alternative nutrient supply for highly oxygenated normal and malignant cells. The acidic microenvironment also causes cancer-associated fibroblasts (CAFs) to produce growth factors, cytokines, feed tumor cells (TCA replenishing metabolites and the use of amino acids as nutrients), and matrix degradation. Additionally, the low pH could play a role in cytokine and enzyme activation, assisting with tumor cell adaptation, matrix remodeling, and tumor cell migration, as part of immunosuppressive and tumor-promoting niches. c The names, sources, metabolic contributions, and the relevance of traditional and non-traditional oncometabolites in various cancers
According to the literature, the most frequent tumorigenic alterations increase lactate production, causing acidification in tumor tissue; this is a well-known metabolic consequence of fast proliferating tumor cells. Based on these studies, lactate is not only a by-product but an important oncometabolite in tumor homeostasis [41]. In summary, elevated lactate concentrations in tumor tissues and blood are correlated with a high risk of cancer progression, metastasis, and mortality.
Lactate has a significant role in metabolic symbiosis; it can be used and oxidized by highly oxygenated cells. In addition, lactate is a signaling molecule responsible for further extra- and intracellular effects in the microenvironment. Elevated lactate levels influence the production of several growth factors by fibroblasts, endothelial cells, and adipocytes (HGF, ILs, TGFβ, IFNs, FGFs, VEGF, etc.). Consequently, this can induce the expression of some regulatory proteins, metabolites, microRNAs (miRs), and their exosomal transport [4, 42, 43]. Lactate can bind NDGR3, preventing association with PHD2, and stabilize HIF1α by inhibiting prolyl hydroxylase (PHD) activity [44]. Moreover, proton-sensitive lactate sensors (GPR4/65/68/132) in the acidic tissue microenvironment can activate intracellular Ca2+-cAMP-ROS and the related MAPK pathways, leading to increased matrix metalloproteinase (MMP) expression and stimulation of EMT in the tumor. Additionally, acidic pH induces the proteolysis of ECM elements and activates MMPs and cathepsins [45]. Acidic conditions in the microenvironment cause redistribution of lysosomes to the cell periphery, e.g., secretion of cathepsin B to the microenvironment. These factors contribute to tissue remodeling and alteration of tumor cell polarity and adhesion properties [46, 47]. Lactate and the acidic microenvironment negatively influence the immune response, affecting tumor-infiltrating immune cells in many ways. These microenvironmental changes induce immunosuppressive functions via inhibition of immune effector cells; reduction of natural killer, cytotoxic T cells infiltration, and monocyte-macrophage differentiation (inducing a shift to M2 macrophages), impairment of antigen presentation, and supporting of regulatory T cells. These all contribute to the tumor immune escape mechanism [37, 41, 48]. Finally, all these alterations promote survival, motility, migration, and invasion of cancer cells (Fig. 2).
Besides the classical oncometabolites (fumarate, succinate, 2HG), lactate is the most important and well-studied oncometabolite. In recent metabolomic studies, several putative oncometabolites have been reported. There is abundant room for further progress in determining additional oncometabolites and their tumorigenic effect in different cancers. For example, in gliomas, increased levels of hypotaurine are strongly correlated with the tumor grade. Additionally, homocysteic acid (an inhibitor of cysteine sulfinic acid decarboxylase — hypotaurine production) can inhibit the proliferation of glioblastoma cell lines, and some in vivo studies have confirmed that hypotaurine could be a targetable oncometabolite in glioblastomas [49, 50]. Kynurenine is known to relate to de novo nicotinic acid and NAD synthesis. Based on these findings, it is thought to have an oncometabolite function, e.g., in colon cancers [51]. Additionally, methylglyoxal involves the formation of advanced glycation end-products, which could have tumor-promoting effects. In conclusion, using the term oncometabolites for methylglyoxal and other potential metabolites requires further experimental and clinical investigations [52] (Fig. 2).
4 ECM as a metabolic niche in tissue microenvironment
Alongside tumor and non-tumorous cells, the functions of the extracellular matrix and their role in carcinogenesis and cancer progression are extensively reported in the literature. As a part of metabolic symbiosis, tumor microenvironment heterogeneity is a proposed hallmark of cancer metabolism and is responsible for the specific metabolic niche in the tumor mass [2, 5, 6]. Consequently, this affects tumor growth in a complex manner [4].
Similarly, to wound healing, the growing tumor also influences stromal elements and matrix proteins and provides nutrients (proteins, amino acids, lipids, and their building blocks). As elements of tissue remodeling, cancer cells reorganize the surrounding cellular milieu, which promotes both tissue regeneration and the production of several additional factors (e.g., cytokines and growth factors). The alterations in danger-associated molecular patterns propagate the differentiation of myofibroblast-like to cancer-associated fibroblasts (CAFs). Tumor cell–derived metabolic shifts directly affect the tumor type–dependent metabolites as well as the cellular nutrient demand and can completely exhaust the matrix and release metabolic products into the ECM [53] (Fig. 1).
The hypoxic or pseudohypoxic changes induce the Warburg effect; therefore, the concentration of glucose, lactate, and other metabolic intermediates (e.g., carbohydrates, ribose, specific lipids, FAs, acetate, and amino acids) can vary in cancer microenvironments. Increased lactate production and/or O2 consumption resulting CO2 release and their transport mechanisms (using MCTs and carbonic anhydrase IX (CAIX) transporters) also contribute to lowered pH in the tumor mass [54, 55].
Glucose and amino acids are the primary carbon sources for proliferating cells in tumor tissues [56,57,58,59]. However, glutamine utilization and its replenishing effect (anaplerosis) in the TCA cycle can support the tumor growth/survival processes under hypoxic conditions [60]. While glutamine uptake maintains nucleotide biosynthesis, glutathione plays a role in the exchange of nonessential amino acids. Therefore, glutamine, serine, and cysteine concentrations are reduced in the ECM. Other utilization processes can be propagated in case of inadequate vascularization and reduce the levels of building block metabolites within the ECM. Acetate- and/or citrate-derived acetyl-CoA could be used in lipid/FA biosynthesis, especially in RAS-transformed and/or acetyl-CoA synthase 2 (ACSS2) overexpressing cells [61]. As mentioned previously, growing tumors are similar to dysregulated wound healing based on their high-rate glucose consumption, lactate production, and depletion of extracellular glutamine [4]. To speed up glucose uptake and support catabolism, tumor cells have advantages due to their oncogenic alterations. The studies presented thus far provide evidence that the glucose concentration is typically lowered by about 90% in tissue fluids during intensive tumor proliferation [62]. Immune effector cells also have high glucose demands; therefore, lowered glucose impairs their functions (e.g., IFNγ production, Th1 differentiation) [63]. Based on these factors, the available glucose concentration could be an important metabolic checkpoint in the downregulation of antitumor immune response (in addition to increasing the lactate levels). The importance of sufficient nutrient sources was described in PD-L1 immune checkpoint therapy. In correlation with this, GLUT1 expression was found to be upregulated in effector T cell population, forcing glucose uptake from tumor microenvironment [63]. It was also described that the efficacy of adaptive T cell therapy was lower in highly glycolytic tumors [64].
Moreover, starving conditions and lowered ATP level induce autophagy of stromal fibroblasts to replenish nutrition for the ECM (amino acids and nonessential amino acids) via the activation of AMPK and subsequent inhibition of the mTOR pathway [65]. Low glucose concentration decreases PFK1 and the proliferation and migration of endothelial cells, and, as a further consequence, this inhibits the vascularization and nutrient supply in the whole tumor mass triggering continuous tissue regeneration. These also contribute to the lactate accumulation in the ECM [41].
Based on these findings, the metabolic plasticity and symbiosis among different cellular elements and their metabolic subtypes highlight the importance of glucose and lactate – as metabolic substrates in the tumor microenvironment [66]. The lactate concentration changes and the appearance of pH alterations are being studied in greater detail, especially regarding their contribution to tumor progression and drug resistance [48].
Amino acids and their uptake could have critical importance in tumor proliferation [67]. Glutamine consumption could be essential in promoting intensive proliferation and refueling of the TCA cycle. Moreover, similar to serine, glutamine is also important for nucleotide synthesis and maintaining redox homeostasis through the production and exchange of glutathione [68]. In amino acid– and nutrient-depleted conditions, the increased expression of transporters and oncogene-driven alteration mechanisms induce macropinocytosis of ECM proteins to support rapidly growing tumor cell populations [69]. The induced autophagy in CAFs can provide other amino acids and dipeptides. Additionally, adipocytes can provide FAs and other nutrients under these conditions. These effects may have a supporting role in cancer cell proliferation [70]. Many cancer types have described the correlation between lipid droplet (LD) accumulation and chemoresistance [71]. Furthermore, the exogenous lipids and obesity-related factors could influence metastasis [72, 73].
It is also known that other major molecular components of the ECM (collagen, laminin, fibronectin, proteoglycans, and elastin) may be degraded by acidosis-activated proteases and MMPs. These ECM-released nutrients could impact the metabolic regulation of tumor growth. Moreover, negatively charged glycose-amino-glycans within the ECM can alter proton and cytokine distribution, playing a role in re-localization in the pre-metastatic niche and metastasis progression. It was also suggested that cell surface proteoglycans can bind low-density lipoproteins and help in their internalization [74, 75]. Exosomes enriched in proteoglycan-bounded lipoproteins could influence cellular communication and vesicle cargo [76]. In addition, nucleic acids, protein, and exosomal metabolites could influence the invasive potential of cancer cells (e.g., melanoma cells) [77].
In summary, these alterations adjust metabolic symbiosis (many cells and biomolecules compete, communicate, and even feed each other as dominated by tumor cells) causing metabolic rewiring of tumor cells, influence crosstalk between tumor and non-tumorous cells, increase tumor tissue heterogeneity, and maintain the homeostasis of the organ/whole organism [78, 79].
5 Dynamic EMT-MET transitions and their metabolic consequences (metabolic plasticity) in cancer tissues; cancer stem cells, dormant cells
Differentiation, dedifferentiation, transdifferentiation, and stemness are central elements of organ development, inflammation, tissue regeneration, and repair. These underline the importance of phenotypic plasticity and its contribution to cancer initiation, development, and progression. Previously, transdifferentiations were believed to be limited to inferior vertebrates (e.g., amphibians); however, it is now understood that mammalian cells can dedifferentiate and transdifferentiate. Several transcription factors could reverse the actual cellular phenotype and initiate the reprogramming of human cells, e.g., fibroblasts to cardiomyocytes, neurons, among other cell types [80]. MyoD, OCT4, SOX2, KLF4, and Myc reprogram mammalian fibroblasts to an embryonic-like state [81,82,83]. The related experimental results highlighted the potential role of cellular plasticity in tumorigenesis. Based on the studies from the last decade, it was accepted that tumors can hijack the differentiation programs of the original cells [84]. The activation of EMT (and its reverse process; mesenchymal-to-epithelial transition — MET) drives differentiation during tumor initiation and progression [85, 86]. In these transitions, cells gain several features such as plasticity and some stem cell–like properties. Further consequences include loss of polarity and adherence to the basement membrane in epithelial cells, reorganization of their cytoskeletal structure, expression of specific mesenchymal cell markers, as well as cellular migration and renewal.
Recent studies on colon and skin carcinogenesis have identified several transitional stages in tumor initiation and progression [87]. Cells within the heterogeneous tumor tissue show different stages of EMT and diverse intermediate (hybrid) phenotypes during epithelial and mesenchymal transition states. It was also described that these different subpopulations had altered tumorigenic properties, colony formation, and metastatic capacity in vivo. The EMT phenotype and mesenchymal characteristics of circulating tumor cells (CTCs) correlate with their metastatic potential and have prognostic significance for cancer patients [88]. Several results suggest the role of EMT in dedifferentiation and altered transcriptional regulatory network in the loss of therapy sensitivity [89, 90].
It was proposed that the amount of CSCs correlates with the tumorigenicity of the cell population (e.g., aggressiveness during in vivo xenotransplantation) [91]. CSCs and dormant cancer cells are often considered the same; however, these are definitively not interchangeable terms. CSCs have special characteristics, including self-renewal features together with their special differentiation state. At the same point of cell maturation, dormant cancer cells are “similar” to their active tumor cell pair [92]. In addition, dormant tumor cells and their tumor counterparts can switch back and forth in correlation with their functions and metabolic states during tumor evolution. To add this complexity, the CSC population is inhomogeneous, and cancer dormancy can have several meanings, depending on the definition used. “Cancer dormancy” was originally used in the clinical setting corresponding with “tumor mass dormancy,” when the tumor was at undetectable level with a balanced tumor cell proliferation and death (clinical dormancy) [93,94,95]. Dormant cancer cells have different and frequently misunderstood meanings. Additionally, non-proliferating, quiescent cancer cells are in the G0-G1 phase at cell cycle arrest. Quiescence and dormancy are almost similar cell states that can be distinguished based on how they re-enter to the cell cycle. Quiescence, also referred to as G0, is a temporary pause of proliferation that will be resumed when conditions are favorable. On the other hand, dormancy is perceived as a deeper arrested state, suggesting that dormancy is more persistent than quiescence. In comparison to this, the stem cells are not arrested in the cell cycle and have a self-renewal (asymmetric division capacity).
The dormant and quiescent cells are different from slow-cycling or other CSCs. However, they have many similarities from a transcription factor expression point of view (e.g., overexpression of SOX2, NANOG, and NRF2). Both dormant cells and CSCs are rare in tumor tissue, and the presence of these cells could be a source of relapse and drug resistance. To characterize these cells, several markers can be used; SOX2, N-cadherin, and CD13 are usually expressed in both cell types. Additionally, there are some other intracellular and adhesion proteins which can help to distinguish these states; e.g., OCT4, increased RORγ transcription factors, ALDH1 enzyme, c-Met, EpCAM, CD44, CD133, CD24, ABCG2, ABC transporter expression, and the marker of cycling cells (Ki67) are characteristic for cancer stem cells. However, the expression pattern highly depends on the tumor type [96,97,98]. To select dormant cells, other marker sets are necessary, for example, increased expression of p21, p27, FBXW7, NR2F1, DEC2, activated p38, and inhibition of TGFβ1-2 and VEGF signaling, with decreased levels of ZEB1 [99]. The original “seed and soil” hypothesis underlines the regulatory role of the special microenvironmental niche in cellular differentiation (dedifferentiation, redifferentiation) and metastasis during tumor evolution.
In cancer development and progression, the above mentioned plasticity can be accounted for by tumorigenic mutations, environmental changes, or response to therapeutic interventions. Accordingly, the activation of EMT in cancer cells is closely related to stem cell–like and dormant cell phenotypes, which also participate in cellular plasticity. These all could contribute to tissue heterogeneity and help the adaptation of tumor cell population during extreme situations [79, 83]. EMT, cancer cell dedifferentiation, stemness, and dormancy require different metabolic activities. These states are connected with the altered sensitivity to microenvironmental conditions and are associated with tumor cell survival.
Metabolic adaptation depends on several factors such as the cell type, mutational profile (oncogenic alterations), and extracellular conditions. This emphasizes the individuality and complexity of each tumor and their development. Therefore, it is not surprising that papers published over the recent decade have many conflicting results about the metabolic characteristics of EMT, stemness, and dormancy.
EMT/MET alterations and their metabolic associations can be determined by transcriptomic and metabolic analyses in different tumors, where induced mitochondrial respiration and decreased Warburg glycolysis dependence in quiescent cells are shown [100]. Furthermore, the increased autophagy and lysosomal degradation as metabolic differences were also reported in many different cancer stem cells (e.g., pancreatic cancer and glioblastoma cells) [101, 102]. On the contrary, transcriptomic and experimental studies showed decreased mitochondrial OXPHOS enzyme expression and mitochondrial mass in different cancers (e.g., hepatocellular and renal cancers) in their dedifferentiated stem cell–like surviving tumor cells. These findings suggest that dormant and stem cell mechanisms are more complex, tumor type, and environmental condition–dependent [103].
A small population of aggressive, less differentiated CSCs with self-renewal capacity were reported to be responsible for therapy resistance, metastasis, and disease relapse. There are several contradictions regarding the metabolic phenotype of CSCs; depending on the stem cell enrichment method, isolation techniques, model systems, and definition of CSCs applied in a given study (characteristic marker sets of CSCs), for example, in lung, breast, and ovarian cancers and glioblastomas [100, 104]. The metabolic activity of normal stem cells relies on glycolysis; however, CSCs can use aerobic glycolysis or OXPHOS mechanisms depending on their tumor type and microenvironment (hypoxia, starvations) [105,106,107].
The original stem cell and CSC studies — the principal hypothesis — suggested that cellular metabolism controls stemness and CSCs could not be directly linked to a specific metabolic phenotype. Several studies demonstrated that metabolic rewiring is a glycolytic shift in CSCs, e.g., hepatocellular, colorectal carcinomas, osteosarcomas, and radioresistant nasopharyngeal carcinomas [104, 105, 108]. Surprisingly, the OXPHOS-dependent metabolism of CSCs is found in other tumor types (glioblastoma, pancreatic ductal adenocarcinoma, breast cancers, and AML). Moreover, increased nutrient utilization (such as glutamine or other amino acids, fatty acids, ketones) and alternative energy supplementation (especially the environment-independent survival benefits) are associated with tumor cell stemness [109,110,111,112]. One of the essential amino acid substrates in cancer cells is glutamine, and glutamine utilization could have particular importance in certain CSCs (e.g., breast and pancreatic cancers). Based on these results, the mitochondrial functions and alternative metabolic pathways (glutamine uptake and de novo synthetic capacity) play an essential role in CSC generation [113, 114]. Consequently, these results suggest that mitochondrial functions could be crucial in the survival of CSCs [109, 115].
There are several limitations which are beyond the scope of our review. Despite significant advancements in the field of cancer cell metabolism, more precise definitions and metabolic characterizations are required to avoid the misuse of the described terms in this chapter [99]. Based on these and the complexity of this subject, our recommendation is to focus on characterizing the metabolic characteristics and metabolic rewiring of stem/dormant and quiescent cells in a separate and comprehensive review.
6 Metabolic plasticity and the main metabolic phenotypes in cancer
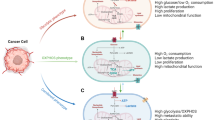
Evidence from ecology and biodiversity studies demonstrates the role of biological diversity in stabilizing the ecosystem (as part of a fluctuating environmental adaptation process). The presence of different, individual strategies is essential for some species’ survival because the absence of environmental compensatory mechanisms leads to their collapse. Diversity is necessary for maintaining stability and productivity of the cellular ecosystem [116]. The available literature in cancer research confirms that tumors generate highly diverse and unique ecosystems in the body, expanding the adaptation possibilities [3, 117]. High plasticity in extreme situations, such as nutrient, O2 starvation, and treatments with high cellular toxicity, could have significant advantages, especially at a cellular metabolism level. Metabolic plasticity, the interconnecting, flexible metabolic pathways, and rewiring cause a therapeutic nightmare during cancer progression. There are several attempts to define the various metabolic phenotypes of different cell types in cancer tissues. Based on database analyses, Yu et al. suggest that a minimum of three main metabolic phenotypes must be distinguished in tumor tissues. Aside from cancer cells with glycolytic and OXPHOS metabolic characteristics (two main types), the most dangerous cells could have the flexibility to utilize both OXPHOS and Warburg glycolysis simultaneously, and as a result, these cells have the highest metabolic plasticity [118] (Fig. 3). This newly defined hybrid state could have several advantages in a developing tumor, but targeting these hybrid cells, and other bioenergetic mechanisms, is not easy without causing potential side effects. Compared to normal cells (immune cells, fibroblast, adipocytes, etc.), the selective advantage of such a hybrid phenotype is the plasticity and rapid adaptation to utilize different bioenergetic sources (from the microenvironment or internal utilization of cells — autophagy) in cellular survival.

Metabolic phenotypes and their continuous transition in tumor tissues. Schematic presentation of Warburg (glycolytic), hybrid, and OXPHOS metabolic phenotypes. The metabolic rewiring (activated metabolic pathways are depicted in bold, in contrast to the less active routes which are faded). Additional phenotypic characteristics are labeled in the lower part of the figure
Apart from these three main metabolic phenotypes, there is a progressive and continuous transition in metabolic reprogramming, similar to epithelial–quasi-mesenchymal–mesenchymal transitions. Cells with different metabolic phenotypes and plasticity are constituents of metabolic symbiosis (promote tumor growth by supplementing each other). Moreover, metabolic heterogeneity could be the basis of tissue heterogeneity and tumor survival. The characterization of these findings and the discovery of potentially favorable master regulators as targets could improve the understanding and clinical outcomes (therapy resistance, relapse, and distant metastasis in advanced cancers) [39].
7 Characterization of metabolic heterogeneity in cancer tissues
Next-generation sequencing is a relatively quick and sensitive technique for analyzing individual genetic variability; additionally, its routine application serves as a tool to identify alterations on genomic level and tailor precision and targeted therapies. Despite the early and, in some instances, more prolonged success with personalized precision oncotherapy (based on genotype mapping), the prognosis for many cancer patients remains poor due to disease relapse and development of resistance to therapy. The advancement and clinical benefits of molecular targeted therapy still have some limitations, and usually, the median progression-free survival (PFS) does not increase more than 6 months [119]. The available targeted therapies usually depend on cytostatic effects that reduce tumor growth and kill many malignant cells. However, a selective survival of highly aggressive tumor cells can occur for several reasons (e.g., the genome-based characterization may not take into consideration the adaptive landscape). Another critical aspect of cancer progression is that if most tumors are successfully eliminated by therapy, one or a few resistant phenotypes may remain. These surviving tumor cells exist in a less-competitive environment at the primary tumor site or distant metastasis (the so-called competitive release). Additional studies are required involving mapping tissue heterogeneity and identifying these potentially surviving cells for more precise targeting [120,121,122]. To discover the source of tissue relapse and evaluate its relevance in the clinical setting, better in situ characterizations by radiologists and pathologists are needed. Mapping metabolic heterogeneity could be an additional tool in this research field. Therefore, we need to integrate metabolic profiling and in situ metabolic characterization in precision oncology. Several factors must be considered: metabolic heterogeneity of the primary tumor tissue (at the time of diagnosis); potential metabolic adaptation mechanisms which are available during tumor growth and development; and treatment regimen inducing alterations, etc. Studying these aspects, potential new metabolic targets need to be identified to interact with the survival mechanisms in surviving and developing cancer tissue.
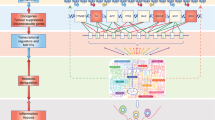
Intratumoral heterogeneity, including metabolic distributions and altering tissue microenvironmental conditions, must be mapped during the diagnostic process. Pathomorphological studies are necessary to apply this in practice with new mass spectrometry and imaging analyses in combination with in vivo imaging technologies. In vivo imaging methods, including the application of PET-CT and MRI [123], are potent tools for the following circumstances: (a) monitor in vivo metabolism, (b) diagnosis, and (c) follow the response rate of the therapy. This review could not summarize the available studies of these in vivo and in situ imaging technologies (MALDI imaging, CEST MRI, MRSI, IR-MALDESIMSI, SpaceM, single-cell spectrometry, Raman spectrometry, MetaSensor, etc. [124]). The overall advantage of these techniques is the ability to measure proliferation, necrosis, and inflammation by applying novel radiotracers and expanding evaluation methods. These methods must be complemented with the accurate pathologist-guided in situ metabolic characterizations of the available biopsies. Metabolic pathway–related expression and activity markers should be examined for in situ investigations of the tissue sections. To validate these markers, additional in vivo and in vitro metabolic profiling and studies are required. Furthermore, we need to synthesize DNA sequencing and transcriptomic data sets with the results of experimental metabolic studies (experimental metabolic profiling analyses of in vitro studies and patient-derived xenograft models) and in vivo metabolic imaging results (Fig. 4).

Metabolic characterization in vivo, in patients’ tumors, and experimental studies. Tumor diagnostic methods: clinical image analysis (PET-CT, MRI), tumor biopsy methods including histological and molecular genetic examinations (pathomorphological, immune, and next-generation sequencing–based phenotyping, and genetic analyses) are shown on the left side of the figure. Additionally, several in vitro and in vivo model systems and experimental studies using cell lines or patient-derived cells are shown on the right side of the figure
Metabolic pattern as a fingerprint is unique in each tumor tissue, and it depends on the tumor-specific cellular content (including non-tumorous cells: blood vessels, immune cells, etc.), genetic and epigenetic alterations, microenvironmental pressures (therapy, nutrient, oxygen stresses, etc.), and the metastatic sites in new organs. There were several attempts to characterize the metabolic activity and local differences in the central metabolic processes using immunohistochemistry (IHC) staining with different antibodies against metabolic enzymes, regulators, and transporters. Our primary interest is to map metabolic heterogeneity at tissue level and combine these data with the pathomorphological diagnosis.
In the last few years, the potential hybrid metabolic phenotype highlighted the interplay between glycolysis and OXPHOS. These results suggested that the high expression of HIF1α and p-AMPK could be the main features of hybrid cells. However, high HIF1α and low p-AMPK expression and their opposite distributions are characteristic for Warburg glycolytic and OXPHOS tumor cells, respectively [125, 126]. It was also proposed that hybrid metabolic cancer cells tend to progress highly aggressively and are associated with frequent metastasis and relapsed cancers [18]. These were examined using metabolomic and transcriptomic patient data sets diagnosed with breast cancer. However, some recent new results confirm that this hybrid metabolic phenotype could exist in both in vitro experimental models and tissue materials (both pathology and experimental studies) [118, 127, 128]. Further, especially pathological, studies are needed in these subjects. Recent studies focus on one aspect of the signaling regulatory failure–related metabolic alterations/characteristics, subtype-specific hyperactivation in specific metabolic processes, and nutrient utilization in different cancers. However, a comprehensive analysis of the whole network is not performed. Additionally, omics databases and integration algorithms can generate cancer-specific genotype, phenotype, and metabolic phenotypes of different cancers. Metabolic genes and their expression as cancer biomarkers can correlate with tissue acidification and migration or other cancer progression–related cellular functions [129, 130]. However, the selection, survival adaptation, and evolution of cancer metabolism at tissue level were suggested to optimize the growth potential of tumor cells in different cancers. The potentially helpful metabolic targets in extracellular and mitochondrial transporter mechanisms, peptide metabolism, FA synthesis, and PPP vary in different cancers [131,132,133]. The results of other pathomorphological and IHC combined biopsy studies are very diverse, and their conclusions depend on the applied scoring systems and whether tissue heterogeneity was investigated.
8 In situ metabolic protein expression alterations and heterogeneity with respect to tumor progression
8.1 Heterogeneity of carbon source and transmembrane trafficking
Tumor cell proliferation, survival, and migration are crucial elements in cancer progression. These processes have energy demands fueled by nutrients and metabolites sourced from additional bioenergetic mechanisms. The main metabolic pathways are connected to glycolysis and OXPHOS, preferentially using glucose as their energy source. Other properties that affect tumor survival and adaptation mechanisms include surrounding tissue microenvironment, biochemical factors, starvation, alternative nutrient utilization, and shifts in cellular metabolism. Healthy and nonmalignant cells usually produce pyruvate to fuel OXPHOS predominantly. During starvation, especially at low O2 concentrations, anaerobic glucose utilization is preferred, and alternative substrates are directed to fuel the TCA cycle. Tumor cells have metabolic plasticity and switch dynamically between lactate-producing Warburg glycolysis and OXPHOS. Tumor cells during significant proliferation use Warburg glycolysis preferentially due to metabolic reprogramming caused by several oncogenic mutations of the signaling network. The metabolic characteristics of cells are determined by the distribution of glucose, carboxylate, or amino acid transporters and the carbon utilization of the tissue. The expression patterns of specific transporters such as GLUT1, MCT1/MCT4, CAIX, and amino acid transporters (e.g., ASCT2 — alanine/serine/cysteine transporter 2, LAT1 — large neutral amino acid transporter 1) help characterize the supplementary nutrient processes within an in situ tumor mass. GLUT1 overexpression is characteristic for several cancers in correlation with their glucose utilization. This feature is useful to detect several relapsed and metastatic tumors with in vivo imaging diagnostic tools (e.g., FDG-PET-CT) depending on the tumor type. MCTs facilitate the lactic acid exchange between the cytoplasm and extracellular space. MCT1 is expressed in all cells and assists with the transportation of lactate and pyruvate. In contrast, MCT4 expression is inducible and responsible for the exportation of lactate under hypoxic conditions, e.g., in glycolytic tissues, especially in many cancer cells.
Experimental and imaging studies have extensively analyzed the expressions of GLUT receptors, MCTs, carbonic anhydrases (CAs), and other proteins (e.g., amino acid transporters). Accordingly, tumors are classified based on their levels of protein expression (high or low levels) [134]. However, the tissue distribution of transporters in various cancers is not yet well characterized. The available results focus on scoring total protein expression (staining intensity) using only high or low categories for GLUT1, MCT1/4, and CAIX. In certain studies, the staining level and pattern were described in correlation with hypoxia, HIF1α expression, and FDG-PET-CT images in various tumor types [135]. IHC documentations demonstrate intratumoral heterogeneity; however, this phenomenon was not evaluated/scored in pathology studies and Protein Atlas documentations.
A vast literature exists about oncogene-driven alterations and the prognostic role of GLUT1 expression. These findings suggest that, in general, difference in GLUT1 expression might be a significant predictor for overall survival (OS), disease-free survival (DFS), PFS, and disease-specific survival (DSS). Poor OS correlation is associated with several cancer types, e.g., in gastric, urinary, ovarian, oral squamous cell, pancreatic, colorectal, lung, gallbladder, and esophageal carcinoma [135]. Multi-tracer characterization studies with FDG-PET-CT have resulted in the ability to show a complete representation of glucose uptake, heterogeneity, and therapy-induced metabolic adaptation of cancers. CT- and MRI-driven imaging technologies can facilitate the mapping of metabolic phenotypes and guide metabolic treatments, e.g., lung cancer [136]. GLUT inhibitors have gained attention in correlation with the growing knowledge of the glucose dependency of cancer, inflammation, and other diseases [137]. Targeting GLUT receptors or glycolysis could be an alternative option in cancer treatment, but several clinical trials were discontinued because of the reported high toxicity and severe side effects (Table 1).
Overexpression of MCT1/4 is a poor prognostic factor for various cancers, including breast, bone, colon, and renal cancers [138]. MCT1 overexpression is associated with a worse prognosis in bladder, endometrial, and clear cell renal cell cancers [139,140,141] and with MCT4 in oral, colorectal, prostate, lung, and clear cell renal cell cancers (ccRCCs) [141,142,143,144,145,146], respectively. MCT4 expression is regulated by AMPK and protein kinase C (PKC), and these signals influence metabolic adaptations via lactate shuttle under specific conditions [147].
CAs — which have 16 different isoforms — are involved in the regulation of intra- and extracellular pH; their overexpression and different localization were described in various tumors. CAIX requires special attention based on its predicted diagnostic, prognostic, and therapeutic biomarker potential in solid tumor pathology [148]. Diffuse CAIX expression, similarly to high GLUT1 and HIF1α expression, is characteristic for ccRCCs (in contrast with papillary renal clear cell carcinoma — RCC). This can help in differential diagnosis; ccRCC has lower CAIX expression [149]. On the contrary, higher expression of CAIX is correlated with poorer prognosis in the majority of breast, lung, ovarian, oral squamous, liver, and bladder cancer cells and glioblastomas [150,151,152,153,154,155,156]. Considering this, CAIX could be therapeutically targeted [148]. In addition, IHC evaluation of MCT1/4 and CAIX — mainly focused on detecting higher or lower expression levels in tissues [66] — showed a correlation with hypoxia. Further research should be done to investigate tissue heterogeneity of CAIX and MCT4 expression, and its impact on the progression of solid tumors requires further analysis [157]. It was described that knockout (KO) or drug targeting of MCT1/4 inhibits the proliferation capacity both in vitro and in vivo [66]. Furthermore, these combined with metformin or phenformin [158] lead to synthetic lethality in tumor models [159]. Some effective small-molecule MCT1-specific inhibitors are in clinical trials (e.g., AZD3965), monoclonal antibodies, and small-molecule inhibitors of CAIX (SLC-0111) showing promising results. Some of these MCT1 inhibitors have entered phase II clinical trials, but the lack of isoform selectivity and potential associated toxicity necessitates designing additional, more selective drugs. Combining these with traditional chemotherapy or other metabolic targeting drugs during anticancer therapy is also being investigated [148, 149, 160] (Table 1).
Cellular uptake, transport, and the role of amino acids (as supplementary mechanisms) are well known in differentiated and proliferating cells. The primary function of amino acid metabolism is to serve protein synthesis. However, the complex metabolic coupling also supports the source of building blocks and other molecular precursors such as nucleotide, glutathione, and polyamine synthesis [161]. Furthermore, the emerging roles of non-proteinogenic amino acid metabolism in cancer should be considered. Nonessential amino acids limit tumor proliferation; in this case, the stromal and extracellular amino acids are often consumed and recycled. Amino acid restriction can be compensated by autophagy, and it could be mentioned that amino acid availability (intracellular concentrations) regulates the growth potential and influences stress responses, e.g., via mTORC1 activity [161,162,163]. Based on increased amino acid demand during intensive proliferation, some nonessential amino acids (e.g., glutamine, asparagine, arginine, and cysteine) become conditionally essential. Glutamine addiction, the importance of glutaminolysis (reversible glutamine-glutamate-αKG metabolic axis), and additional “by-products” provide building blocks for fast proliferating cells. It should be considered that glutamine can be nitrogen and carbon sources in amino acid, nucleic acid, and lipid synthesis. There are several glutamine exchangers (ASCTs and Na+-coupled neutral amino acid transporters (SNATs)) that are often overexpressed by tumor cells (e.g., ASCT2 — SLC1A5; SNAT1 — SLC38A1, SNAT2 — SLC38A, and SNAT5 — SLC38A5) [164]. There were promising results for inhibiting tumor growth with applying ASCT2 inhibitors in gastric, prostate, lung, and breast cancers [165, 166]. However, many compensatory mechanisms were detected (e.g., SNAT1 overexpression) negating the effect of these inhibitors [167].
Therefore, the generation of novel targeted therapies and new inhibitors is under development. Many other amino acid transport (symport, antiport) mechanisms have been reported and tested in the last few decades. Serine/glycine-linked metabolic network and the related transporters can be involved in different mechanisms in cancer progression. These transports are dysregulated via epigenetic alterations (e.g., methylation) or de novo ATP synthesis. In addition, the modifications in serine-glycine extracellular level could influence the proliferation and function of cells in the immune microenvironment (T cell expansion) [168]. The transport of leucine, isoleucine, and valine — essential branched-chain amino acids — can also be upregulated in common cancers (e.g., lung, breast, and prostate). They have a role in maintaining amino acid pools in tumors [169]. Recently, small-molecule inhibitors and their derivatives have been applied as actionable targets, e.g., LAT1 (SLC7A5). Additionally, LAT1 targeted inhibitors were tested in vitro and in vivo, where decreasing leucine level and inactivation of mTORC1 are correlated with reduced tumor growth. [170]. Some of these (e.g., JPH203) have recently been introduced to clinical trials in advanced solid malignancies [171]. The de novo biosynthesis of arginine occurs in the urea cycle during ammonia detoxification. Arginine is referred to as a conditionally essential amino acid that serves as a precursor for polyamines and generates NO, creatine, and other amino acids. Additionally, arginine can be taken up by cationic amino acid transporters. Overexpression of SLC7A1 cationic amino acid transporter and its role in tumor cell survival were described in arginine-dependent breast cancers and cases with multikinase inhibitor resistance. The characterization of amino acid transporter expression and their alterations have been described in different cancers at protein and tissue levels. Limited data are available about the heterogeneous staining of LAT1, ASCT2, or other transporters. Some recent studies have combined these with in vivo PET-CT images [172, 173]. This highlights that metabolic heterogeneity could be scored by in vivo analyses and by special pathomorphological studies of these transport mechanisms in the future.
More recent attention has focused on the provision of the FA, acetate, and citrate utilization processes which have renewed interest; FA and lipids contribute to new biological membrane synthesis during the intensive proliferation of doubling cells. FA can be served by exogenous uptake and de novo synthesis. During tumor development, lipid metabolism alterations occur such as FA transport, lipid storage (e.g., LD), de novo lipogenesis, and β-oxidation–mediated energy production [174]. FA translocases (FATs), CD36, FA transport proteins (SLC27 members — FATP1-6), and other FA-binding proteins in membranes (e.g., FABPpm) have been intensively studied. These transporters were described to be overexpressed in breast, gastric, pancreatic, hepatocellular, and prostate cancers [175,176,177]. It was also suggested that CD36 plays an essential role in metabolite exchange and symbiosis of the tissue microenvironment, facilitating lipid uptake of tumor cells [72, 178]. The occurrence of metastasis in visceral adipose tissue could be correlated with FA uptake and utilization. During progression, the adipose tissue generates an immunogenic inflammatory microenvironment and modulates paracrine-endocrine signals [179]. Lipid storage elevates LD formation, which dynamically contributes to lipid homeostasis by preventing lipid toxicity and providing acetyl-CoA, ATP, and NADH through β-oxidation [174]. Acetyl-CoA produced by β-oxidation from LDs assists the ATP generation in high amount via TCA cycle and electron transport chain. This process is highly effective since it gives a much higher amount of bioenergy than other mechanisms from carbohydrates. In parallel, the generated NADH is also helpful for ROS detoxification [180]. Besides lipid uptake, de novo lipid synthesis is characteristic for cancer tissues using LDs or other lipid sources [181]. The primary source is cytoplasmic acetyl-CoA from carbohydrates (through acetate and citrate) and amino acids (including glutamine), converted to FAs. Under metabolic stress (e.g., hypoxia and lipid deficit), cancer cells convert acetate and citrate using ACSS and ACLY. Additionally, acetyl-CoA carboxylases (ACCs) and FA synthase (FASN) facilitate palmitate generation in lipid synthesis.
In correlation with the importance of balanced lipid homeostasis, the complex pan-cancer analyses of the prognostic role of CD36 overexpression have been published recently and highlighted the tumor type–dependent increase in CD36 expression. Unfortunately, intratumoral heterogeneity was not evaluated in these studies; therefore, further analyses are necessary by pathologists in different tumor types and their metastases [182].
Many transport proteins and different channels are necessary to connect mitochondrial and cytosolic metabolism, providing the integrity, metabolic cargo, and maintenance of mitochondrial homeostasis. Most of the 53 mitochondrial carriers are localized in the inner mitochondrial membrane. Mitochondrial carrier family (SLC25) members are the most critical transporters; however, others such as pyruvate carriers and ABC transporters also have essential functions [183]. These amino acids, nucleotides/dinucleotides, carboxylates, ketoacids, and additional carriers use several exchange mechanisms. Pi carriers (PiC) are used for phosphorylation of ADP by ATP synthase. The overexpression of SLC25A1, mitochondrial citrate transporter, was described in primary lung cancers and their metastases, contributing to therapy resistance. SLC25A10 transports malate and succinate, which is linked to NADPH synthesis and the maintenance of redox homeostasis [184, 185]. For example, aspartate and glutamate mitochondrial carrier (SLC25A12) overexpression (via unlimited amino acid shuttle) supports aspartate utilization in hepatocellular carcinoma (HCC) [186]. Changes in mitochondrial metabolite transporter expression and their role in cancer progression correlated with ROS accumulation in central nerve system (CNS) neoplasms (overexpression of SLC25A30). On the contrary, SLC25A43 loss was detected in HER2 + breast cancers [187]. Based on these results, the alterations in mitochondrial carrier expression (see below), their potential impact on tumor growth, and inhibitor therapy are under investigation (e.g., SLC25A10 inhibitors, which disturb redox homeostasis).
Different metabolite transporters and their role in cancer progression are a current topic of interest. The expression patterns of the above-discussed transporters could expand therapeutic targets concerning the dynamic and complex adaptation mechanisms in cellular stress, altered extracellular nutrient concentration, and growing/survival demands.
8.2 Tissue heterogeneity of certain metabolic enzymes and their significance in metabolic adaptation
There are many networks, data analysis, and IHC studies focusing on the changes in metabolic enzyme expressions, at tissue level. Most of these studies analyzed the changes (increase, decrease, or loss of some central metabolic enzymes) in various catabolic and anabolic processes (glycolysis, TCA enzymes, lipid synthesis or oxidation, amino acids, electron transport chain proteins, etc.). However, the description of staining heterogeneity in correlation with the clinical data are usually not evaluated.
In the past, the metabolic state of certain tumors was analyzed in granulomas of sarcoidosis, some rare tumors, and other diseases by applying traditional enzyme histochemistry to detect enzyme activity. For example, cyclooxygenase (COX), NADH, SDH, and ATPase activity could be shown by enzyme histochemistry on native slides; however, routine biopsy materials are mainly formalin-fixed. Nowadays, this staining has been replaced by specific IHC reactions in routine pathology. Instead of testing real metabolic enzyme activity in tissues in situ, the current approach evaluates the enzymes and their active forms.
Several studies describe the alterations of enzyme expression in correlation with hypoxia, increased HIF1α, and related altered tissue distribution of prolyl hydroxylases (PHDs) [188]. This highlights the role of the tissue microenvironment and its influence on metabolic alterations in various cancers and cancer progression. In situ HK2, LDHA, PKM2, PDKs, PDHs, factor inhibiting hypoxia (FIH), and different PHDs were studied in tissues over the last few decades. The upregulation of PKM2 expression, which is often associated with increased glycolytic enzymes or glucose transporters (e.g., HK2, GLUT), was described at tissue level in various cancers (e.g., in breast, lung, ovarian, bladder, colon cancers). The unfavorable prognosis (lower OS and DFS) was correlated with higher PKM2 expression, e.g., in breast, hepatocellular cancer, as well as in tongue and esophagus squamous cell carcinoma [189,190,191,192]. In correlation with glucose utilization, elevated HK2 was recently described in patients’ biopsies of esophageal, renal, lung, cervical, colon, and breast cancers [193,194,195]. The analyses of LDHA and PDK1 in situ protein expression also showed association with poor survival and therapy resistance in many malignancies (colon, breast, uterine, pancreas, lung, renal, gastric cancers, CNS malignancies, etc.). Considering the increasing serum lactate level of end-stage cachexia patients, these observations are not surprising [196,197,198,199,200,201].
Glutamine and glutamate utilization are essential supporters of TCA fulfilling mechanisms and redox homeostasis. GLS expression and tissue distribution were also intensively examined. These studies highlight that GLS expression is often increased during metabolic adaptation in malignant progression of prostate, breast, colon, ovarian, pancreatic cancers, gliomas, rhabdomyosarcomas, etc. These could be potential targets in rare malignant cancers (e.g., sarcoma, lymphangioleiomyomatosis, or pheochromocytoma) [202,203,204,205,206,207]. Other amino acids and their roles in metabolic network regulation are also crucial. Amino acid (e.g., leucine and arginine) sensing, transporters, carriers, and their cytoplasmic or lysosomal levels influence the intracellular anabolic and catabolic balance. This will be discussed below among mTOR activity regulation and its metabolic functions.
TCA cycle, OXPHOS, the mitochondrial electron transport chain enzymes, and their activity are also under investigation. However, their functional alterations are more common than changes in expression level. It was found that COX enzymes are overexpressed in many cancers; e.g., overexpression of COX2 is characteristic for breast, gastric, and prostate cancer; COX4/COX5B can be detected in glioma and renal and breast cancers. Additionally, increased COX2 level was described to correlate with worse prognosis in cholangiocarcinoma, gynecologic, and gastric cancers. However, there were contradictory COX expression results in other cancers (e.g., breast and cervical cancers) [208,209,210,211,212] regarding the differences in metabolic rewiring (tissue-specific). The ATP synthases level in various tumor tissues suggests that lowered ATP synthase expression and the reduced mitochondrial electron transport chain activity may have a predictive role in progression and prognosis (worse outcome in ccRCC, NSCLC, and colon cancer patients) [213,214,215]. An increased need for substrates places stress on the dynamic network, in which a back flux of protons can attenuate into the mitochondrial matrix (uncoupling). Mitochondrial uncoupling proteins (UCP1-5) have a role in nucleotide and FA cycling. As important mitochondrial transport mechanisms, these link mitochondrial respiration and ATP synthesis and decrease superoxide formation. UCP2-mediated metabolite antiport (aspartate, oxaloacetate, and malate) could cause special metabolism-rewiring in cancer cells which confer chemoresistance as well [216, 217]. Induction of UCP2 has been related to metabolic switch leading to accelerated glycolysis and reduced mitochondrial activity, e.g., in colon, pancreas, lung, breast, prostate, and head and neck cancers [218].
Lipids (triglycerides, phospholipids, sphingolipids, and cholesterol) can be used as an energy sources, building blocks of membrane components, and precursors for steroid hormone, bile acids, vitamins, etc. Based on these, cancer cells can completely rewire their lipid metabolism with increased de novo lipid synthesis, FA uptake, and FA oxidation and finally alter cancer-associated adipose tissues [174]. These central changes influence FA, cholesterol, arachidonic metabolism, and peroxisome proliferator–activated receptor (PPAR) signals. Elevated CD36 expression facilitates free FA uptake that supplies new membrane building and influences signaling events via activating, e.g., Wnt and TGFβ pathways [175, 176]. De novo lipid synthesis can be supported by acetyl-CoA production from glucose and glutamine via glycolysis or the truncated TCA cycle, respectively. Furthermore, hyperactivated FA synthesis is in correlation with an increased amount of crucial lipogenic enzymes (ACLY, ACC, CoA carboxylase — ACACA, FA synthase — FASN, and stearoyl-CoA — SCD). ACC and FASN are generally upregulated in growing tumors (breast, renal, gastric, colon, esophagus, lung, ovarian, prostate cancers, melanoma, gastrointestinal stromal tumors — GISTs, gliomas, etc.) [176, 219, 220]. Additionally, SREBPs activate the transcription of several enzymes participating in FA, cholesterol, and phospholipid synthesis in cancers (e.g., in NSCLC, pancreas, colon, endometrial, and breast cancers). The levels of many previously mentioned enzymes (ACLY, FASN, ACC) are increasing after SREBP1 activation propagated by PI3K/Akt/mTOR or Ras/Raf/Mek/Erk signaling pathways [221,222,223,224]. Furthermore, some studies indicate that a high level of SREBP1 protein expression was detected in specific tumor types by IHC, which could be an independent prognostic marker in breast, thyroid, and pancreatic cancers. Carnitine palmitoyltransferases (CPTs), localized in either the inner or outer mitochondrial membranes, and carnitine-acyl transferase (CAT) support the rate-limiting steps of FA β-oxidation via FAO, which can produce a high amount of ATP. Shifts among switching on or off, the oxidation of newly synthesized FAs, or their accumulation in LDs are regulated by extracellular tissue environment and local conditions. Moreover, starving conditions can provoke autophagy both in tumors and neighboring cells [225]. PPARγ participates in storing FAs as triglycerides in lipid droplets; therefore, this mechanism could rescue the cells from the toxic increase of endogenous palmitate [226]. LDH cholesterols are also essential components for membrane building in lipid rafts. Cholesterol homeostasis is balanced by de novo synthesis, uptake, and removal; the upregulated cholesterol synthesis is influenced by SREBP2 and orchestrated by oncogenic signals (PI3K/Akt, RAS/MAPK) [227]. The prognostic value of dysregulated FA acid oxidation, synthesis, lipid homeostasis, and expression alterations of contributing metabolic enzymes are known from several IHC studies. This highlights that lipid metabolism–related enzyme expression changes could be targeted (e.g., ACC, CPT1A, ACLY, or FASN) since these alterations could correlate with patients’ prognosis [128, 202, 205, 228,229,230,231]. Recent in situ human tumor tissue studies have investigated only some elements of these networks; therefore, its complexity is not well examined, and a deeper characterization is necessary.
In case of lipid metabolism regulation, the role of altered oncogene-addicted metabolic rewiring was described in many details. The consequences of activation and frequent mutations of RAS and other cellular signaling network elements (e.g., growth factor receptor kinases, TSC1/2, PTEN, PI3KCA in colon and breast cancers, and NSCLCs) contribute to the Erk1/2 and mTORC1/C2 hyperactivity by many steps (regulation of mitochondrial biosynthesis and lipogenesis, SREBP1 maturation) [232, 233]. Additionally, mTOR phosphorylates and inactivates lipin1 to liberate SREBP1. It is well-known that mTOR inhibitor therapies or Raptor KO decrease the expression of lipogenic enzymes FASN, ACLY, and ACC [234, 235]. Moreover, mTORC1 activation has translational effects by activating ribosomal S6K and ribosome genesis and influencing spliceosome formation [236]. The recent publications and ongoing studies suggest that mTORC2 can also affect lipogenesis. mTORC2 phosphorylates PKC isoforms, SGK1, and ACLY, therefore increasing activity of ChREBP, histone acetylation, and lipogenic gene expression in adipocytes [237]. These underline the specific regulatory role of mTOR signal activity in lipid metabolism.
Metastasis-driven cellular alterations (EMT-invasion-intravasation-extravasation-MET-metastatic growing) are cooperating in a complex manner during metastatic progression. Various molecular mechanisms influence these programs; these related cellular processes reprogram metabolic signals and regulate the expression of related enzymes. Additionally, metabolites produced by cancer cells/tissues stimulate migration, survival of circulating cells, colonization, and regrowth of tumor cells in a specific new microenvironment. Several experimental and clinical data underline the role of glycolysis (Warburg glycolytic process) in metastatic progression. In animal models, c-Src tyrosine kinase–mediated HK1/2 increase was documented in lung metastasis of colon cancers [238]. In other experiments, the silencing of HK2 reduced lung metastasis in several cancer models [239].
Moreover, the accumulation of glycolytic methylglyoxal and the glycated proteins activate EMT and migration in breast and colon cancer metastasis [240]. Increased amount and nuclear localization of PKM2 are correlated with the metastatic properties and behavior of many epithelial cancers [241]. Glycolytic lactate has additional critical regulatory functions in metastasis formation and progression (e.g., β-integrin, MCT expression alterations, or increasing MMPs). As a part of glucose usage, PPP is a primary source of NADPH that can neutralize ROS in detached and regrowing cells. In contrast, cells with lost anchorage have reduced proliferation, glucose uptake, and PPP pathway activity, increasing the ROS level. In this context, it was described that metabolic rewiring could be microenvironment-dependent in breast cancer metastasis; G6PDH and glutathione reductase levels can be increased in brain metastasis. However, in bone metastasis, the opposite is observed [242]. It was published that PPP can be upregulated in metastatic versus primary tumors in melanoma, renal, and pancreatic cancers [243]. Oxidation of glutamine, FAs, and glucose in TCA is also linked to cancer cell progression and metastasis formation [244, 245]. Glutamine and FA oxidation were described as characteristic features of specific metastatic cancer cells, e.g., melanoma, prostate, and breast cancer [246,247,248,249].
PGC1a could have tumor type–dependent effect in mitochondrial oxidation processes: PGC1a was described as a metastatic promoter expressed in circulating breast cancer cells [250]; however, its KO could increase the invasiveness of melanoma cells through focal adhesion kinase signaling, modulating cell–cell, cell–matrix connection, and support dissemination [251]. Even in starving conditions, the low amount of ATP (bioenergy) liberated from nutrients can serve migration and invasion, protect the cells from apoptosis and immune system attacks, and promote survival in a new organ. In correlation with these, some TCA intermediates could alter epigenetic regulation. For example, it was suggested that increased αKG could play an essential role in maintaining stemness [252]. Regarding this, ketoglutarate dehydrogenase inhibition can reduce metastasis in various models [253]. These examples underline that glycolytic and mitochondrial alterations can have anti- or pro-metastatic effects, depending on the tumor type and cellular microenvironment. These strongly emphasize the role of altered metabolism during metastasis [254]. Defining the metabolic profile in various cellular states (during the metastatic cascade) could expand therapeutic targets and precision-based therapy. Based on this growing evidence, metabolic enzyme inhibitors have been tested in many preclinical and clinical studies. These ongoing studies focus on conventional treatments where metabolic adaptation could have a role in developing resistance (Fig. 5). Based on the results from these trials, therapeutic options can be successfully tailored to each patient individually using metabolic inhibitors in combination with conventional therapies. However, undesirable side effects from combined treatment require special care by clinicians.

A simplified summary of the main targetable metabolic pathways and related compounds. The schematic figure shows the main metabolic pathways and inhibitor therapy reported in different phase trials (marketed and tested drugs are labeled with green or red color, respectively). OXPHOS inhibitors, metformin (marketed in type 2 diabetes), fenofibrate (marketed in different cardiovascular diseases), atovaquone (marketed in malaria), arsenic trioxide (marketed in acute promyelocytic leukemia), mIBG (marketed in paraganglioma and pheochromocytoma), enasidenib mesylate (marketed in relapsed acute myeloid leukemia), leflunomide (marketed in psoriatic and rheumatoid arthritis), miltefosine (marketed in leishmaniasis), tetracycline, doxycycline, minocycline, tigecycline, azithromycin (marketed in bacterial infections), pyrvinium pamoate (FDA-approved anthelmintic drug) and drugs in clinical trials are vorasidenib (NCT04603001 — phase III in advanced hematologic malignancies), emvododstat (NCT03761069 — phase I in relapsed acute leukemias), FN-1501 (NCT03690154 — phase I in advanced solid tumors and leukemias), HMPL-306 (NCT04764474 — phase I in IDH gene mutated cancers), and SH-1573 — phase I in acute myelogenous leukemia); glycolysis inhibitors, nedosiran (NCT04042402 — phase III in primary hyperoxaluria), GP-2250 (NCT03854110 — phase II in pancreatic cancer), tuvatexib (NCT03538951 — phase II in actinic keratosis), PS101/3BP (NCT04021277 — phase I in hepatic metastases); lipid metabolism inhibitors, icosapent ethyl (marketed in different cardiovascular diseases), TVB-2640 (NCT03179904 — phase II in advanced breast cancers); glutaminolysis inhibitors, DRP-104 (NCT04471415 — phase II in advanced solid tumors), CB-839 (NCT03047993 — phase II in advanced myelodysplastic syndrome); transporter inhibitors, AZD-3965 (progressing to phase II trials), SLC-0111 (NCT03450018 — phase II in pancreatic ductal cancers), JPH-203 (pre-registration in bile-duct cancer)
9 mTOR as a master regulator in metabolic adaptation and mTOR hyperactivity in cancer
Examining metabolic symbiosis in situ is and correlating this data with patient survival and therapy response can be challenging. mTOR has been considered a master regulator in metabolic adaptation, and mTOR hyperactivity is characteristic for the majority of cancers. Over the last decades, there have been several attempts to characterize the main drivers of metabolic rewiring and their impact on cancer progression. Recently published studies are limited, and tissue heterogeneity was less studied in these works. Many studies have focused on some elements of PI3K/Akt/mTOR pathway. According to these results, it was suggested that this axis could have a special role in regulating metabolic symbiosis, maintaining bioenergetics, and providing building blocks for tumor growth at the primary and metastatic sites. Since mTOR kinase is at the central crossroad of the cellular signaling network, it has many metabolic sensory and effector functions. Accordingly, it is considered the master checkpoint in metabolic adaptation.
Almost 50 years ago, a small molecule was purified from Streptomyces hygroscopicus after an expedition to Easter Island (Rapa Nui) [255]. The discovered “rapamycin” gained more interest in the 1980s when its immunosuppressive effects were beginning to be investigated and applied in kidney transplantation [256]. The cellular binding partners and targets of this compound were identified in the 1990s by studying the rapamycin-resistant Saccharomyces cerevisiae [257]. Additionally, the mammalian/mechanistic target of rapamycin (mTOR) kinase and cellular targets were also isolated after M.N. Hall’s discovery. Their functions were characterized with the help of several scientists, and it was demonstrated that mTOR kinase exists in two different protein complexes, which have other cellular processes and inhibitor sensitivity [233, 258,259,260,261,262,263,264].
Emerging data provide additional links of mTOR complexes to many major signaling pathways and physiological functions maintaining cellular homeostasis. These confirm the central role of mTOR in regulating cellular survival, growth, and differentiation in the continuously changing surrounding microenvironment.
In parallel, it was described that rapamycin or rapalog (derivatives of rapamycin) treatment reduce tumor growth in renal cancers developing in kidney transplanted patients. Later, it was observed that the incidence of posttransplant malignancy decreased [265] in rapalog converted immunosuppression. Identifying the contribution of mTOR activity in cyclin D1 overexpression in mantle cell lymphomas highlighted the possible use of mTOR inhibitors (mTORIs) in lymphomas and other malignancies [266]. To date, mTORIs have been introduced into the therapy of several cancers: renal cancers, mantle cell lymphomas, advanced breast cancers (hormone receptor-positive), pancreatic, gastrointestinal, pulmonary neuroendocrine cancers, and sporadic lymphangiomatosis. The application and development of mTORIs are ongoing; currently, oncologists prescribe them as a personalized therapy regimen or combined therapies.
9.1 mTOR complexes and their sensing functions in cellular fitness and growth potential
The two functionally and structurally different mTOR complexes (mTORC1 and mTORC2) consist of two common proteins, mTOR serine/threonine kinase, and mLST8. The specific subunits are composed of unique scaffold proteins, Raptor in mTORC1 and Rictor in mTORC2, respectively. The mTORC2 complex has additional elements such as mSIN1, MAPK-interacting protein 1, which determine the subcellular localization of the complex and help in substrate recruitment. Additionally, the endogenous common inhibitory protein (DEPTOR) can bind and inhibit mTOR kinase in both complexes, while PROTOR1/2 can associate with Rictor and inhibit mTORC2 specifically [267]. Crystallographic analysis revealed that dimerized protein complexes have different structures in different cellular localizations (membrane, lysosomes, endoplasmic reticulum — ER, etc.). These offer binding sites for several other regulators (Rheb GTPase), substrates, and protein complexes [268]. mTOR complexes integrate different external and internal information (signals) based on monitoring cellular stress conditions; these influence “cellular decisions” (proliferation/differentiation or growth/survival). These decisions are assisted by mTOR-related signaling crosstalk, networking with many pathways, cellular state sensing, feedback, and feed-forward regulatory mechanisms [269].
The environment and the actual condition of cells influence the cellular programs and functions. mTORC1 and mTORC2 complexes integrate several signals, including growth factors, nutrients, starving conditions, energy, and stress levels, to regulate their activity. mTORC1 should “turn on and off” in correlation with cellular bioenergetics, genome integrity status, energy, O2, and growth factor levels [264]. To monitor bioenergetics and the activity of growth signals, two small G protein families, the Rheb and Rag GTPases, alter mTOR kinase phosphorylation status, activity, and the cytoplasmic/lysosomal localization of mTORC1 complex [270]. Signals of cytokines and endocrine hormones maintain Rheb in active GTP-bounded forms and determine the lysosomal localization of Rheb. The translocation of the mTORC1 complex to lysosomes mainly depends on the presence of amino acids and glucose. Their appropriate concentrations activate Rag GTPases and induce lysosomal localization of the mTORC1 complex [271]. Adequate levels of lysosomal cholesterol and arginine influence the lysosomal sensor (SLC38A9) and generate the activity of Rag GTPases [272]. GATOR1 usually shifts to mTORC1-Rag-GDPs; therefore, inhibited GATOR1 serves the RagA/B-GTP isoform and localization of mTORC1 to the lysosome [273, 274]. Additionally, GATOR1 functions can be inhibited by the folliculin sensor (high total amino acids level) and GATOR2 amino acid sensors (high level of arginine and leucine). Low levels of leucine, arginine, and their metabolites can activate Sestrin1/2/3, SAMTOR (S-adenosyl methionine regulator), and SARB1, leading to the inhibition of GATOR2. Therefore, active GATOR1 shifts RagC/D to GDP-bound forms, and mTORC1 loses the lysosomal membrane localization [275,276,277]. Moreover, lowering amino acids through Sestrin1/3 can alter the expressions of uncoupling proteins which switch off mTORC1 activity amino acid sensor pathways, independently [278]. This nutrient and growth factor sensor regulation provides the balance between anabolism and catabolism depending on nutrient conditions. Additionally, the mTOR activating function of lysosomal Rheb GTPases is negatively regulated by TSC1/2. The inhibitory effect of TSC1/2 can be maintained (reactivated) by hypoxia, energetic stress (e.g., low glucose and ATP level) — AMPK activation, DNA damage, or Wnt signaling. However, it can be suspended by several growth factors (e.g., IGFR, EGFR) [279,280,281].
To date, the regulation of mTORC2 complex activity has been less described. Usually, the mTORC2 complex is positively regulated by growth factors mediated by PI3K pathway [282]. mSIN1 pleckstrin domain autoinhibits mTORC2 activity; this can be released by insulin- or serum-induced PI3K activation and increased PIP3. After these alterations, PIP3, mTORC2, and Akt are recruited into the cell membrane. mTORC2 is mainly active in the plasma membrane, but its active forms were also detected in the outer mitochondrial membrane and endosomes [283]. Evidence showed that growth factors regulate mTORC2; however, amino acids and other metabolites could also have some regulatory effects. Several new data highlighted that Rac1, Rap1, and Ras GTPases had potential roles in mTORC1 and mTORC2 activation and membrane localization processes in the last few years. These, especially oncogenic Ras, can directly induce mTORC2 kinase activation, promoting proliferation, and cell survival, which are associated with poor patient outcomes [284]. It was reported that lowered glutamine concentration can activate mTORC2 and modulate glutamine homeostasis by increasing glutamine-fructose 6 phosphate amidotransferase 1 and cellular catabolism [285]. mTORC2 complex activity can also be regulated through Rictor phosphorylation and acetylation. For example, GSK3 phosphorylates Rictor, initiating the FBXW7 ubiquitin–proteasome degradation of Rictor. It was also described that Rictor acetylation increases mTORC2 activity. Additionally, glucose stress and starvation activate AMPK, increasing mTORC2 activity in cells (instead of inhibiting mTORC1). These mechanisms underline the importance of mTORC2 functions in cell survival mechanisms under the conditions of starvation and acute bioenergetic stress (Fig. 6).

mTOR as a metabolic crossroad in the signaling network (simplified figures). a mTOR kinase and the mTORC1 and C2 complexes are metabolic and “cell-state” sensors in the signaling network. b The effector mechanisms and targets of mTORC1 and C2 complexes and their cellular regulatory functions. The activating (black) and inhibiting (red) effects are shown in the schematic diagram.
9.2 Cellular fitness regulated by mTOR activity, controls proliferation, growth, and metabolic rewiring
“mTOR controls almost everything” — this statement highlights that mTOR plays crucial roles as it not only promotes cellular growth by stimulating proteins and initiates the synthesis of building blocks (from various sources: nucleotides and lipids) but inhibits autophagy as well.
The switch from anabolism to catabolism and vice versa is primarily regulated by mTORC1 activity. For example, in in vitro cell cultures, the growing cells are bathed in nutrients (growth factors, glucose, and proteins). In correlation with this, tumor cell lines have higher mTOR activity than their original in vivo tumor tissue counterparts (xenograft or human cancers in situ). However, in in vivo tumors, the fluctuation between fasting and feeding and the deregulated mTOR cause metabolic alterations under various environmental conditions [286, 287]. Several data confirm the importance of mTORC1-dependent metabolic regulatory functions in the whole organ and organism as well. After a 24-h fasting period in mouse models, it was observed that liver cell mass was reduced by 25% mTORC1 dependently (in case of Raptor KO mice, this reduction did not occur) [288]. Additional experiments described that mTORC1 activity delays autophagy under perinatal fasting conditions. The disturbance of these pathways results in uncompensated glucose reduction and mortality in animal models [289]. Moreover, hundreds of results underline the central role of mTORC1 in dysregulated metabolic signaling during overfeeding, obesity, and type II diabetes. It is not surprising that mTOR inhibitory treatments could decrease symptoms of obesity and diabetes [233]. mTORC1 activity is a well-known promoter of cellular protein, nucleotide, and lipid synthesis. These are in correlation with the already described effector mechanism of mTORC1: (a) phosphorylates eukaryotic initiation factor 4E–binding proteins (4EBP1) releasing eIF4E and enhances cap-dependent mRNA translation; (b) phosphorylates p70 S6 kinase 1 (S6K1) and subsequently ribosomal S6, which enhances the translational efficiency by influencing the expression and function of several ribosomal proteins (SKAR, RNA polymerases) and protein synthesis [290,291,292]; (c) phosphorylates and inactivates lipin1 and induces the translocation of SREBP1 from ER to the nucleus in an S6K1-dependent manner, initiating the expression of enzymes in lipid and cholesterol syntheses [293]; (d) phosphorylates ATF4 (activating transcription factor 4) and consequently induces MTHFD2 (mitochondrial tetrahydrofolate dehydrogenase) enhancing purine biosynthesis [294]; (e) phosphorylates and activates carbamoyl phosphate synthase (CAD), the rate-limiting enzyme in pyrimidine synthesis [295]; (f) influences several further transcription factors regulating hypoxia, energetic stress responses, or lysosomal biogenesis (HIF1α, PGC1a, TFEB) [296]. In parallel, mTORC1 activity decreases the catabolism in tumor cells by inhibiting the phosphorylation of autophagosome proteins ULK1 kinase and Atg13 autophagosome compartments, arresting autophagosome functions, and inducing autophagosome accumulation. Furthermore, it inhibits autophagosome maturation through phosphorylating other proteins by reducing lysosome biogenesis (UVRAG, Rab7, TFEB, TF3) [297, 298].
mTORC2 complex has been less characterized. However, its effector mechanisms were described in correlation with kinase activity, phosphorylation of AGC kinases, Akt, PKC members, and SGK1. The best-characterized kinase activity of mTORC2 is Ser473-Akt phosphorylation which is complex and context-dependent. The Ser473-Akt phosphorylation is required to phosphorylate some additional Akt substrates, including PRAS40, FOXO1/3a, and TSC2. It was suggested that this Ser473 phosphorylation is more critical for maximal activity of Akt than substrate specificity. Other phospho-Akts (Ser477, Thr479) resulting from mTORC2 induce the degradation of IRS1 by stabilizing FBW8 ubiquitin ligase activity [299]. mTORC2 is also stated to promote ACLY phosphorylation which increases ChREBP activity and influences histone acetylation [237]. mTORC2-mediated Akt phosphorylation could further promote c-Myc expression and, in turn, increase glycolytic HK2 expression and glucose-6-phosphate level. PKC phosphorylation by mTORC2 was suggested to regulate actin polymerization, which could orchestrate EMT, migration, and metastatic events [300]. Moreover, TGFβ, Wnt, and YAP/Hippo can also activate mTORC2, which could influence EMT-associated migration and invasion [282]. In a mouse model study, the role of mTORC2 in lipid metabolism was also highlighted. This detailed analysis confirmed mTORC2-mediated promotion of FA and lipid metabolism (sphingolipid and cardiolipin biosyntheses), causing steatosis and tumor development in the liver. Regarding these results, elevated mTORC2 and correlated lipogenesis were detected in human HCC cases. Besides the well-known regulatory functions of mTORC1 in autophagy, mTORC2 activity influences SGK-1– and Akt-mediated alterations of autophagy-related proteins (Atg7, beclin-1, FOXO3, VDAC1, etc.). In correlation with this autophagy regulating function the inactivation of mTORC2 and lowered SGK-1 activity, disrupting the autophagy and the normal differentiation in an animal model (Caenorhabditis elegans) [301, 302] (Fig. 6).
mTOR hyperactivity has multifunctional tumor growth–promoting effects. High mTOR activity (both complexes) through a broad spectrum of mechanisms contributes to cellular adaptation. mTOR hyperactivity has a central regulatory role in integrating complex pathway networks, especially in adaptation to bioenergetic demands and survival of migrating cells which can metastasize and be reactivated at distant sites. Therefore, it is not surprising that mTORC1 and mTORC2 complex overexpression and hyperactivity were described in many cancers, especially in patients with unfavorable prognoses [303, 304].
9.3 Characterization of mTOR hyperactivity and its correlation with cancer progression and metastasis
Higher mTOR activity scores are associated with a worse prognosis in several tumor types (metastasis, leading to metastasis-related cancer mortality). During cancer progression, malignant cells lose their adherence capacity, gain invasive nature, survive under extreme conditions (e.g., in circulation), spread, and find new sites to form additional tumors. Tumor cells need to adapt to the new microenvironment and rearrange their resources, dependencies, and cellular connections during all these steps. Therefore, the mTOR activity–provided EMT-MET could contribute to the migratory and invasive potential of tumor cells in the metastatic cascade. Several publications confirmed that small GTPases support actin-cytoskeletal rearrangement, migration, and invasion. The contribution of mTOR signaling elements was described in correlation with TGFβ-mediated protein expression changes of EMT. Based on this observation, rapamycin can inhibit the TGFβ-dependent PI3K/Akt/mTOR activation and influence the migratory and invasive features of cancer cells. It was also described that mTORC2 complex–mediated functions are required in EMT, migration, and metastasis [305]. In prostate cancer, Rho and Rac1 GTPases were proposed to be regulated by mTOR [306]. Additionally, dual mTOR inhibitors were shown to arrest hypoxia- and TGFβ-mediated EMT in cancer cell lines. Besides these effects, mTOR activity–dependent alterations can induce protein expression (e.g., that of BMP2 and Sestrin2) and promote EMT and metastasis in different cancer models (ovarian, nasopharyngeal, pancreatic cancer, NSCLC, and HCC) [307]. Both mTORC1 and mTORC2 could contribute to modifying the metastatic properties of cells. It was described that IGF induces mTOR/Raptor/S6K1–dependent F-actin reorganization and phosphorylation of focal adhesion protein [308]. It was also published that the knockdown of mTOR/Rictor/mLST8 resulted in malformation of F-actin fibers and prevented paxillin phosphorylation, leading to the recruitment of focal adhesion complexes in starved fibroblasts [309,310,311]. The dynamics of cytoskeleton reorganization were found to be mediated by mTOR, mTORC2 via the Rho family of small GTPases (Rho A, Cdc42, and Rac1) in RH30 and HeLa cells. It was also proposed that mTORC2 controls the migration of neutrophils via Rac/Cdc42 [312, 313]. Recently, it was confirmed that mTOR KO caused dis-morphogenesis (documented by morphology analyses) in correlation with actin synthesis and assembly (F-actin and Cdc42 expression) in tooth epithelia [313].
Genetically modified Rictor expression influences the metastasis formation of HER2 overexpressing mouse breast cancers. The studied metastasis model system documented that mTORC2 can activate Rac1 into two ways, either via decreasing the expression of RhoGDI2 inhibitor by PKCa or via Akt-activated Rac-GEF Tiam1 [314]. Additionally, data obtained from mTOR/Rictor/PKCa silencing studies confirmed the role of mTORC2 in PKCa-mediated actin filament organization [315]. Following these results, mTOR inhibitor therapy decreases cell motility and metastatic potential in several preclinical and clinical studies (OS and DFS studies were performed).
Receptor tyrosine kinases (EGFR, HER2, PDGFRa, and FGFR in colorectal, breast, head and neck, gastric cancer, NSCLC, glioblastomas, GISTs, melanoma, etc.), PI3KCA and RAS mutation-mediated PI3K hyperactivity, as well as the loss of PTEN or TSC1/2, leading to mTOR hyperactivity in many cancers. Additionally, amplification and mutations of mTOR kinase and Rictor could also influence mTOR deregulation in cancers. PI3KCA, AKT1, PTEN, TSC1/2, or LKB1 modifications and PI3KCA alterations are frequently described in breast (> 20%), colon, and gynecologic cancers. PTEN mutations occur in colorectal cancers and about 10% of central nervous system malignancies. TSC1/2 loss can be detected in 4–6% of endometrial, urothelial, cervical, liver, and lung cancers. On the contrary, loss-of-function mutations occur less frequently in the prostate, endometrium, breast cancers, melanomas, glioblastomas, and renal cancers (in less aggressive hamartomas, rhabdomyoma, and angiofibroma). The deregulation of these growth signaling pathways correlates with mTOR hyperactivity. mTOR mutations and RICTOR amplification are among the most significant and frequent oncogenic mutations. Approximately, 5% of solid cancers carry disease-related mTOR kinase activating mutations (e.g., in melanomas, colon, renal, lung, endometrial, and gastric cancers) (Fig. 7). The importance of RICTOR amplification and overexpression was described in small cell lung (SCLC), breast, gastric, and head and neck cancers [316,317,318,319]. As a result of cellular signaling network–related oncogenic alterations, increased mTOR activity can be detected in ~ 80% of human malignancies. Consequently, mTOR hyperactivity and its outcome on a cellular level can influence the therapeutic sensitivity and progression (in a tumor type–dependent manner). mTOR hyperactivity can overwrite the metabolic checkpoints, contributing to cancer growth nutrient and growth factors independently.

Genetic alterations of MTOR gene and their frequency in human cancer (point mutations, structural alterations — fusions, amplifications, deletions, or multiple alterations) are shown based on TCGA databases
Studying the mTOR hyperactivity of malignancies at the protein level was initiated in 1998. Studies examined p-mTOR and p-p70S6K proteins in malignant cell lines were initially conducted by Western blot analyses [320]. The development of phospho-protein antibodies and further examination of mTORC1 and mTORC2 targets could be possible with in situ characterization of human biopsy materials (formalin-fixed tissues).
Experimental data found that the pattern of mTOR hyperactivity could be altered at the tissue level during therapy [321]. Additionally, the progression and metastatic potential and the preference of metastatic sites were analyzed in correlation with mTOR hyperactivity in different cancers [322]. However, higher mTOR activity was described in many various cancers; the in situ staining pattern (heterogeneity) and the mTORC1/C2 complex distribution were not studied in detail. More precise analyses of tissue and mTORC1/C2 complex–related mTOR activity distribution, special markers, the use of well-defined complex scoring systems, and excellent pathologists are required. Moreover, active (phosphorylated) forms of mTOR kinase, other mTORC1, and mTORC2-specific elements (e.g., Raptor and Rictor) and active forms of mTOR complex target proteins (p-S6K1, p-4EBP1, pS6, p-Ser473-Akt, p-SGK1, etc.) must be analyzed.
Many studies on mTOR hyperactivity and its correlation with prognosis have been published; however, mTORC1 and mTORC2 complex distribution and additional metabolic regulatory proteins and their correlation with tumor progression have been less studied [323]. In these studies, strict conclusions were not possible due to lower case numbers, a broad diversity of tumor subtypes (with higher incidence), and intertumoral differences. In the early 2010s, our and others’ results confirmed that elevated mTOR activity is characteristic for human lymphoma and leukemia cells. Based on IHC and flow cytometry data, higher mTOR activity, especially in the presence of a high amount of mTORC2 complex, was correlated with worse prognosis and lower OS in lymphoma/leukemia patients [324,325,326,327].
In the case of solid tumors, similar observations were published in the last decade. For example, almost all of the published studies underline the role of mTOR hyperactivity in the progression and metastasis formation in hormone receptor–positive (HR +) or triple-negative (TN) breast cancers. Recent preclinical studies described that mTORC1 and mTORC2 inhibition could have significant growth inhibitory effects in these malignancies [328,329,330]. Moreover, in some reports, it was also highlighted that high mTOR activity could be associated with the presence of metabolic plasticity (higher expression level of alternative metabolic enzymes at tissue level, e.g., simultaneously increased expressions of LDHA, GLS, and carnitine palmitoyltransferase 1A — CPT1A) in breast and other cancers [128, 202, 331,332,333]. Our findings suggest that metabolic plasticity (high mTOR activity and the overexpression of at least two alternative metabolic pathway–related enzymes) could be a subtype-independent risk factor in breast cancers, certain lung tumors rhabdomyosarcomas, and pheochromocytomas [128, 207, 334, 335]. In general, mTOR hyperactivity and the expression of mTORC2 complex elements were analyzed and characterized in correlation with prognosis and metastasis of other malignancies. Most of these results confirm the significant role of mTOR hyperactivity in malignant progression.
The site-specific metastatic progression was suggested to be influenced by mTORC1 and mTORC2 activity in liver, lung, bone, brain, colon, breast, and pancreatic cancers or melanomas. The incidence of PIK3CA mutations, the activation of PI3K/Akt/mTOR signaling axis, and, in correlation with these, the increased mTORC2 activity (p-Ser473-Akt)–related HER2 expression were found to be involved in the liver metastases of breast cancers, which are occurring more frequently [336]. However, another study emphasized the potential role of PI3K/Akt/mTOR axis hyperactivity in brain metastases of melanomas. mTOR-related hyperactivity correlated with metastases of other cancers was reported in a few lower case number studies (e.g., breast, lung, and renal carcinomas) [334, 337]. There is a strong correlation between high mTOR activity and poor prognosis, significantly increasing mTORC2 complex expression analyzed by IHC follow-up studies.
Regarding these observations, Rictor overexpression results and RICTOR amplification were found to negatively influence the progression of different cancers [304]. This could help in patient selection for mTORC1 and mTORC2 or dual inhibitor therapies (Table 2). Several experimental studies described that dual inhibitors could reduce brain metastasis formation and growth of PI3KCA-mutated cancers [338]. Additionally, in colon carcinomas and highly aggressive pancreatic cancers, dual mTOR inhibitors could reduce the occurrence of liver and lung/liver metastases, respectively [339].
Based on these results and the detected high mTOR activity in many cancers, mTOR inhibitor therapies have been tested in highly metastatic and aggressive cancer types characterized by high mTOR activity (e.g., pancreas, relapsed HER2 + , and TN breast cancers, EGFRI-resistant or cisplatin-, radiotherapy-resistant cancers). mTORIs have been approved in the therapy of renal cancers; mantle cell lymphomas; advanced breast cancers (hormone receptor–positive); pancreatic, gastrointestinal, and pulmonary neuroendocrine cancers; and sporadic lymphangioleiomyomatosis. Despite the correlation between high mTOR activity and worse prognosis, it might be surprising that clinical phase trials usually have lower beneficial therapeutic outcomes; mTORIs also have a lower success rate in monotherapy. Therefore, better mTOR and more specific mTORC1 and mTORC2 inhibitors are needed to be developed. Next-generation mTOR inhibitors include dual mTOR and ATP competitive mTOR kinase inhibitors (2nd generation) [340]. The 3rd-generation new synthetic mTOR (e.g., JR-AB2-011, RapaLinks) and specific mTORC2 complex inhibitors (Rictor si-NPs: nanoparticles harboring Rictor siRNA sequences) are under preclinical and clinical investigations [330, 340]. Additionally, more and more mTOR and PI3K/Akt/mTOR pathways inhibiting natural compounds have been discovered and tested in clinical trials. For example, indole-3-carbinol (an alkaloid from Brassica genus — NCT00579332), berberine (an alkaloid from European barberry — NCT03281096, NCT02226185), curcumin (diarylheptanoid from Curcuma genus — NCT03769766), epicatechin and other catechins (flavonoids from green tea — NCT02029352), and genistein (isoflavone from soybean — NCT00584532) are in phase III trials, and these have favorable results and are available as dietary supplements. Many different PI3K/Akt/mTOR inhibitors have been under development in clinical trials. These all have promising effects in preclinical studies (Table 3). Unfortunately, the application of mTORI monotherapy has often lower final survival advantages than expected, as is the unfortunate case of targeted monotherapies in general. Therefore, the selection of optimal partner drugs needs to be considered for achieving more effective combination therapies. The combination of mTORIs with targeted or traditional chemotherapeutics/radiotherapy could be applied and initiated in clinical trials (Table 4). However, the possible higher toxicity could limit the success of these combined treatments. Based on the highlighted role of metabolic profiles, other antimetabolic drugs with traditional therapies or mTORIs should be considered to find additional therapeutic options. Some new preclinical studies highlight that mTORIs combined with metabolic (e.g., GLS, OXPHOS, lipid metabolism) inhibitors could have additive or synergistic effects on tumor growth in advanced cancer, in in vitro and in vivo models [341,342,343].
Such combined antimetabolic treatments can induce synthetic lethality [389]. For example, inhibiting both OXPHOS and glycolysis (e.g., with metformin + 2-deoxy-D-glucose/GLUT/MCTI) can effectively induce the collapse of balanced energy metabolism, which was detected to be effective in many different cell line and xenograft models (e.g., breast, prostate, ovarian, and hepatocellular cancers) [159, 390, 391]. In the case tumors depend on glutamine utilization and anaplerosis, this metabolic rewiring provides a potential opportunity for rational dual targeting of glutaminolysis and glycolysis. Simultaneous glutaminolysis blockade with CB-839 and 3-bromopyruvate (3BP) could also induce tumor regression in the renal cancer model [392]. These preclinical combination studies initiated a phase I study using ritonavir and metformin in the management of multiple myeloma and chronic lymphocytic leukemia (CLL) (NCT02948283). Further preclinical data and key clinical trials using metabolic inhibitors are available; including mTORIs combined with conventional tumor type–dependent and targeted therapy (including immune checkpoint inhibitor therapy) [393]. There are several promising preclinical observations regarding the potential use of antimetabolic (OXPHOS or glutamine metabolism inhibitors) and targeted kinase inhibitory therapy in various cancer types (e.g., melanoma, GIST, etc.) (NCT03026517, NCT03831932) [394,395,396]. These have propagated further clinical studies (lung RCC NCT03158324, NCT03071705, and NCT02071862). According to this promising preclinical data, early-phase trials of rapalog and metformin combinations were set off. These suggest that further examinations of temsirolimus and metformin is necessary since this combination reported good tolerance and promising effects in advanced cancers (NCT01529593, NCT03163667, and NCT00659568). Moreover, immune checkpoint inhibitors or other monoclonal antibody therapies promoted the attempts of glutamine utilization inhibitor (including mTORIs) combinations with pembrolizumab, nivolumab, or daratumumab in advanced solid cancers (NCT04265534 and NCT02771626) [397]. Other agents and drugs (e.g., antibiotics, cardiovascular drugs, antidepressants) [398] with antimetabolic (off-target) properties can be applied in an off-label use in these combinations in future studies [399,400,401].
All these data suggest that there is an urgent need to clarify the role of metabolic adaptations in tumor resistance, with respect to recently used therapies (metabolic rewiring at tissue level). Future implications in this field of research are to expand the clinically available metabolic targets (drugs) to improve the response rate and patient survival in recently used treatments.
10 Concluding remarks
Albert Szent-Györgyi (who won the Nobel Prize for discovering the components and reactions in TCA in 1937 and isolated vitamin C) said — “Nature is huge, man is tiny. Therefore, man's existence depends on what kind of interactions he can make with nature, how much he understands it, and how he uses its resources for his benefit.” In the last decades, we have discovered many various mechanisms and their alterations, complexity, and tumor type dependency in the genetic, epigenetic, and metabolic deregulation of tumor growth. Now, we have ideas how tumors reprogram their proliferation, survival mechanisms, and the microenvironment, the organ, the ecosystem, and even the whole body. Finally, we understand that “Man is huge, the tumor cell is tiny. Therefore, tumor’s existence depends on what kind of interactions it can make with the environment, how much tumor “understands” this, and how it uses the environmental resources for its benefit.” The complexity of heterogeneous tumors, including different cell populations and their symbiosis, further complicates our understanding of how tumors can develop. Consequently, the local conditions are altering (hypoxic, acidic, and nutrient-deprived environment) and promoting metastasis (“escape”) to find another niche to grow and invade the whole body and use its sources. Finally, the metabolic plasticity and all the related forces in the human body serve the survival of the tumor population. During this, the tumor disrupts the homeostasis of the host, which leads to the death of the patient. After the scientific revolution in different research fields, primarily due to oncogenomic and molecular carcinogenesis studies, we could describe and comprehend many pieces of carcinogenesis and progression. However, to overcome cancer therapy failures, we need to understand oncogenic networks, including the regulatory shortcomings of metabolic adaptation in this complex and dynamic cancer ecosystem.
References
Warburg, O., & Minami, S. (1923). Versuche an Überlebendem Carcinom-gewebe. Klinische Wochenschrift, 2(17), 776–777. https://doi.org/10.1007/BF01712130
Pavlova, N. N., & Thompson, C. B. (2016). The emerging hallmarks of cancer metabolism. Cell Metabolism, 23(1), 27–47. https://doi.org/10.1016/j.cmet.2015.12.006
Martinez-Outschoorn, U. E., Bartrons, M., & Bartrons, R. (2019). Editorial: Cancer ecosystems. Frontiers in Oncology, 9, 718. https://doi.org/10.3389/fonc.2019.00718
Dvorak, H. F. (1986). Tumors: Wounds that do not heal Similarities between tumor stroma generation and wound healing. N Engl J Med, 315(26), 1650–1659. https://doi.org/10.1056/nejm198612253152606
Baltazar, F., Afonso, J., Costa, M., & Granja, S. (2020). Lactate beyond a waste metabolite: Metabolic affairs and signaling in malignancy. [Mini Review]. Frontiers in Oncology, 10(231), https://doi.org/10.3389/fonc.2020.00231.
Yoshida, G. J. (2021). The harmonious interplay of amino acid and monocarboxylate transporters induces the robustness of cancer cells. Metabolites, 11(1), https://doi.org/10.3390/metabo11010027.
Levan, A., & Hauschka, T. S. (1953). Endomitotic reduplication mechanisms in ascites tumors of the mouse. Journal of the National Cancer Institute, 14(1), 1–43.
Klein, G., & Klein, E. (1956). Conversion of solid neoplasms into ascites tumors. Annals of the New York Academy of Sciences, 63(5), 640–661. https://doi.org/10.1111/j.1749-6632.1956.tb50883.x
Makino, S. (1956). Further evidence favoring the concept of the stem cell in ascites tumors of rats. Annals of the New York Academy of Sciences, 63(5), 818–830. https://doi.org/10.1111/j.1749-6632.1956.tb50894.x
Fidler, I. J. (1978). Tumor heterogeneity and the biology of cancer invasion and metastasis. Cancer Research, 38(9), 2651–2660.
Miller, F. R. (1982). Intratumor immunologic heterogeneity. Cancer and Metastasis Reviews, 1(4), 319–334. https://doi.org/10.1007/bf00124215
Heppner, G. H., & Miller, B. E. (1983). Tumor heterogeneity: Biological implications and therapeutic consequences. Cancer and Metastasis Reviews, 2(1), 5–23. https://doi.org/10.1007/bf00046903
Heppner, G. H. (1984). Tumor heterogeneity. Cancer Research, 44(6), 2259–2265.
Brastianos, P. K., Carter, S. L., Santagata, S., Cahill, D. P., Taylor-Weiner, A., Jones, R. T., et al. (2015). Genomic characterization of brain metastases reveals branched evolution and potential therapeutic targets. Cancer Discovery, 5(11), 1164–1177. https://doi.org/10.1158/2159-8290.Cd-15-0369
Stine, Z. E., Walton, Z. E., Altman, B. J., Hsieh, A. L., & Dang, C. V. (2015). MYC, metabolism, and cancer. Cancer Discovery, 5(10), 1024–1039. https://doi.org/10.1158/2159-8290.Cd-15-0507
Kinsey, C. G., Camolotto, S. A., Boespflug, A. M., Guillen, K. P., Foth, M., Truong, A., et al. (2019). Protective autophagy elicited by RAF→MEK→ERK inhibition suggests a treatment strategy for RAS-driven cancers. Nature Medicine, 25(4), 620–627. https://doi.org/10.1038/s41591-019-0367-9
Takács, T., Kudlik, G., Kurilla, A., Szeder, B., Buday, L., & Vas, V. (2020). The effects of mutant Ras proteins on the cell signalome. Cancer and Metastasis Reviews, 39(4), 1051–1065. https://doi.org/10.1007/s10555-020-09912-8
White, E. (2019). Blockade of RAF and autophagy is the one-two punch to take out Ras. Proc Natl Acad Sci U S A, 116(10), 3965–3967. https://doi.org/10.1073/pnas.1900800116
Thoreen, C. C., & Sabatini, D. M. (2005). AMPK and p53 help cells through lean times. Cell Metabolism, 1(5), 287–288. https://doi.org/10.1016/j.cmet.2005.04.009
Jones, R. G., Plas, D. R., Kubek, S., Buzzai, M., Mu, J., Xu, Y., et al. (2005). AMP-activated protein kinase induces a p53-dependent metabolic checkpoint. Molecular Cell, 18(3), 283–293. https://doi.org/10.1016/j.molcel.2005.03.027
Lee, C.-W., Wong, L.L.-Y., Tse, E.Y.-T., Liu, H.-F., Leong, V.Y.-L., Lee, J.M.-F., et al. (2012). AMPK promotes p53 acetylation via phosphorylation and inactivation of SIRT1 in liver cancer cells. Cancer Research, 72(17), 4394–4404. https://doi.org/10.1158/0008-5472.Can-12-0429
Harami-Papp, H., Pongor, L. S., Munkácsy, G., Horváth, G., Nagy, Á. M., Ambrus, A., et al. (2016). TP53 mutation hits energy metabolism and increases glycolysis in breast cancer. Oncotarget, 7(41), 67183–67195.
Hashimoto, N., Nagano, H., & Tanaka, T. (2019). The role of tumor suppressor p53 in metabolism and energy regulation, and its implication in cancer and lifestyle-related diseases. Endocrine Journal, 66(6), 485–496. https://doi.org/10.1507/endocrj.EJ18-0565
Liu, J., Zhang, C., Hu, W., & Feng, Z. (2019). Tumor suppressor p53 and metabolism. Journal of Molecular Cell Biology, 11(4), 284–292. https://doi.org/10.1093/jmcb/mjy070
Labuschagne, C. F., Zani, F., & Vousden, K. H. (2018). Control of metabolism by p53 - Cancer and beyond. Biochimica et Biophysica Acta - Reviews on Cancer, 1870(1), 32–42. https://doi.org/10.1016/j.bbcan.2018.06.001
Najdi, R., Holcombe, R. F., & Waterman, M. L. (2011). Wnt signaling and colon carcinogenesis: Beyond APC. Journal of carcinogenesis, 10, 5–5. https://doi.org/10.4103/1477-3163.78111
Jiang, W. G., Sanders, A. J., Katoh, M., Ungefroren, H., Gieseler, F., Prince, M., et al. (2015). Tissue invasion and metastasis: Molecular, biological and clinical perspectives. Seminars in Cancer Biology, 35, S244–S275. https://doi.org/10.1016/j.semcancer.2015.03.008
Cha, Y. H., Yook, J. I., Kim, H. S., & Kim, N. H. (2015). Catabolic metabolism during cancer EMT. Archives of Pharmacal Research, 38(3), 313–320. https://doi.org/10.1007/s12272-015-0567-x
Jerby, L., Wolf, L., Denkert, C., Stein, G. Y., Hilvo, M., Oresic, M., et al. (2012). Metabolic associations of reduced proliferation and oxidative stress in advanced breast cancer. Cancer Research, 72(22), 5712–5720. https://doi.org/10.1158/0008-5472.Can-12-2215
Jeon, S. M., Chandel, N. S., & Hay, N. (2012). AMPK regulates NADPH homeostasis to promote tumour cell survival during energy stress. Nature, 485(7400), 661–665. https://doi.org/10.1038/nature11066
Guo, Y., Zhu, H., Yao, Z., Liu, F., & Yang, D. (2021). The diagnostic and predictive efficacy of (18)F-FDG PET/CT metabolic parameters for EGFR mutation status in non-small-cell lung cancer: A meta-analysis. European Journal of Radiology, 141, 109792. https://doi.org/10.1016/j.ejrad.2021.109792
Magometschnigg, H., Pinker, K., Helbich, T., Brandstetter, A., Rudas, M., Nakuz, T., et al. (2019). PIK3CA mutational status is associated with high glycolytic activity in ER+/HER2- early invasive breast cancer: A molecular imaging study using [(18)F]FDG PET/CT. Molecular Imaging and Biology, 21(5), 991–1002. https://doi.org/10.1007/s11307-018-01308-z
Jin, N., Bi, A., Lan, X., Xu, J., Wang, X., Liu, Y., et al. (2019). Identification of metabolic vulnerabilities of receptor tyrosine kinases-driven cancer. Nature Communications, 10(1), 2701. https://doi.org/10.1038/s41467-019-10427-2
Whi, W., Ha, S., Bae, S., Choi, H., Paeng, J. C., Cheon, G. J., et al. (2020). Relationship of EGFR mutation to glucose metabolic activity and asphericity of metabolic tumor volume in lung adenocarcinoma. Nuclear medicine and molecular imaging, 54(4), 175–182. https://doi.org/10.1007/s13139-020-00646-7
Qian, X.-J., Li, Y.-T., Yu, Y., Yang, F., Deng, R., ji, J., et al. (2015). Inhibition of DNA methyltransferase as a novel therapeutic strategy to overcome acquired resistance to dual PI3K/mTOR inhibitors. Oncotarget, 6, https://doi.org/10.18632/oncotarget.3016.
Beyoğlu, D., & Idle, J. R. (2021). Metabolic rewiring and the characterization of oncometabolites. Cancers, 13(12), 2900.
Erez, A., & DeBerardinis, R. J. (2015). Metabolic dysregulation in monogenic disorders and cancer - Finding method in madness. Nature Reviews Cancer, 15(7), 440–448. https://doi.org/10.1038/nrc3949
Balss, J., Meyer, J., Mueller, W., Korshunov, A., Hartmann, C., & von Deimling, A. (2008). Analysis of the IDH1 codon 132 mutation in brain tumors. Acta Neuropathologica, 116(6), 597–602. https://doi.org/10.1007/s00401-008-0455-2
Nowicki, S., & Gottlieb, E. (2015). Oncometabolites: Tailoring our genes. FEBS Journal, 282(15), 2796–2805. https://doi.org/10.1111/febs.13295
Morin, A., Letouzé, E., Gimenez-Roqueplo, A. P., & Favier, J. (2014). Oncometabolites-driven tumorigenesis: From genetics to targeted therapy. International Journal of Cancer, 135(10), 2237–2248. https://doi.org/10.1002/ijc.29080
Certo, M., Tsai, C.-H., Pucino, V., Ho, P.-C., & Mauro, C. (2021). Lactate modulation of immune responses in inflammatory versus tumour microenvironments. Nature Reviews Immunology, 21(3), 151–161. https://doi.org/10.1038/s41577-020-0406-2
Parks, S. K., Mueller-Klieser, W., & Pouysségur, J. (2020). Lactate and acidity in the cancer microenvironment. Annual Review of Cancer Biology, 4(1), 141–158. https://doi.org/10.1146/annurev-cancerbio-030419-033556
Li, F., & Simon, M. C. (2020). Cancer cells don’t live alone: Metabolic communication within tumor microenvironments. Developmental Cell, 54(2), 183–195. https://doi.org/10.1016/j.devcel.2020.06.018
Lee, D. C., Sohn, H. A., Park, Z. Y., Oh, S., Kang, Y. K., Lee, K. M., et al. (2015). A lactate-induced response to hypoxia. Cell, 161(3), 595–609. https://doi.org/10.1016/j.cell.2015.03.011
Parks, S. K., & Pouysségur, J. (2017). Targeting pH regulating proteins for cancer therapy-Progress and limitations. Seminars in Cancer Biology, 43, 66–73. https://doi.org/10.1016/j.semcancer.2017.01.007
Kobayashi, H., Moniwa, N., Sugimura, M., Shinohara, H., Ohi, H., & Terao, T. (1993). Effects of membrane-associated cathepsin B on the activation of receptor-bound prourokinase and subsequent invasion of reconstituted basement membranes. Biochimica et Biophysica Acta, 1178(1), 55–62. https://doi.org/10.1016/0167-4889(93)90109-3
Glunde, K., Guggino, S. E., Solaiyappan, M., Pathak, A. P., Ichikawa, Y., & Bhujwalla, Z. M. (2003). Extracellular acidification alters lysosomal trafficking in human breast cancer cells. Neoplasia, 5(6), 533–545. https://doi.org/10.1016/s1476-5586(03)80037-4
Lau, A. N., & Heiden, M. G. V. (2020). Metabolism in the tumor microenvironment. Annual Review of Cancer Biology, 4(1), 17–40. https://doi.org/10.1146/annurev-cancerbio-030419-033333
Chinnaiyan, P., Kensicki, E., Bloom, G., Prabhu, A., Sarcar, B., Kahali, S., et al. (2012). The metabolomic signature of malignant glioma reflects accelerated anabolic metabolism. Cancer Research, 72(22), 5878–5888. https://doi.org/10.1158/0008-5472.Can-12-1572-t
Shen, D., Tian, L., Yang, F., Li, J., Li, X., Yao, Y., et al. (2021). ADO/hypotaurine: A novel metabolic pathway contributing to glioblastoma development. Cell Death Discov, 7(1), 21. https://doi.org/10.1038/s41420-020-00398-5
Venkateswaran, N., Lafita-Navarro, M. C., Hao, Y. H., Kilgore, J. A., Perez-Castro, L., Braverman, J., et al. (2019). MYC promotes tryptophan uptake and metabolism by the kynurenine pathway in colon cancer. Genes & Development, 33(17–18), 1236–1251. https://doi.org/10.1101/gad.327056.119
Leone, A., Nigro, C., Nicolò, A., Prevenzano, I., Formisano, P., Beguinot, F., et al. (2021). The dual-role of methylglyoxal in tumor progression – Novel therapeutic approaches. [Review]. Frontiers in Oncology, 11(822), https://doi.org/10.3389/fonc.2021.645686.
Karta, J., Bossicard, Y., Kotzamanis, K., Dolznig, H., & Letellier, E. (2021). Mapping the metabolic networks of tumor cells and cancer-associated fibroblasts. Cells, 10(2), 304.
Benej, M., Svastova, E., Banova, R., Kopacek, J., Gibadulinova, A., Kery, M., et al. (2020). CA IX stabilizes intracellular pH to maintain metabolic reprogramming and proliferation in hypoxia. [Original Research]. Frontiers in Oncology, 10(1462), https://doi.org/10.3389/fonc.2020.01462.
Gillies, R. J. (2021). Cancer heterogeneity and metastasis: Life at the edge. Clinical & Experimental Metastasis. https://doi.org/10.1007/s10585-021-10101-2
Keibler, M. A., Wasylenko, T. M., Kelleher, J. K., Iliopoulos, O., Vander Heiden, M. G., & Stephanopoulos, G. (2016). Metabolic requirements for cancer cell proliferation. Cancer & Metabolism, 4(1), 16. https://doi.org/10.1186/s40170-016-0156-6
DeBerardinis, R. J., & Chandel, N. S. (2016). Fundamentals of cancer metabolism. Sci Adv, 2(5), e1600200. https://doi.org/10.1126/sciadv.1600200
Zhu, J., & Thompson, C. B. (2019). Metabolic regulation of cell growth and proliferation. Nature Reviews Molecular Cell Biology, 20(7), 436–450. https://doi.org/10.1038/s41580-019-0123-5
Lane, A. N., Higashi, R. M., & Fan, T. W. M. (2019). Metabolic reprogramming in tumors: Contributions of the tumor microenvironment. Genes & diseases, 7(2), 185–198. https://doi.org/10.1016/j.gendis.2019.10.007
Lane, A., Higashi, R., & Fan, T. (2019). Metabolic reprogramming in tumors: Contributions of the tumor microenvironment. Genes & diseases, 7, https://doi.org/10.1016/j.gendis.2019.10.007
Huang, Z., Zhang, M., Plec, A. A., Estill, S. J., Cai, L., Repa, J. J., et al. (2018). ACSS2 promotes systemic fat storage and utilization through selective regulation of genes involved in lipid metabolism. Proc Natl Acad Sci U S A, 115(40), E9499-e9506. https://doi.org/10.1073/pnas.1806635115
Gullino, P. M., Clark, S. H., & Grantham, F. H. (1964). The interstitial fluid of solid tumors. Cancer Research, 24, 780–794.
Angelin, A., Gil-de-Gómez, L., Dahiya, S., Jiao, J., Guo, L., Levine, M. H., et al. (2017). Foxp3 reprograms T cell metabolism to function in low-glucose, high-lactate environments. Cell Metabolism, 25(6), 1282-1293.e1287. https://doi.org/10.1016/j.cmet.2016.12.018
Chang, C. H., Qiu, J., O’Sullivan, D., Buck, M. D., Noguchi, T., Curtis, J. D., et al. (2015). Metabolic competition in the tumor microenvironment is a driver of cancer progression. Cell, 162(6), 1229–1241. https://doi.org/10.1016/j.cell.2015.08.016
Sousa, C. M., Biancur, D. E., Wang, X., Halbrook, C. J., Sherman, M. H., Zhang, L., et al. (2016). Pancreatic stellate cells support tumour metabolism through autophagic alanine secretion. Nature, 536(7617), 479–483. https://doi.org/10.1038/nature19084
Payen, V. L., Mina, E., Van Hée, V. F., Porporato, P. E., & Sonveaux, P. (2020). Monocarboxylate transporters in cancer. Mol Metab, 33, 48–66. https://doi.org/10.1016/j.molmet.2019.07.006
Hosios, A. M., Hecht, V. C., Danai, L. V., Johnson, M. O., Rathmell, J. C., Steinhauser, M. L., et al. (2016). Amino acids rather than glucose account for the majority of cell mass in proliferating mammalian cells. Developmental Cell, 36(5), 540–549. https://doi.org/10.1016/j.devcel.2016.02.012
Valencia, T., Kim, J. Y., Abu-Baker, S., Moscat-Pardos, J., Ahn, C. S., Reina-Campos, M., et al. (2014). Metabolic reprogramming of stromal fibroblasts through p62-mTORC1 signaling promotes inflammation and tumorigenesis. Cancer Cell, 26(1), 121–135. https://doi.org/10.1016/j.ccr.2014.05.004
Su, H., Yang, F., Sun, B., & Karin, M. (2021). Macropinocytosis: The big drinker behind cancer cell self-consumption. Autophagy, 17(5), 1290–1291. https://doi.org/10.1080/15548627.2021.1919969
Meyer, K. A., Neeley, C. K., Baker, N. A., Washabaugh, A. R., Flesher, C. G., Nelson, B. S., et al. (2016). Adipocytes promote pancreatic cancer cell proliferation via glutamine transfer. Biochem Biophys Rep, 7, 144–149. https://doi.org/10.1016/j.bbrep.2016.06.004
Cotte, A. K., Aires, V., Fredon, M., Limagne, E., Derangère, V., Thibaudin, M., et al. (2018). Lysophosphatidylcholine acyltransferase 2-mediated lipid droplet production supports colorectal cancer chemoresistance. Nature Communications, 9(1), 322. https://doi.org/10.1038/s41467-017-02732-5
Ladanyi, A., Mukherjee, A., Kenny, H. A., Johnson, A., Mitra, A. K., Sundaresan, S., et al. (2018). Adipocyte-induced CD36 expression drives ovarian cancer progression and metastasis. Oncogene, 37(17), 2285–2301. https://doi.org/10.1038/s41388-017-0093-z
Rajesh, Y., & Sarkar, D. (2021). Association of adipose tissue and adipokines with development of obesity-induced liver cancer. Int J Mol Sci, 22(4), https://doi.org/10.3390/ijms22042163.
Menard, J. A., Cerezo-Magaña, M., & Belting, M. (2018). Functional role of extracellular vesicles and lipoproteins in the tumour microenvironment. Philos Trans R Soc Lond B Biol Sci, 373(1737), https://doi.org/10.1098/rstb.2016.0480.
Christianson, H. C., & Belting, M. (2014). Heparan sulfate proteoglycan as a cell-surface endocytosis receptor. Matrix Biology, 35, 51–55. https://doi.org/10.1016/j.matbio.2013.10.004
Gyukity-Sebestyén, E., Harmati, M., Dobra, G., Németh, I. B., Mihály, J., Zvara, Á., et al. (2019). Melanoma-derived exosomes induce PD-1 overexpression and tumor progression via mesenchymal stem cell oncogenic reprogramming. Frontiers in Immunology, 10, 2459. https://doi.org/10.3389/fimmu.2019.02459
Peppicelli, S., Bianchini, F., Torre, E., & Calorini, L. (2014). Contribution of acidic melanoma cells undergoing epithelial-to-mesenchymal transition to aggressiveness of non-acidic melanoma cells. Clinical & Experimental Metastasis, 31(4), 423–433. https://doi.org/10.1007/s10585-014-9637-6
Elia, I., & Haigis, M. C. (2021). Metabolites and the tumour microenvironment: From cellular mechanisms to systemic metabolism. Nature Metabolism, 3(1), 21–32. https://doi.org/10.1038/s42255-020-00317-z
Seth Nanda, C., Venkateswaran, S. V., Patani, N., & Yuneva, M. (2020). Defining a metabolic landscape of tumours: Genome meets metabolism. British Journal of Cancer, 122(2), 136–149. https://doi.org/10.1038/s41416-019-0663-7
Lambert, A. W., & Weinberg, R. A. (2021). Linking EMT programmes to normal and neoplastic epithelial stem cells. Nature Reviews Cancer, 21(5), 325–338. https://doi.org/10.1038/s41568-021-00332-6
Takahashi, K., & Yamanaka, S. (2006). Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell, 126(4), 663–676. https://doi.org/10.1016/j.cell.2006.07.024
Patel, M., & Yang, S. (2010). Advances in reprogramming somatic cells to induced pluripotent stem cells. Stem Cell Rev Rep, 6(3), 367–380. https://doi.org/10.1007/s12015-010-9123-8
Pastushenko, I., & Blanpain, C. (2019). EMT transition states during tumor progression and metastasis. Trends in Cell Biology, 29(3), 212–226. https://doi.org/10.1016/j.tcb.2018.12.001
Brabletz, T., Kalluri, R., Nieto, M. A., & Weinberg, R. A. (2018). EMT in cancer. Nature Reviews Cancer, 18(2), 128–134. https://doi.org/10.1038/nrc.2017.118
Stone, R. C., Pastar, I., Ojeh, N., Chen, V., Liu, S., Garzon, K. I., et al. (2016). Epithelial-mesenchymal transition in tissue repair and fibrosis. Cell and Tissue Research, 365(3), 495–506. https://doi.org/10.1007/s00441-016-2464-0
Kovacic, J. C., Mercader, N., Torres, M., Boehm, M., & Fuster, V. (2012). Epithelial-to-mesenchymal and endothelial-to-mesenchymal transition: From cardiovascular development to disease. Circulation, 125(14), 1795–1808. https://doi.org/10.1161/circulationaha.111.040352
Pastushenko, I., Brisebarre, A., Sifrim, A., Fioramonti, M., Revenco, T., Boumahdi, S., et al. (2018). Identification of the tumour transition states occurring during EMT. Nature, 556(7702), 463–468. https://doi.org/10.1038/s41586-018-0040-3
Yu, M., Bardia, A., Wittner, B. S., Stott, S. L., Smas, M. E., Ting, D. T., et al. (2013). Circulating breast tumor cells exhibit dynamic changes in epithelial and mesenchymal composition. Science, 339(6119), 580–584. https://doi.org/10.1126/science.1228522
Shibue, T., & Weinberg, R. A. (2017). EMT, CSCs, and drug resistance: The mechanistic link and clinical implications. Nature Reviews. Clinical Oncology, 14(10), 611–629. https://doi.org/10.1038/nrclinonc.2017.44
Bornes, L., Belthier, G., & van Rheenen, J. (2021). Epithelial-to-mesenchymal transition in the light of plasticity and hybrid E/M states. J Clin Med, 10(11), https://doi.org/10.3390/jcm10112403.
Varga, J., & Greten, F. R. (2017). Cell plasticity in epithelial homeostasis and tumorigenesis. Nature Cell Biology, 19(10), 1133–1141. https://doi.org/10.1038/ncb3611
Phan, T. G., & Croucher, P. I. (2020). The dormant cancer cell life cycle. Nature Reviews Cancer, 20(7), 398–411. https://doi.org/10.1038/s41568-020-0263-0
Holmgren, L., O’Reilly, M. S., & Folkman, J. (1995). Dormancy of micrometastases: Balanced proliferation and apoptosis in the presence of angiogenesis suppression. Nature Medicine, 1(2), 149–153. https://doi.org/10.1038/nm0295-149
Pan, H., Gray, R., Braybrooke, J., Davies, C., Taylor, C., McGale, P., et al. (2017). 20-year risks of breast-cancer recurrence after stopping endocrine therapy at 5 years. New England Journal of Medicine, 377(19), 1836–1846. https://doi.org/10.1056/NEJMoa1701830
The spread of tumours in the human body (1953). Postgraduate Medical Journal, 29(329), 160–160.
Kim, W. T., & Ryu, C. J. (2017). Cancer stem cell surface markers on normal stem cells. BMB Reports, 50(6), 285–298. https://doi.org/10.5483/bmbrep.2017.50.6.039
Zhao, W., Li, Y., & Zhang, X. (2017). Stemness-related markers in cancer. Cancer Transl Med, 3(3), 87–95. https://doi.org/10.4103/ctm.ctm_69_16
Zhang, X., Powell, K., & Li, L. (2020). Breast cancer stem cells: Biomarkers, identification and isolation methods, regulating mechanisms, cellular origin, and beyond. Cancers (Basel), 12(12), https://doi.org/10.3390/cancers12123765.
Talukdar, S., Bhoopathi, P., Emdad, L., Das, S., Sarkar, D., & Fisher, P. B. (2019). Dormancy and cancer stem cells: An enigma for cancer therapeutic targeting. Advances in Cancer Research, 141, 43–84. https://doi.org/10.1016/bs.acr.2018.12.002
Sotgia, F., Fiorillo, M., & Lisanti, M. P. (2019). Hallmarks of the cancer cell of origin: Comparisons with "energetic" cancer stem cells (e-CSCs). Aging, 11(3), 1065–1068, https://doi.org/10.18632/aging.101822.
Yang, M.-C., Wang, H.-C., Hou, Y.-C., Tung, H.-L., Chiu, T.-J., & Shan, Y.-S. (2015). Blockade of autophagy reduces pancreatic cancer stem cell activity and potentiates the tumoricidal effect of gemcitabine. Molecular Cancer, 14(1), 179. https://doi.org/10.1186/s12943-015-0449-3
Seyfried, T. N., & Chinopoulos, C. (2021). Can the mitochondrial metabolic theory explain better the origin and management of cancer than can the somatic mutation theory? Metabolites, 11(9), 572. https://doi.org/10.3390/metabo11090572
Yasuda, T., Ishimoto, T., & Baba, H. (2021). Conflicting metabolic alterations in cancer stem cells and regulation by the stromal niche. Regenerative Therapy, 17, 8–12. https://doi.org/10.1016/j.reth.2021.01.005
Sancho, P., Barneda, D., & Heeschen, C. (2016). Hallmarks of cancer stem cell metabolism. British Journal of Cancer, 114(12), 1305–1312. https://doi.org/10.1038/bjc.2016.152
Yadav, U. P., Singh, T., Kumar, P., Sharma, P., Kaur, H., Sharma, S., et al. (2020). Metabolic adaptations in cancer stem cells. Frontiers in Oncology, 10, 1010. https://doi.org/10.3389/fonc.2020.01010
Jagust, P., de Luxán-Delgado, B., Parejo-Alonso, B., & Sancho, P. (2019). Metabolism-based therapeutic strategies targeting cancer stem cells. Frontiers in pharmacology, 10, 203–203. https://doi.org/10.3389/fphar.2019.00203
de Beauchamp, L., Himonas, E., & Helgason, G. V. (2021). Mitochondrial metabolism as a potential therapeutic target in myeloid leukaemia. Leukemia. https://doi.org/10.1038/s41375-021-01416-w
Bighetti-Trevisan, R. L., Sousa, L. O., Castilho, R. M., & Almeida, L. O. (2019). Cancer stem cells: Powerful targets to improve current anticancer therapeutics. Stem Cells Int, 2019, 9618065. https://doi.org/10.1155/2019/9618065
Martinez-Outschoorn, U., Sotgia, F., & Lisanti, M. P. (2014). Tumor microenvironment and metabolic synergy in breast cancers: Critical importance of mitochondrial fuels and function. Seminars in Oncology, 41(2), 195–216. https://doi.org/10.1053/j.seminoncol.2014.03.002
Wan Kamarul Zaman, W. S., Nurul, A. A., & Nordin, F. (2021). Stem cells and cancer stem cells: The Jekyll and Hyde scenario and their implications in stem cell therapy. Biomedicines, 9(9), 1245
Bonnay, F., Veloso, A., Steinmann, V., Köcher, T., Abdusselamoglu, M. D., Bajaj, S., et al. (2020). Oxidative metabolism drives immortalization of neural stem cells during tumorigenesis. Cell, 182(6), 1490-1507.e1419. https://doi.org/10.1016/j.cell.2020.07.039
Corbet, C. (2017). Stem cell metabolism in cancer and healthy tissues: Pyruvate in the limelight. Frontiers in pharmacology, 8, 958. https://doi.org/10.3389/fphar.2017.00958
De Francesco, E. M., Sotgia, F., & Lisanti, M. P. (2018). Cancer stem cells (CSCs): Metabolic strategies for their identification and eradication. The Biochemical Journal, 475(9), 1611–1634. https://doi.org/10.1042/bcj20170164
Pacifico, F., Badolati, N., Mellone, S., Stornaiuolo, M., Leonardi, A., & Crescenzi, E. (2021). Glutamine promotes escape from therapy-induced senescence in tumor cells. Aging, 13(17), 20962-20991, https://doi.org/10.18632/aging.203495
Loureiro, R., Mesquita, K. A., Magalhães-Novais, S., Oliveira, P. J., & Vega-Naredo, I. (2017). Mitochondrial biology in cancer stem cells. Seminars in Cancer Biology, 47, 18–28. https://doi.org/10.1016/j.semcancer.2017.06.012
Amend, S. R., & Pienta, K. J. (2015). Ecology meets cancer biology: The cancer swamp promotes the lethal cancer phenotype. Oncotarget, 6(12), 9669–9678, https://doi.org/10.18632/oncotarget.3430.
McGranahan, N., & Swanton, C. (2017). Clonal heterogeneity and tumor evolution: Past, present, and the future. Cell, 168(4), 613–628. https://doi.org/10.1016/j.cell.2017.01.018
Yu, L., Lu, M., Jia, D., Ma, J., Ben-Jacob, E., Levine, H., et al. (2017). Modeling the genetic regulation of cancer metabolism: Interplay between glycolysis and oxidative phosphorylation. Cancer Research, 77(7), 1564–1574. https://doi.org/10.1158/0008-5472.Can-16-2074
Vander Linden, C., & Corbet, C. (2020). Reconciling environment-mediated metabolic heterogeneity with the oncogene-driven cancer paradigm in precision oncology. Seminars in Cell & Developmental Biology, 98, 202–210. https://doi.org/10.1016/j.semcdb.2019.05.016
Enriquez-Navas, P. M., Wojtkowiak, J. W., & Gatenby, R. A. (2015). Application of evolutionary principles to cancer therapy. Cancer Research, 75(22), 4675–4680. https://doi.org/10.1158/0008-5472.Can-15-1337
Gillies, R. J., Verduzco, D., & Gatenby, R. A. (2012). Evolutionary dynamics of carcinogenesis and why targeted therapy does not work. Nature Reviews Cancer, 12(7), 487–493. https://doi.org/10.1038/nrc3298
Meads, M. B., Gatenby, R. A., & Dalton, W. S. (2009). Environment-mediated drug resistance: A major contributor to minimal residual disease. Nature Reviews Cancer, 9(9), 665–674. https://doi.org/10.1038/nrc2714
Romeo, V., Accardo, G., Perillo, T., Basso, L., Garbino, N., Nicolai, E., et al. (2021). Assessment and prediction of response to neoadjuvant chemotherapy in breast cancer: A comparison of imaging modalities and future perspectives. Cancers (Basel), 13(14), https://doi.org/10.3390/cancers13143521.
Sengupta, D., & Pratx, G. (2016). Imaging metabolic heterogeneity in cancer. Molecular Cancer, 15(1), 4. https://doi.org/10.1186/s12943-015-0481-3
Jia, D., Park, J. H., Jung, K. H., Levine, H., & Kaipparettu, B. A. (2018). Elucidating the metabolic plasticity of cancer: Mitochondrial reprogramming and hybrid metabolic states. Cells, 7(3), https://doi.org/10.3390/cells7030021.
Paudel, B. B., & Quaranta, V. (2019). Metabolic plasticity meets gene regulation. Proc Natl Acad Sci U S A, 116(9), 3370–3372. https://doi.org/10.1073/pnas.1900169116
Jia, D., Lu, M., Jung, K. H., Park, J. H., Yu, L., Onuchic, J. N., et al. (2019). Elucidating cancer metabolic plasticity by coupling gene regulation with metabolic pathways. Proc Natl Acad Sci U S A, 116(9), 3909–3918. https://doi.org/10.1073/pnas.1816391116
Petővári, G., Dankó, T., Tőkés, A.-M., Vetlényi, E., Krencz, I., Raffay, R., et al. (2020). In situ metabolic characterisation of breast cancer and its potential impact on therapy. Cancers, 12(9), 2492.
Angione, C. (2019). Human systems biology and metabolic modelling: A review-from disease metabolism to precision medicine. BioMed Research International, 2019, 8304260. https://doi.org/10.1155/2019/8304260
Di Filippo, M., Colombo, R., Damiani, C., Pescini, D., Gaglio, D., Vanoni, M., et al. (2016). Zooming-in on cancer metabolic rewiring with tissue specific constraint-based models. Computational Biology and Chemistry, 62, 60–69. https://doi.org/10.1016/j.compbiolchem.2016.03.002
Yizhak, K., Le Dévédec, S. E., Rogkoti, V. M., Baenke, F., de Boer, V. C., Frezza, C et al (2014) A computational study of the Warburg effect identifies metabolic targets inhibiting cancer migration. Mol Syst Biol, 10 (8) 744
Uhlen, M., Zhang, C., Lee, S., Sjöstedt, E., Fagerberg, L., Bidkhori, G., et al. (2017). A pathology atlas of the human cancer transcriptome. Science, 357(6352), https://doi.org/10.1126/science.aan2507.
Jalili, M., Scharm, M., Wolkenhauer, O., Damaghi, M., & Salehzadeh-Yazdi, A. (2021). Exploring the metabolic heterogeneity of cancers: A benchmark study of context-specific models. J Pers Med, 11(6), https://doi.org/10.3390/jpm11060496.
Barron, C. C., Bilan, P. J., Tsakiridis, T., & Tsiani, E. (2016). Facilitative glucose transporters: Implications for cancer detection, prognosis and treatment. Metabolism, 65(2), 124–139. https://doi.org/10.1016/j.metabol.2015.10.007
Ancey, P. B., Contat, C., & Meylan, E. (2018). Glucose transporters in cancer - From tumor cells to the tumor microenvironment. FEBS Journal, 285(16), 2926–2943. https://doi.org/10.1111/febs.14577
Martinez, C. A., & Scafoglio, C. (2020). Heterogeneity of glucose transport in lung cancer. Biomolecules, 10(6), https://doi.org/10.3390/biom10060868.
Reckzeh, E. S., & Waldmann, H. (2020). Development of glucose transporter (GLUT) inhibitors. European journal of organic chemistry, 2020(16), 2321–2329. https://doi.org/10.1002/ejoc.201901353
Park, S. J., Smith, C. P., Wilbur, R. R., Cain, C. P., Kallu, S. R., Valasapalli, S., et al. (2018). An overview of MCT1 and MCT4 in GBM: Small molecule transporters with large implications. American journal of cancer research, 8(10), 1967–1976.
Kim, Y., Choi, J. W., Lee, J. H., & Kim, Y. S. (2015). Expression of lactate/H+ symporters MCT1 and MCT4 and their chaperone CD147 predicts tumor progression in clear cell renal cell carcinoma: Immunohistochemical and the Cancer Genome Atlas data analyses. Human Pathology, 46(1), 104–112. https://doi.org/10.1016/j.humpath.2014.09.013
Zhang, G., Zhang, Y., Dong, D., Wang, F., Ma, X., Guan, F., et al. (2018). MCT1 regulates aggressive and metabolic phenotypes in bladder cancer. [Research Paper]. Journal of Cancer, 9(14), 2492–2501, https://doi.org/10.7150/jca.25257.
Latif, A., Chadwick, A. L., Kitson, S. J., Gregson, H. J., Sivalingam, V. N., Bolton, J., et al. (2017). Monocarboxylate Transporter 1 (MCT1) is an independent prognostic biomarker in endometrial cancer. BMC clinical pathology, 17, 27–27. https://doi.org/10.1186/s12907-017-0067-7
Simões-Sousa, S., Granja, S., Pinheiro, C., Fernandes, D., Longatto-Filho, A., Laus, A. C., et al. (2016). Prognostic significance of monocarboxylate transporter expression in oral cavity tumors. Cell cycle (Georgetown, Tex.), 15(14), 1865–1873, https://doi.org/10.1080/15384101.2016.1188239.
Nakayama, Y., Torigoe, T., Inoue, Y., Minagawa, N., Izumi, H., Kohno, K., et al. (2012). Prognostic significance of monocarboxylate transporter 4 expression in patients with colorectal cancer. Experimental and therapeutic medicine, 3(1), 25–30. https://doi.org/10.3892/etm.2011.361
Zhu, J., Wu, Y. N., Zhang, W., Zhang, X. M., Ding, X., Li, H. Q., et al. (2014). Monocarboxylate transporter 4 facilitates cell proliferation and migration and is associated with poor prognosis in oral squamous cell carcinoma patients. PLoS ONE, 9(1), e87904. https://doi.org/10.1371/journal.pone.0087904
Pértega-Gomes, N., Vizcaíno, J. R., Miranda-Gonçalves, V., Pinheiro, C., Silva, J., Pereira, H., et al. (2011). Monocarboxylate transporter 4 (MCT4) and CD147 overexpression is associated with poor prognosis in prostate cancer. BMC Cancer, 11, 312–312. https://doi.org/10.1186/1471-2407-11-312
Ruan, Y., Zeng, F., Cheng, Z., Zhao, X., Fu, P., & Chen, H. (2017). High expression of monocarboxylate transporter 4 predicts poor prognosis in patients with lung adenocarcinoma. Oncology letters, 14(5), 5727–5734. https://doi.org/10.3892/ol.2017.6964
Kobayashi, M. (2015). Role of monocarboxylate transporter in statin-induced cytotoxicity. Yakugaku Zasshi, 135(11), 1227–1233. https://doi.org/10.1248/yakushi.15-00199
Aldera, A., & Govender, D. (2021). Carbonic anhydrase IX: A regulator of pH and participant in carcinogenesis. Journal of Clinical Pathology, 74, 1–5. https://doi.org/10.1136/jclinpath-2020-207073
Szendrői, A., Szász, A. M., Kardos, M., Tőkés, A.-M., Idan, R., Szűcs, M., et al. (2016). Opposite prognostic roles of HIF1α and HIF2α expressions in bone metastatic clear cell renal cell cancer. Oncotarget, 7(27), 42086–42098, https://doi.org/10.18632/oncotarget.9669.
Lou, Y., McDonald, P. C., Oloumi, A., Chia, S., Ostlund, C., Ahmadi, A., et al. (2011). Targeting tumor hypoxia: Suppression of breast tumor growth and metastasis by novel carbonic anhydrase IX inhibitors. Cancer Research, 71(9), 3364–3376. https://doi.org/10.1158/0008-5472.Can-10-4261
Ilie, M., Mazure, N. M., Hofman, V., Ammadi, R. E., Ortholan, C., Bonnetaud, C., et al. (2010). High levels of carbonic anhydrase IX in tumour tissue and plasma are biomarkers of poor prognostic in patients with non-small cell lung cancer. British Journal of Cancer, 102(11), 1627–1635. https://doi.org/10.1038/sj.bjc.6605690
Choschzick, M., Oosterwijk, E., Müller, V., Woelber, L., Simon, R., Moch, H., et al. (2011). Overexpression of carbonic anhydrase IX (CAIX) is an independent unfavorable prognostic marker in endometrioid ovarian cancer. Virchows Archiv, 459(2), 193–200. https://doi.org/10.1007/s00428-011-1105-y
Klatte, T., Seligson, D. B., Rao, J. Y., Yu, H., de Martino, M., Kawaoka, K., et al. (2009). Carbonic anhydrase IX in bladder cancer: A diagnostic, prognostic, and therapeutic molecular marker. Cancer, 115(7), 1448–1458. https://doi.org/10.1002/cncr.24163
Eckert, A. W., Horter, S., Bethmann, D., Kotrba, J., Kaune, T., Rot, S., et al. (2019). Investigation of the prognostic role of carbonic anhydrase 9 (CAIX) of the cellular mRNA/protein level or soluble CAIX protein in patients with oral squamous cell carcinoma. International journal of molecular sciences, 20(2), 375. https://doi.org/10.3390/ijms20020375
Cetin, B., Gonul, I. I., Gumusay, O., Bilgetekin, I., Algin, E., Ozet, A., et al. (2018). Carbonic anhydrase IX is a prognostic biomarker in glioblastoma multiforme. Neuropathology, 38(5), 457–462. https://doi.org/10.1111/neup.12485
Srivastava, S., Thakkar, B., Yeoh, K. G., Ho, K. Y., Teh, M., Soong, R., et al. (2015). Expression of proteins associated with hypoxia and Wnt pathway activation is of prognostic significance in hepatocellular carcinoma. Virchows Archiv, 466(5), 541–548. https://doi.org/10.1007/s00428-015-1745-4
Afonso, J., Santos, L. L., Morais, A., Amaro, T., Longatto-Filho, A., & Baltazar, F. (2016). Metabolic coupling in urothelial bladder cancer compartments and its correlation to tumor aggressiveness. Cell Cycle, 15(3), 368–380. https://doi.org/10.1080/15384101.2015.1121329
Marchiq, I., Le Floch, R., Roux, D., Simon, M. P., & Pouyssegur, J. (2015). Genetic disruption of lactate/H+ symporters (MCTs) and their subunit CD147/BASIGIN sensitizes glycolytic tumor cells to phenformin. Cancer Research, 75(1), 171–180. https://doi.org/10.1158/0008-5472.Can-14-2260
Benjamin, D., Robay, D., Hindupur, S. K., Pohlmann, J., Colombi, M., El-Shemerly, M. Y., et al. (2018). Dual inhibition of the lactate transporters MCT1 and MCT4 is synthetic lethal with metformin due to NAD+ depletion in cancer cells. Cell Reports, 25(11), 3047-3058.e3044. https://doi.org/10.1016/j.celrep.2018.11.043
Carta, F., Vullo, D., Osman, S. M., AlOthman, Z., & Supuran, C. T. (2017). Synthesis and carbonic anhydrase inhibition of a series of SLC-0111 analogs. Bioorganic & Medicinal Chemistry, 25(9), 2569–2576. https://doi.org/10.1016/j.bmc.2017.03.027
Vettore, L., Westbrook, R. L., & Tennant, D. A. (2020). New aspects of amino acid metabolism in cancer. British Journal of Cancer, 122(2), 150–156. https://doi.org/10.1038/s41416-019-0620-5
Lieu, E. L., Nguyen, T., Rhyne, S., & Kim, J. (2020). Amino acids in cancer. Experimental & Molecular Medicine, 52(1), 15–30. https://doi.org/10.1038/s12276-020-0375-3
Bernfeld, E., & Foster, D. A. (2019). Glutamine as an essential amino acid for KRas-driven cancer cells. Trends in Endocrinology and Metabolism, 30(6), 357–368. https://doi.org/10.1016/j.tem.2019.03.003
Bhutia, Y. D., Babu, E., Ramachandran, S., & Ganapathy, V. (2015). Amino acid transporters in cancer and their relevance to “glutamine addiction”: Novel targets for the design of a new class of anticancer drugs. Cancer Research, 75(9), 1782–1788. https://doi.org/10.1158/0008-5472.Can-14-3745
Osanai-Sasakawa, A., Hosomi, K., Sumitomo, Y., Takizawa, T., Tomura-Suruki, S., Imaizumi, M., et al. (2018). An anti-ASCT2 monoclonal antibody suppresses gastric cancer growth by inducing oxidative stress and antibody dependent cellular toxicity in preclinical models. American journal of cancer research, 8(8), 1499–1513.
Saarinen, I., Jambor, I., Kim, M., Kuisma, A., Kemppainen, J., Merisaari, H., et al. (2019). Correlation between 18F-1-amino-3-fluorocyclobutane-1-carboxylic acid (18F-fluciclovine) uptake and expression of alanine-serine-cysteine-transporter 2 (ASCT2) and L-type amino acid transporter 1 (LAT1) in primary prostate cancer. EJNMMI Research, 9, https://doi.org/10.1186/s13550-019-0518-5
Bröer, A., Fairweather, S., & Bröer, S. (2018). Disruption of amino acid homeostasis by novel ASCT2 inhibitors involves multiple targets. Frontiers in pharmacology, 9, 785. https://doi.org/10.3389/fphar.2018.00785
Wei, Z., Liu, X., Cheng, C., Yu, W., & Yi, P. (2021). Metabolism of amino acids in cancer. Frontiers in cell and developmental biology, 8, 603837–603837. https://doi.org/10.3389/fcell.2020.603837
Sivanand, S., & Vander Heiden, M. G. (2020). Emerging roles for branched-chain amino acid metabolism in cancer. Cancer Cell, 37(2), 147–156. https://doi.org/10.1016/j.ccell.2019.12.011
Satoh, K., Yachida, S., Sugimoto, M., Oshima, M., Nakagawa, T., Akamoto, S., et al. (2017). Global metabolic reprogramming of colorectal cancer occurs at adenoma stage and is induced by MYC. Proc Natl Acad Sci U S A, 114(37), E7697-e7706. https://doi.org/10.1073/pnas.1710366114
Okano, N., Hana, K., Naruge, D., Kawai, K., Kobayashi, T., Nagashima, F., et al. (2020). Biomarker analyses in patients with advanced solid tumors treated with the LAT1 inhibitor JPH203. In vivo (Athens, Greece), 34(5), 2595–2606, https://doi.org/10.21873/invivo.12077.
Krys, D., Mattingly, S., Glubrecht, D., Wuest, M., & Wuest, F. (2020). PET imaging of L-type amino acid transporter (LAT1) and cystine-glutamate antiporter (x(c)(-)) with [(18)F]FDOPA and [(18)F]FSPG in breast cancer models. Molecular Imaging and Biology, 22(6), 1562–1571. https://doi.org/10.1007/s11307-020-01529-1
Malviya, G., Patel, R., Salji, M., Martinez, R. S., Repiscak, P., Mui, E., et al. (2020). 18F-Fluciclovine PET metabolic imaging reveals prostate cancer tumour heterogeneity associated with disease resistance to androgen deprivation therapy. EJNMMI research, 10(1), 143–143. https://doi.org/10.1186/s13550-020-00728-9
Koundouros, N., & Poulogiannis, G. (2020). Reprogramming of fatty acid metabolism in cancer. British Journal of Cancer, 122(1), 4–22. https://doi.org/10.1038/s41416-019-0650-z
Kubo, M., Gotoh, K., Eguchi, H., Kobayashi, S., Iwagami, Y., Tomimaru, Y., et al. (2019). Impact of CD36 on chemoresistance in pancreatic ductal adenocarcinoma. Annals of Surgical Oncology, 27, 610–619.
Nath, A., Li, I., Roberts, L. R., & Chan, C. (2015). Elevated free fatty acid uptake via CD36 promotes epithelial-mesenchymal transition in hepatocellular carcinoma. Science and Reports, 5, 14752. https://doi.org/10.1038/srep14752
Tanase, C., Gheorghisan-Galateanu, A.-A., Popescu, I. D., Mihai, S., Codrici, E., Albulescu, R., et al. (2020). CD36 and CD97 in pancreatic cancer versus other malignancies. International journal of molecular sciences, 21(16), 5656. https://doi.org/10.3390/ijms21165656
Mukherjee, A., Chiang, C.-Y., Daifotis, H. A., Nieman, K. M., Fahrmann, J. F., Lastra, R. R., et al. (2020). Adipocyte-induced FABP4 expression in ovarian cancer cells promotes metastasis and mediates carboplatin resistance. Cancer Research, 80(8), 1748–1761. https://doi.org/10.1158/0008-5472.Can-19-1999
Park, J., Euhus, D. M., & Scherer, P. E. (2011). Paracrine and endocrine effects of adipose tissue on cancer development and progression. Endocrine reviews, 32(4), 550–570. https://doi.org/10.1210/er.2010-0030
Fang, D., & Maldonado, E. N. (2018). VDAC regulation: A mitochondrial target to stop cell proliferation. Advances in Cancer Research, 138, 41–69. https://doi.org/10.1016/bs.acr.2018.02.002
Röhrig, F., & Schulze, A. (2016). The multifaceted roles of fatty acid synthesis in cancer. Nature Reviews Cancer, 16(11), 732–749. https://doi.org/10.1038/nrc.2016.89
Chen, Y.-J., Liao, W.-X., Huang, S.-Z., Yu, Y.-F., Wen, J.-Y., Chen, J., et al. (2021). Prognostic and immunological role of CD36: A pan-cancer analysis. [Research Paper]. Journal of Cancer, 12(16), 4762–4773, https://doi.org/10.7150/jca.50502.
Rochette, L., Meloux, A., Zeller, M., Malka, G., Cottin, Y., & Vergely, C. (2020). Mitochondrial SLC25 carriers: Novel targets for cancer therapy. Molecules (Basel, Switzerland), 25(10), 2417. https://doi.org/10.3390/molecules25102417
Fernandez, H. R., Gadre, S. M., Tan, M., Graham, G., Mosaoa, R., Ongkeko, M. S., et al. (2018). The mitochondrial citrate carrier, SLC25A1, drives stemness and therapy resistance in non-small cell lung cancer. Cell Death and Differentiation, 25, 1239–1258.
Hlouschek, J., Ritter, V., Wirsdörfer, F., Klein, D., Jendrossek, V., & Matschke, J. (2018). Targeting SLC25A10 alleviates improved antioxidant capacity and associated radioresistance of cancer cells induced by chronic-cycling hypoxia. Cancer Letters, 439, 24–38. https://doi.org/10.1016/j.canlet.2018.09.002
Sullivan, L. B., Luengo, A., Danai, L. V., Bush, L. N., Diehl, F. F., Hosios, A. M., et al. (2018). Aspartate is an endogenous metabolic limitation for tumour growth. Nature Cell Biology, 20(7), 782–788. https://doi.org/10.1038/s41556-018-0125-0
Tina, E., Lindqvist, B. M., Gabrielson, M., Lubovac, Z., Wegman, P., & Wingren, S. (2012). The mitochondrial transporter SLC25A43 is frequently deleted and may influence cell proliferation in HER2-positive breast tumors. BMC Cancer, 12, 350–350. https://doi.org/10.1186/1471-2407-12-350
Andersen, S., Donnem, T., Stenvold, H., Al-Saad, S., Al-Shibli, K., Busund, L.-T., et al. (2011). Overexpression of the HIF hydroxylases PHD1, PHD2, PHD3 and FIH are individually and collectively unfavorable prognosticators for NSCLC survival. PLoS ONE, 6(8), e23847. https://doi.org/10.1371/journal.pone.0023847
Xie, X., Huang, X., Tang, H., Ye, F., Yang, L., Guo, X., et al. (2018). Diallyl disulfide inhibits breast cancer stem cell progression and glucose metabolism by targeting CD44/PKM2/AMPK signaling. Current Cancer Drug Targets, 18(6), 592–599. https://doi.org/10.2174/1568009617666171024165657
Li, T.-E., Wang, S., Shen, X.-T., Zhang, Z., Chen, M., Wang, H., et al. (2020). PKM2 drives hepatocellular carcinoma progression by inducing immunosuppressive microenvironment. Frontiers in immunology, 11, 589997–589997. https://doi.org/10.3389/fimmu.2020.589997
Suzuki, A., Puri, S., Leland, P., Puri, A., Moudgil, T., Fox, B. A., et al. (2019). Subcellular compartmentalization of PKM2 identifies anti-PKM2 therapy response in vitro and in vivo mouse model of human non-small-cell lung cancer. PLoS ONE, 14(5), e0217131. https://doi.org/10.1371/journal.pone.0217131
Yan, X. L., Zhang, X. B., Ao, R., & Guan, L. (2017). Effects of shRNA-mediated silencing of PKM2 gene on aerobic glycolysis, cell migration, cell invasion, and apoptosis in colorectal cancer cells. Journal of Cellular Biochemistry, 118(12), 4792–4803. https://doi.org/10.1002/jcb.26148
Duan, K., Gucer, H., Kefeli, M., Asa, S. L., Winer, D. A., & Mete, O. (2020). Immunohistochemical analysis of the metabolic phenotype of adrenal cortical carcinoma. Endocrine Pathology, 31(3), 231–238. https://doi.org/10.1007/s12022-020-09624-3
Wang, Y., Pan, S., He, X., Wang, Y., Huang, H., Chen, J., et al. (2021). CPNE1 enhances colorectal cancer cell growth, glycolysis, and drug resistance through regulating the AKT-GLUT1/HK2 pathway. OncoTargets and therapy, 14, 699–710. https://doi.org/10.2147/OTT.S284211
Liu, W., Yu, X., Zhou, L., Li, J., Li, M., Li, W., et al. (2020). Sinomenine inhibits non-small cell lung cancer via downregulation of hexokinases II-mediated aerobic glycolysis. OncoTargets and therapy, 13, 3209–3221. https://doi.org/10.2147/OTT.S243212
Wang, Y., Nie, H., Liao, Z., He, X., Xu, Z., Zhou, J., et al. (2021). Expression and clinical significance of lactate dehydrogenase a in colon adenocarcinoma. [Original Research]. Frontiers in Oncology, 11(2645), https://doi.org/10.3389/fonc.2021.700795.
Yang, Y., Chong, Y., Chen, M., Dai, W., Zhou, X., Ji, Y., et al. (2021). Targeting lactate dehydrogenase a improves radiotherapy efficacy in non-small cell lung cancer: From bedside to bench. Journal of Translational Medicine, 19(1), 170. https://doi.org/10.1186/s12967-021-02825-2
Mohammad, G. H., Olde Damink, S. W. M., Malago, M., Dhar, D. K., & Pereira, S. P. (2016). Pyruvate kinase M2 and lactate dehydrogenase A are overexpressed in pancreatic cancer and correlate with poor outcome. PLoS ONE, 11(3), e0151635. https://doi.org/10.1371/journal.pone.0151635
Pennington, Z., Goodwin, M. L., Westbroek, E. M., Cottrill, E., Ahmed, A. K., & Sciubba, D. M. (2019). Lactate and cancer: Spinal metastases and potential therapeutic targets (part 2). Annals of translational medicine, 7(10), 221–221, https://doi.org/10.21037/atm.2019.01.85.
Koukourakis, M. I., Giatromanolaki, A., Sivridis, E., Gatter, K. C., & Harris, A. L. (2005). Pyruvate dehydrogenase and pyruvate dehydrogenase kinase expression in non small cell lung cancer and tumor-associated stroma. Neoplasia, 7(1), 1–6. https://doi.org/10.1593/neo.04373
Dumas, J. F., Brisson, L., Chevalier, S., Mahéo, K., Fromont, G., Moussata, D., et al. (2017). Metabolic reprogramming in cancer cells, consequences on pH and tumour progression: Integrated therapeutic perspectives with dietary lipids as adjuvant to anticancer treatment. Seminars in Cancer Biology, 43, 90–110. https://doi.org/10.1016/j.semcancer.2017.03.004
Petővári, G., Dankó, T., Krencz, I., Hujber, Z., Rajnai, H., Vetlényi, E., et al. (2020). Inhibition of metabolic shift can decrease therapy resistance in human high-grade glioma cells. Pathology & Oncology Research, 26(1), 23–33. https://doi.org/10.1007/s12253-019-00677-2
Peterse, E. F. P., Niessen, B., Addie, R. D., de Jong, Y., Cleven, A. H. G., Kruisselbrink, A. B., et al. (2018). Targeting glutaminolysis in chondrosarcoma in context of the IDH1/2 mutation. British Journal of Cancer, 118(8), 1074–1083. https://doi.org/10.1038/s41416-018-0050-9
Bhattacharya, P., & Maity, P. (2000). Localization of phosphate dependent glutaminase in ascites fluid of ovarian cancer patient. Pathology Oncology Research, 6(3), 217–223. https://doi.org/10.1007/bf03032376
Krencz, I., Sebestyen, A., Papay, J., Jeney, A., Hujber, Z., Burger, C. D., et al. (2018). In situ analysis of mTORC1/2 and cellular metabolism-related proteins in human lymphangioleiomyomatosis. Human Pathology, 79, 199–207. https://doi.org/10.1016/j.humpath.2018.05.018
Pan, T., Gao, L., Wu, G., Shen, G., Xie, S., Wen, H., et al. (2015). Elevated expression of glutaminase confers glucose utilization via glutaminolysis in prostate cancer. Biochemical and Biophysical Research Communications, 456(1), 452–458. https://doi.org/10.1016/j.bbrc.2014.11.105
Felkai, L., Krencz, I., Kiss, D. J., Nagy, N., Petővári, G., Dankó, T., et al. (2020). Characterization of mTOR activity and metabolic profile in pediatric rhabdomyosarcoma. Cancers, 12(7), 1947. https://doi.org/10.3390/cancers12071947
Doll, C. M., Winter, K., Gaffney, D. K., Ryu, J. K., Jhingran, A., Dicker, A. P., et al. (2013). COX-2 expression and survival in patients with locally advanced cervical cancer treated with chemoradiotherapy and celecoxib: A quantitative immunohistochemical analysis of RTOG C0128. International Journal of Gynecological Cancer, 23(1), 176–183. https://doi.org/10.1097/IGC.0b013e3182791efc
Honjo, S., Kase, S., Osaki, M., Ardyanto, T. D., Kaibara, N., & Ito, H. (2004). Cyclooxygenase-2 expression in human gastric tubular adenomas and carcinomas; correlation with intratumoral microvessel density and apoptotic index. Anticancer Research, 24(3a), 1439–1444.
Grupp, K., Jedrzejewska, K., Tsourlakis, M. C., Koop, C., Wilczak, W., Adam, M., et al. (2013). High mitochondria content is associated with prostate cancer disease progression. Molecular Cancer, 12(1), 145. https://doi.org/10.1186/1476-4598-12-145
Stein, J., Tenbrock, J., Kristiansen, G., Müller, S. C., & Ellinger, J. (2019). Systematic expression analysis of the mitochondrial respiratory chain protein subunits identifies COX5B as a prognostic marker in clear cell renal cell carcinoma. International Journal of Urology, 26(9), 910–916. https://doi.org/10.1111/iju.14040
Hu, T., & Xi, J. (2017). Identification of COX5B as a novel biomarker in high-grade glioma patients. OncoTargets and therapy, 10, 5463–5470. https://doi.org/10.2147/OTT.S139243
Brüggemann, M., Gromes, A., Poss, M., Schmidt, D., Klümper, N., Tolkach, Y., et al. (2017). Systematic analysis of the expression of the mitochondrial ATP synthase (complex V) subunits in clear cell renal cell carcinoma. Translational Oncology, 10, 661–668.
Kobayashi, M., Funayama, R., Ohnuma, S., Unno, M., & Nakayama, K. (2016). Wnt-β-catenin signaling regulates ABCC3 (MRP3) transporter expression in colorectal cancer. Cancer Science, 107(12), 1776–1784. https://doi.org/10.1111/cas.13097
Gao, Y. X., Chen, L., Hu, X. G., Wu, H. B., Cui, Y. H., Zhang, X., et al. (2016). ATPase inhibitory factor 1 expression is an independent prognostic factor in non-small cell lung cancer. American Journal of Cancer Research, 6(5), 1141–1148.
Aguilar, E., Esteves, P., Sancerni, T., Lenoir, V., Aparicio, T., Bouillaud, F., et al. (2019). UCP2 deficiency increases colon tumorigenesis by promoting lipid synthesis and depleting NADPH for antioxidant defenses. Cell reports, 28(9), 2306-2316.e2305. https://doi.org/10.1016/j.celrep.2019.07.097
Su, W. P., Lo, Y. C., Yan, J. J., Liao, I. C., Tsai, P. J., Wang, H. C., et al. (2012). Mitochondrial uncoupling protein 2 regulates the effects of paclitaxel on Stat3 activation and cellular survival in lung cancer cells. Carcinogenesis, 33(11), 2065–2075. https://doi.org/10.1093/carcin/bgs253
Donadelli, M., Dando, I., Dalla Pozza, E., & Palmieri, M. (2015). Mitochondrial uncoupling protein 2 and pancreatic cancer: A new potential target therapy. World journal of gastroenterology, 21(11), 3232–3238. https://doi.org/10.3748/wjg.v21.i11.3232
Hilvo, M., Denkert, C., Lehtinen, L., Müller, B., Brockmöller, S., Seppänen-Laakso, T., et al. (2011). Novel theranostic opportunities offered by characterization of altered membrane lipid metabolism in breast cancer progression. Cancer Research, 71(9), 3236–3245. https://doi.org/10.1158/0008-5472.Can-10-3894
Xu, W., Hu, X., Anwaier, A., Wang, J., Liu, W., Tian, X., et al. (2021). Fatty acid synthase correlates with prognosis-related abdominal adipose distribution and metabolic disorders of clear cell renal cell carcinoma. [Original Research]. Frontiers in Molecular Biosciences, 7(453), https://doi.org/10.3389/fmolb.2020.610229.
Gouw, A. M., Margulis, K., Liu, N. S., Raman, S. J., Mancuso, A., Toal, G. G., et al. (2019). The MYC oncogene cooperates with sterol-regulated element-binding protein to regulate lipogenesis essential for neoplastic growth. Cell Metabolism, 30(3), 556-572.e555. https://doi.org/10.1016/j.cmet.2019.07.012
Zhou, C., Qian, W., Li, J., Ma, J., Chen, X., Jiang, Z., et al. (2019). High glucose microenvironment accelerates tumor growth via SREBP1-autophagy axis in pancreatic cancer. Journal of Experimental & Clinical Cancer Research, 38(1), 302. https://doi.org/10.1186/s13046-019-1288-7
Bao, J., Zhu, L., Zhu, Q., Su, J., Liu, M., & Huang, W. (2016). SREBP-1 is an independent prognostic marker and promotes invasion and migration in breast cancer. Oncology Letters, 12(4), 2409–2416. https://doi.org/10.3892/ol.2016.4988
Shao, F., Bian, X., Wang, J., Xu, D., Guo, W., Jiang, H., et al. (2021). Prognostic impact of PCK1 protein kinase activity-dependent nuclear SREBP1 activation in non-small-cell lung carcinoma. [Original Research]. Frontiers in Oncology, 11(945), https://doi.org/10.3389/fonc.2021.561247.
Ferro, F., Servais, S., Besson, P., Roger, S., Dumas, J. F., & Brisson, L. (2020). Autophagy and mitophagy in cancer metabolic remodelling. Seminars in Cell & Developmental Biology, 98, 129–138. https://doi.org/10.1016/j.semcdb.2019.05.029
Mal, S., Dwivedi, A. R., Kumar, V., Kumar, N., Kumar, B., & Kumar, V. (2021). Role of peroxisome proliferator-activated receptor gamma (PPARγ) in different disease states: Recent updates. Current Medicinal Chemistry, 28(16), 3193–3215. https://doi.org/10.2174/0929867327666200716113136
Mullen, P. J., Yu, R., Longo, J., Archer, M. C., & Penn, L. Z. (2016). The interplay between cell signalling and the mevalonate pathway in cancer. Nature Reviews Cancer, 16(11), 718–731. https://doi.org/10.1038/nrc.2016.76
Su, Y. W., Wu, P. S., Lin, S. H., Huang, W. Y., Kuo, Y. S., & Lin, H. P. (2020). Prognostic value of the overexpression of fatty acid metabolism-related enzymes in squamous cell carcinoma of the head and neck. Int J Mol Sci, 21(18), https://doi.org/10.3390/ijms21186851.
Tan, Z., Zou, Y., Zhu, M., Luo, Z., Wu, T., Zheng, C., et al. (2021). Carnitine palmitoyl transferase 1A is a novel diagnostic and predictive biomarker for breast cancer. BMC Cancer, 21(1), 409–409. https://doi.org/10.1186/s12885-021-08134-7
Wang, L., Li, C., Song, Y., & Yan, Z. (2020). Inhibition of carnitine palmitoyl transferase 1A-induced fatty acid oxidation suppresses cell progression in gastric cancer. Archives of Biochemistry and Biophysics, 696, 108664. https://doi.org/10.1016/j.abb.2020.108664
Vernieri, C., Pusceddu, S., Fucà, G., Indelicato, P., Centonze, G., Castagnoli, L., et al. (2019). Impact of systemic and tumor lipid metabolism on everolimus efficacy in advanced pancreatic neuroendocrine tumors (pNETs). International Journal of Cancer, 144(7), 1704–1712. https://doi.org/10.1002/ijc.32042
Hadad, S. M., Baker, L., Quinlan, P. R., Robertson, K. E., Bray, S. E., Thomson, G., et al. (2009). Histological evaluation of AMPK signalling in primary breast cancer. BMC Cancer, 9(1), 307. https://doi.org/10.1186/1471-2407-9-307
Mossmann, D., Park, S., & Hall, M. N. (2018). mTOR signalling and cellular metabolism are mutual determinants in cancer. Nature Reviews Cancer, 18(12), 744–757. https://doi.org/10.1038/s41568-018-0074-8
Porstmann, T., Santos, C. R., Griffiths, B., Cully, M., Wu, M., Leevers, S., et al. (2008). SREBP activity is regulated by mTORC1 and contributes to Akt-dependent cell growth. Cell Metabolism, 8(3), 224–236. https://doi.org/10.1016/j.cmet.2008.07.007
Ricoult, S. J., Yecies, J. L., Ben-Sahra, I., & Manning, B. D. (2016). Oncogenic PI3K and K-Ras stimulate de novo lipid synthesis through mTORC1 and SREBP. Oncogene, 35(10), 1250–1260. https://doi.org/10.1038/onc.2015.179
Lee, G., Zheng, Y., Cho, S., Jang, C., England, C., Dempsey, J. M., et al. (2017). Post-transcriptional regulation of de novo lipogenesis by mTORC1-S6K1-SRPK2 signaling. Cell, 171(7), 1545-1558.e1518. https://doi.org/10.1016/j.cell.2017.10.037
Martinez Calejman, C., Trefely, S., Entwisle, S. W., Luciano, A., Jung, S. M., Hsiao, W., et al. (2020). mTORC2-AKT signaling to ATP-citrate lyase drives brown adipogenesis and de novo lipogenesis. Nature Communications, 11(1), 575. https://doi.org/10.1038/s41467-020-14430-w
Zhang, J., Wang, S., Jiang, B., Huang, L., Ji, Z., Li, X., et al. (2017). c-Src phosphorylation and activation of hexokinase promotes tumorigenesis and metastasis. Nature Communications, 8, 13732. https://doi.org/10.1038/ncomms13732
Botzer, L. E., Maman, S., Sagi-Assif, O., Meshel, T., Nevo, I., Yron, I., et al. (2016). Hexokinase 2 is a determinant of neuroblastoma metastasis. British Journal of Cancer, 114(7), 759–766. https://doi.org/10.1038/bjc.2016.26
Nokin, M.-J., Bellier, J., Durieux, F., Peulen, O., Rademaker, G., Gabriel, M., et al. (2019). Methylglyoxal, a glycolysis metabolite, triggers metastasis through MEK/ERK/SMAD1 pathway activation in breast cancer. Breast cancer research : BCR, 21(1), 11–11. https://doi.org/10.1186/s13058-018-1095-7
Pan, Y., Wang, W., Huang, S., Ni, W., Wei, Z., Cao, Y., et al. (2019). Beta-elemene inhibits breast cancer metastasis through blocking pyruvate kinase M2 dimerization and nuclear translocation. Journal of Cellular and Molecular Medicine, 23(10), 6846–6858. https://doi.org/10.1111/jcmm.14568
Cha, Y. J., Jung, W. H., & Koo, J. S. (2017). Differential site-based expression of pentose phosphate pathway-related proteins among breast cancer metastases. Disease Markers, 2017, 7062517. https://doi.org/10.1155/2017/7062517
Piskounova, E., Agathocleous, M., Murphy, M. M., Hu, Z., Huddlestun, S. E., Zhao, Z., et al. (2015). Oxidative stress inhibits distant metastasis by human melanoma cells. Nature, 527(7577), 186–191. https://doi.org/10.1038/nature15726
Porporato, P. E., Payen, V. L., Baselet, B., & Sonveaux, P. (2016). Metabolic changes associated with tumor metastasis, part 2: Mitochondria, lipid and amino acid metabolism. Cellular and Molecular Life Sciences, 73(7), 1349–1363. https://doi.org/10.1007/s00018-015-2100-2
Corbet, C., & Feron, O. (2017). Cancer cell metabolism and mitochondria: Nutrient plasticity for TCA cycle fueling. Biochimica et Biophysica Acta - Reviews on Cancer, 1868(1), 7–15. https://doi.org/10.1016/j.bbcan.2017.01.002
Rodrigues, M. F., Obre, E., de Melo, F. H., Santos, G. C., Jr., Galina, A., Jasiulionis, M. G., et al. (2016). Enhanced OXPHOS, glutaminolysis and β-oxidation constitute the metastatic phenotype of melanoma cells. The Biochemical Journal, 473(6), 703–715. https://doi.org/10.1042/bj20150645
Luis, G., Godfroid, A., Nishiumi, S., Cimino, J., Blacher, S., Maquoi, E., et al. (2021). Tumor resistance to ferroptosis driven by stearoyl-CoA desaturase-1 (SCD1) in cancer cells and fatty acid binding protein-4 (FABP4) in tumor microenvironment promote tumor recurrence. Redox biology, 43, 102006–102006. https://doi.org/10.1016/j.redox.2021.102006
Torrano, V., Valcarcel-Jimenez, L., Cortazar, A. R., Liu, X., Urosevic, J., Castillo-Martin, M., et al. (2016). The metabolic co-regulator PGC1α suppresses prostate cancer metastasis. Nature Cell Biology, 18(6), 645–656. https://doi.org/10.1038/ncb3357
Samanta, D., Park, Y., Andrabi, S. A., Shelton, L. M., Gilkes, D. M., & Semenza, G. L. (2016). PHGDH expression is required for mitochondrial redox homeostasis, breast cancer stem cell maintenance, and lung metastasis. Cancer Research, 76(15), 4430–4442. https://doi.org/10.1158/0008-5472.Can-16-0530
LeBleu, V. S., O'Connell, J. T., Gonzalez Herrera, K. N., Wikman, H., Pantel, K., Haigis, M. C., et al. (2014). PGC-1α mediates mitochondrial biogenesis and oxidative phosphorylation in cancer cells to promote metastasis. Nat Cell Biol, 16(10), 992–1003, 1001–1015, https://doi.org/10.1038/ncb3039.
Luo, C., Lim, J. H., Lee, Y., Granter, S. R., Thomas, A., Vazquez, F., et al. (2016). A PGC1α-mediated transcriptional axis suppresses melanoma metastasis. Nature, 537(7620), 422–426. https://doi.org/10.1038/nature19347
Carey, B. W., Finley, L. W., Cross, J. R., Allis, C. D., & Thompson, C. B. (2015). Intracellular α-ketoglutarate maintains the pluripotency of embryonic stem cells. Nature, 518(7539), 413–416. https://doi.org/10.1038/nature13981
Atlante, S., Visintin, A., Marini, E., Savoia, M., Dianzani, C., Giorgis, M., et al. (2018). α-Ketoglutarate dehydrogenase inhibition counteracts breast cancer-associated lung metastasis. Cell death & disease, 9(7), 756–756. https://doi.org/10.1038/s41419-018-0802-8
Hipólito, A., Martins, F., Mendes, C., Lopes-Coelho, F., & Serpa, J. (2021). Molecular and metabolic reprogramming: Pulling the strings toward tumor metastasis. [Review]. Frontiers in Oncology, 11(2036), https://doi.org/10.3389/fonc.2021.656851.
Sehgal, S. N., Baker, H., & Vézina, C. (1975). Rapamycin (AY-22,989), a new antifungal antibiotic. II. Fermentation, isolation and characterization. J Antibiot (Tokyo), 28(10), 727–732, https://doi.org/10.7164/antibiotics.28.727.
Martel, R. R., Klicius, J., & Galet, S. (1977). Inhibition of the immune response by rapamycin, a new antifungal antibiotic. Canadian Journal of Physiology and Pharmacology, 55(1), 48–51. https://doi.org/10.1139/y77-007
Heitman, J., Movva, N. R., & Hall, M. N. (1991). Targets for cell cycle arrest by the immunosuppressant rapamycin in yeast. Science, 253(5022), 905–909. https://doi.org/10.1126/science.1715094
Kunz, J., Henriquez, R., Schneider, U., Deuter-Reinhard, M., Movva, N. R., & Hall, M. N. (1993). Target of rapamycin in yeast, TOR2, is an essential phosphatidylinositol kinase homolog required for G1 progression. Cell, 73(3), 585–596. https://doi.org/10.1016/0092-8674(93)90144-f
Brown, E. J., Albers, M. W., Shin, T. B., Ichikawa, K., Keith, C. T., Lane, W. S., et al. (1994). A mammalian protein targeted by G1-arresting rapamycin-receptor complex. Nature, 369(6483), 756–758. https://doi.org/10.1038/369756a0
Sabatini, D. M., Pierchala, B. A., Barrow, R. K., Schell, M. J., & Snyder, S. H. (1995). The rapamycin and FKBP12 target (RAFT) displays phosphatidylinositol 4-kinase activity. Journal of Biological Chemistry, 270(36), 20875–20878. https://doi.org/10.1074/jbc.270.36.20875
Chung, J., Kuo, C. J., Crabtree, G. R., & Blenis, J. (1992). Rapamycin-FKBP specifically blocks growth-dependent activation of and signaling by the 70 kd S6 protein kinases. Cell, 69(7), 1227–1236. https://doi.org/10.1016/0092-8674(92)90643-q
Guertin, D. A., & Sabatini, D. M. (2005). An expanding role for mTOR in cancer. Trends in Molecular Medicine, 11(8), 353–361. https://doi.org/10.1016/j.molmed.2005.06.007
Dazert, E., & Hall, M. N. (2011). mTOR signaling in disease. Current Opinion in Cell Biology, 23(6), 744–755. https://doi.org/10.1016/j.ceb.2011.09.003
Saxton, R. A., & Sabatini, D. M. (2017). mTOR signaling in growth, metabolism, and disease. Cell, 168(6), 960–976. https://doi.org/10.1016/j.cell.2017.02.004
Manuelli, M., De Luca, L., Iaria, G., Tatangelo, P., Sforza, D., Perrone, L., et al. (2010). Conversion to rapamycin immunosuppression for malignancy after kidney transplantation. Transplantation Proceedings, 42(4), 1314–1316. https://doi.org/10.1016/j.transproceed.2010.03.051
Hess, G., Smith, S. M., Berkenblit, A., & Coiffier, B. (2009). Temsirolimus in mantle cell lymphoma and other non-Hodgkin lymphoma subtypes. Seminars in Oncology, 36(Suppl 3), S37-45. https://doi.org/10.1053/j.seminoncol.2009.10.012
Aylett, C. H., Sauer, E., Imseng, S., Boehringer, D., Hall, M. N., Ban, N., et al. (2016). Architecture of human mTOR complex 1. Science, 351(6268), 48–52. https://doi.org/10.1126/science.aaa3870
Yang, H., Jiang, X., Li, B., Yang, H. J., Miller, M., Yang, A., et al. (2017). Mechanisms of mTORC1 activation by RHEB and inhibition by PRAS40. Nature, 552(7685), 368–373. https://doi.org/10.1038/nature25023
Liu, G. Y., & Sabatini, D. M. (2020). mTOR at the nexus of nutrition, growth, ageing and disease. Nature Reviews Molecular Cell Biology, 21(4), 183–203. https://doi.org/10.1038/s41580-019-0199-y
Rogala, K. B., Gu, X., Kedir, J. F., Abu-Remaileh, M., Bianchi, L. F., Bottino, A. M. S., et al. (2019). Structural basis for the docking of mTORC1 on the lysosomal surface. Science, 366(6464), 468–475. https://doi.org/10.1126/science.aay0166
Kim, E., Goraksha-Hicks, P., Li, L., Neufeld, T. P., & Guan, K. L. (2008). Regulation of TORC1 by Rag GTPases in nutrient response. Nature Cell Biology, 10(8), 935–945. https://doi.org/10.1038/ncb1753
Castellano, B. M., Thelen, A. M., Moldavski, O., Feltes, M., van der Welle, R. E., Mydock-McGrane, L., et al. (2017). Lysosomal cholesterol activates mTORC1 via an SLC38A9-Niemann-Pick C1 signaling complex. Science, 355(6331), 1306–1311. https://doi.org/10.1126/science.aag1417
Tsun, Z. Y., Bar-Peled, L., Chantranupong, L., Zoncu, R., Wang, T., Kim, C., et al. (2013). The folliculin tumor suppressor is a GAP for the RagC/D GTPases that signal amino acid levels to mTORC1. Molecular Cell, 52(4), 495–505. https://doi.org/10.1016/j.molcel.2013.09.016
Chantranupong, L., Wolfson, R. L., Orozco, J. M., Saxton, R. A., Scaria, S. M., Bar-Peled, L., et al. (2014). The Sestrins interact with GATOR2 to negatively regulate the amino-acid-sensing pathway upstream of mTORC1. Cell Reports, 9(1), 1–8. https://doi.org/10.1016/j.celrep.2014.09.014
Wolfson, R. L., Chantranupong, L., Saxton, R. A., Shen, K., Scaria, S. M., Cantor, J. R., et al. (2016). Sestrin2 is a leucine sensor for the mTORC1 pathway. Science, 351(6268), 43–48. https://doi.org/10.1126/science.aab2674
Chen, J., Ou, Y., Luo, R., Wang, J., Wang, D., Guan, J., et al. (2021). SAR1B senses leucine levels to regulate mTORC1 signalling. Nature, 596(7871), 281–284. https://doi.org/10.1038/s41586-021-03768-w
Park, J. H., Lee, G., & Blenis, J. (2020). Structural insights into the activation of mTORC1 on the lysosomal surface. Trends in Biochemical Sciences, 45(5), 367–369. https://doi.org/10.1016/j.tibs.2020.02.004
Lear, T. B., Lockwood, K. C., Ouyang, Y., Evankovich, J. W., Larsen, M. B., Lin, B., et al. (2019). The RING-type E3 ligase RNF186 ubiquitinates Sestrin-2 and thereby controls nutrient sensing. Journal of Biological Chemistry, 294(45), 16527–16534. https://doi.org/10.1074/jbc.AC119.010671
Huang, J., & Manning, B. D. (2008). The TSC1-TSC2 complex: A molecular switchboard controlling cell growth. The Biochemical Journal, 412(2), 179–190. https://doi.org/10.1042/bj20080281
Dibble, C. C., & Cantley, L. C. (2015). Regulation of mTORC1 by PI3K signaling. Trends in Cell Biology, 25(9), 545–555. https://doi.org/10.1016/j.tcb.2015.06.002
Laplante, M., & Sabatini, D. M. (2013). Regulation of mTORC1 and its impact on gene expression at a glance. Journal of Cell Science, 126(Pt 8), 1713–1719. https://doi.org/10.1242/jcs.125773
Fu, W., & Hall, M. N. (2020). Regulation of mTORC2 signaling. Genes, 11(9), 1045.
Ebner, M., Sinkovics, B., Szczygieł, M., Ribeiro, D. W., & Yudushkin, I. (2017). Localization of mTORC2 activity inside cells. Journal of Cell Biology, 216(2), 343–353. https://doi.org/10.1083/jcb.201610060
Kovalski, J. R., Bhaduri, A., Zehnder, A. M., Neela, P. H., Che, Y., Wozniak, G. G., et al. (2019). The functional proximal proteome of oncogenic Ras includes mTORC2. Molecular Cell, 73(4), 830-844.e812. https://doi.org/10.1016/j.molcel.2018.12.001
Moloughney, J. G., Kim, P. K., Vega-Cotto, N. M., Wu, C.-C., Zhang, S., Adlam, M., et al. (2016). mTORC2 responds to glutamine catabolite levels to modulate the hexosamine biosynthesis enzyme GFAT1. Molecular Cell, 63(5), 811–826. https://doi.org/10.1016/j.molcel.2016.07.015
Kazyken, D., Magnuson, B., Bodur, C., Acosta-Jaquez, H. A., Zhang, D., Tong, X., et al. (2019). AMPK directly activates mTORC2 to promote cell survival during acute energetic stress. Sci Signal, 12(585), https://doi.org/10.1126/scisignal.aav3249.
Lampada, A., O’Prey, J., Szabadkai, G., Ryan, K. M., Hochhauser, D., & Salomoni, P. (2017). mTORC1-independent autophagy regulates receptor tyrosine kinase phosphorylation in colorectal cancer cells via an mTORC2-mediated mechanism. Cell Death and Differentiation, 24(6), 1045–1062. https://doi.org/10.1038/cdd.2017.41
Sengupta, S., Peterson, T. R., Laplante, M., Oh, S., & Sabatini, D. M. (2010). mTORC1 controls fasting-induced ketogenesis and its modulation by ageing. Nature, 468(7327), 1100–1104. https://doi.org/10.1038/nature09584
Kuma, A., Hatano, M., Matsui, M., Yamamoto, A., Nakaya, H., Yoshimori, T., et al. (2004). The role of autophagy during the early neonatal starvation period. Nature, 432(7020), 1032–1036. https://doi.org/10.1038/nature03029
Holz, M. K., Ballif, B. A., Gygi, S. P., & Blenis, J. (2005). mTOR and S6K1 mediate assembly of the translation preinitiation complex through dynamic protein interchange and ordered phosphorylation events. Cell, 123(4), 569–580. https://doi.org/10.1016/j.cell.2005.10.024
Ma, X. M., Yoon, S. O., Richardson, C. J., Jülich, K., & Blenis, J. (2008). SKAR links pre-mRNA splicing to mTOR/S6K1-mediated enhanced translation efficiency of spliced mRNAs. Cell, 133(2), 303–313. https://doi.org/10.1016/j.cell.2008.02.031
Hsieh, A. C., Liu, Y., Edlind, M. P., Ingolia, N. T., Janes, M. R., Sher, A., et al. (2012). The translational landscape of mTOR signalling steers cancer initiation and metastasis. Nature, 485(7396), 55–61. https://doi.org/10.1038/nature10912
Peterson, T. R., Sengupta, S. S., Harris, T. E., Carmack, A. E., Kang, S. A., Balderas, E., et al. (2011). mTOR complex 1 regulates lipin 1 localization to control the SREBP pathway. Cell, 146(3), 408–420. https://doi.org/10.1016/j.cell.2011.06.034
Ben-Sahra, I., Hoxhaj, G., Ricoult, S. J. H., Asara, J. M., & Manning, B. D. (2016). mTORC1 induces purine synthesis through control of the mitochondrial tetrahydrofolate cycle. Science, 351(6274), 728–733. https://doi.org/10.1126/science.aad0489
Robitaille, A. M., Christen, S., Shimobayashi, M., Cornu, M., Fava, L. L., Moes, S., et al. (2013). Quantitative phosphoproteomics reveal mTORC1 activates de novo pyrimidine synthesis. Science, 339(6125), 1320–1323. https://doi.org/10.1126/science.1228771
Cunningham, J. T., Rodgers, J. T., Arlow, D. H., Vazquez, F., Mootha, V. K., & Puigserver, P. (2007). mTOR controls mitochondrial oxidative function through a YY1-PGC-1alpha transcriptional complex. Nature, 450(7170), 736–740. https://doi.org/10.1038/nature06322
Hosokawa, N., Hara, T., Kaizuka, T., Kishi, C., Takamura, A., Miura, Y., et al. (2009). Nutrient-dependent mTORC1 association with the ULK1-Atg13-FIP200 complex required for autophagy. Molecular Biology of the Cell, 20(7), 1981–1991. https://doi.org/10.1091/mbc.e08-12-1248
Settembre, C., Zoncu, R., Medina, D. L., Vetrini, F., Erdin, S., Erdin, S., et al. (2012). A lysosome-to-nucleus signalling mechanism senses and regulates the lysosome via mTOR and TFEB. EMBO Journal, 31(5), 1095–1108. https://doi.org/10.1038/emboj.2012.32
Kim, S. J., DeStefano, M. A., Oh, W. J., Wu, C. C., Vega-Cotto, N. M., Finlan, M., et al. (2012). mTOR complex 2 regulates proper turnover of insulin receptor substrate-1 via the ubiquitin ligase subunit Fbw8. Molecular Cell, 48(6), 875–887. https://doi.org/10.1016/j.molcel.2012.09.029
Ikenoue, T., Inoki, K., Yang, Q., Zhou, X., & Guan, K. L. (2008). Essential function of TORC2 in PKC and Akt turn motif phosphorylation, maturation and signalling. EMBO Journal, 27(14), 1919–1931. https://doi.org/10.1038/emboj.2008.119
Aspernig, H., Heimbucher, T., Qi, W., Gangurde, D., Curic, S., Yan, Y., et al. (2019). Mitochondrial perturbations couple mTORC2 to autophagy in C. elegans. Cell Rep, 29(6), 1399–1409.e1395, https://doi.org/10.1016/j.celrep.2019.09.072.
Ballesteros-Álvarez, J., & Andersen, J. K. (2021). mTORC2: The other mTOR in autophagy regulation. Aging Cell, 20(8), e13431. https://doi.org/10.1111/acel.13431
Gremke, N., Polo, P., Dort, A., Schneikert, J., Elmshäuser, S., Brehm, C., et al. (2020). mTOR-mediated cancer drug resistance suppresses autophagy and generates a druggable metabolic vulnerability. Nature Communications, 11(1), 4684. https://doi.org/10.1038/s41467-020-18504-7
Gkountakos, A., Pilotto, S., Mafficini, A., Vicentini, C., Simbolo, M., Milella, M., et al. (2018). Unmasking the impact of Rictor in cancer: Novel insights of mTORC2 complex. Carcinogenesis, 39(8), 971–980. https://doi.org/10.1093/carcin/bgy086
Lin, G., Gai, R., Chen, Z., Wang, Y., Liao, S., Dong, R., et al. (2014). The dual PI3K/mTOR inhibitor NVP-BEZ235 prevents epithelial-mesenchymal transition induced by hypoxia and TGF-β1. European Journal of Pharmacology, 729, 45–53. https://doi.org/10.1016/j.ejphar.2014.02.011
Lamouille, S., Connolly, E., Smyth, J. W., Akhurst, R. J., & Derynck, R. (2012). TGF-β-induced activation of mTOR complex 2 drives epithelial-mesenchymal transition and cell invasion. Journal of Cell Science, 125(Pt 5), 1259–1273. https://doi.org/10.1242/jcs.095299
Shin, J., Bae, J., Park, S., Kang, H.-G., Shin, S. M., Won, G., et al. (2020). mTOR-dependent role of Sestrin2 in regulating tumor progression of human endometrial cancer. Cancers, 12(9), 2515.
Liu, L., Chen, L., Chung, J., & Huang, S. (2008). Rapamycin inhibits F-actin reorganization and phosphorylation of focal adhesion proteins. Oncogene, 27(37), 4998–5010. https://doi.org/10.1038/onc.2008.137
Jacinto, E., Loewith, R., Schmidt, A., Lin, S., Rüegg, M. A., Hall, A., et al. (2004). Mammalian TOR complex 2 controls the actin cytoskeleton and is rapamycin insensitive. Nature Cell Biology, 6(11), 1122–1128. https://doi.org/10.1038/ncb1183
Schaller, M. D. (2001). Paxillin: A focal adhesion-associated adaptor protein. Oncogene, 20(44), 6459–6472. https://doi.org/10.1038/sj.onc.1204786
Zhao, H., Chen, G., Ye, L., Yu, H., Li, S., & Jiang, W. G. (2019). DOK7V1 influences the malignant phenotype of lung cancer cells through PI3K/AKT/mTOR and FAK/paxillin signaling pathways. International Journal of Oncology, 54(1), 381–389. https://doi.org/10.3892/ijo.2018.4624
He, Y., Li, D., Cook, S. L., Yoon, M. S., Kapoor, A., Rao, C. V., et al. (2013). Mammalian target of rapamycin and Rictor control neutrophil chemotaxis by regulating Rac/Cdc42 activity and the actin cytoskeleton. Molecular Biology of the Cell, 24(21), 3369–3380. https://doi.org/10.1091/mbc.E13-07-0405
Sarbassov, D. D., Ali, S. M., Kim, D.-H., Guertin, D. A., Latek, R. R., Erdjument-Bromage, H., et al. (2004). Rictor, a novel binding partner of mTOR, defines a rapamycin-insensitive and raptor-independent pathway that regulates the cytoskeleton. Current biology : CB, 14(14), 1296–1302. https://doi.org/10.1016/j.cub.2004.06.054
Morrison Joly, M., Williams, M. M., Hicks, D. J., Jones, B., Sanchez, V., Young, C. D., et al. (2017). Two distinct mTORC2-dependent pathways converge on Rac1 to drive breast cancer metastasis. Breast Cancer Research, 19(1), 74. https://doi.org/10.1186/s13058-017-0868-8
Asrani, K., Sood, A., Torres, A., Georgess, D., Phatak, P., Kaur, H., et al. (2017). mTORC1 loss impairs epidermal adhesion via TGF-β/Rho kinase activation. The Journal of Clinical Investigation, 127(11), 4001–4017. https://doi.org/10.1172/jci92893
Tian, F., Dong, L., Zhou, Y., Shao, Y., Li, W., Zhang, H., et al. (2014). Rapamycin-induced apoptosis in HGF-stimulated lens epithelial cells by AKT/mTOR, ERK and JAK2/STAT3 pathways. International Journal of Molecular Sciences, 15(8), 13833–13848.
Cheng, H., Zou, Y., Ross, J. S., Wang, K., Liu, X., Halmos, B., et al. (2015). RICTOR amplification defines a novel subset of patients with lung cancer who may benefit from treatment with mTORC1/2 inhibitors. Cancer Discovery, 5(12), 1262–1270. https://doi.org/10.1158/2159-8290.Cd-14-0971
Morrison Joly, M., Hicks, D. J., Jones, B., Sanchez, V., Estrada, M. V., Young, C., et al. (2016). Rictor/mTORC2 drives progression and therapeutic resistance of HER2-amplified breast cancers. Cancer Research, 76(16), 4752–4764. https://doi.org/10.1158/0008-5472.Can-15-3393
Balko, J. M., Giltnane, J. M., Wang, K., Schwarz, L. J., Young, C. D., Cook, R. S., et al. (2014). Molecular profiling of the residual disease of triple-negative breast cancers after neoadjuvant chemotherapy identifies actionable therapeutic targets. Cancer Discovery, 4(2), 232–245. https://doi.org/10.1158/2159-8290.Cd-13-0286
Hosoi, H., Dilling, M. B., Liu, L. N., Danks, M. K., Shikata, T., Sekulic, A., et al. (1998). Studies on the mechanism of resistance to rapamycin in human cancer cells. Molecular Pharmacology, 54(5), 815–824. https://doi.org/10.1124/mol.54.5.815
Allen, E., Miéville, P., Warren, C. M., Saghafinia, S., Li, L., Peng, M. W., et al. (2016). Metabolic symbiosis enables adaptive resistance to anti-angiogenic therapy that is dependent on mTOR signaling. Cell Reports, 15(6), 1144–1160. https://doi.org/10.1016/j.celrep.2016.04.029
Pachmayr, E., Treese, C., & Stein, U. (2017). Underlying mechanisms for distant metastasis - Molecular biology. Visc Med, 33(1), 11–20. https://doi.org/10.1159/000454696
Tian, T., Li, X., & Zhang, J. (2019). mTOR signaling in cancer and mTOR inhibitors in solid tumor targeting therapy. Int J Mol Sci, 20(3), https://doi.org/10.3390/ijms20030755.
Nemes, K., Sebestyén, A., Márk, A., Hajdu, M., Kenessey, I., Sticz, T., et al. (2013). Mammalian target of rapamycin (mTOR) activity dependent phospho-protein expression in childhood acute lymphoblastic leukemia (ALL). PLoS ONE, 8(4), e59335. https://doi.org/10.1371/journal.pone.0059335
Sebestyén, A., Sticz, T. B., Márk, A., Hajdu, M., Timár, B., Nemes, K., et al. (2012). Activity and complexes of mTOR in diffuse large B-cell lymphomas–A tissue microarray study. Modern Pathology, 25(12), 1623–1628. https://doi.org/10.1038/modpathol.2012.141
Khanna, A., Bhushan, B., Chauhan, P. S., Saxena, S., Gupta, D. K., & Siraj, F. (2018). High mTOR expression independently prognosticates poor clinical outcome to induction chemotherapy in acute lymphoblastic leukemia. Clinical and Experimental Medicine, 18(2), 221–227. https://doi.org/10.1007/s10238-017-0478-x
Ulińska, E., Mycko, K., Sałacińska-Łoś, E., Pastorczak, A., Siwicka, A., Młynarski, W., et al. (2016). Impact of mTOR expression on clinical outcome in paediatric patients with B-cell acute lymphoblastic leukaemia - Preliminary report. Contemp Oncol (Pozn), 20(4), 291–296. https://doi.org/10.5114/wo.2016.61848
du Rusquec, P., Blonz, C., Frenel, J. S., & Campone, M. (2020). Targeting the PI3K/Akt/mTOR pathway in estrogen-receptor positive HER2 negative advanced breast cancer. Therapeutic advances in medical oncology, 12, 1758835920940939–1758835920940939. https://doi.org/10.1177/1758835920940939
Jhan, J.-R., & Andrechek, E. R. (2017). Triple-negative breast cancer and the potential for targeted therapy. Pharmacogenomics, 18(17), 1595–1609. https://doi.org/10.2217/pgs-2017-0117
Werfel, T. A., Wang, S., Jackson, M. A., Kavanaugh, T. E., Joly, M. M., Lee, L. H., et al. (2018). Selective mTORC2 inhibitor therapeutically blocks breast cancer cell growth and survival. Cancer Research, 78(7), 1845–1858. https://doi.org/10.1158/0008-5472.can-17-2388
Hayashi, K., Nakazato, Y., Ouchi, M., Fujita, T., Endou, H., & Chida, M. (2021). Antitumor effect of dimethyl itaconate on thymic carcinoma by targeting LDHA-mTOR axis. Life Sciences, 282, 119847. https://doi.org/10.1016/j.lfs.2021.119847
Lampa, M., Arlt, H., He, T., Ospina, B., Reeves, J., Zhang, B., et al. (2017). Glutaminase is essential for the growth of triple-negative breast cancer cells with a deregulated glutamine metabolism pathway and its suppression synergizes with mTOR inhibition. PLoS ONE, 12(9), e0185092. https://doi.org/10.1371/journal.pone.0185092
Wysham, W. Z., Roque, D. R., Han, J., Zhang, L., Guo, H., Gehrig, P. A., et al. (2016). Effects of fatty acid synthase inhibition by orlistat on proliferation of endometrial cancer cell lines. Targeted Oncology, 11(6), 763–769. https://doi.org/10.1007/s11523-016-0442-9
Krencz, I., Sebestyén, A., Fábián, K., Márk, Á., Moldvay, J., Khoor, A., et al. (2017). Expression of mTORC1/2-related proteins in primary and brain metastatic lung adenocarcinoma. Human Pathology, 62, 66–73. https://doi.org/10.1016/j.humpath.2016.12.012
Pinato, D. J., Ramachandran, R., Toussi, S. T. K., Vergine, M., Ngo, N., Sharma, R., et al. (2013). Immunohistochemical markers of the hypoxic response can identify malignancy in phaeochromocytomas and paragangliomas and optimize the detection of tumours with VHL germline mutations. British journal of cancer, 108(2), 429–437. https://doi.org/10.1038/bjc.2012.538
Pierobon, M., Ramos, C., Wong, S., Hodge, K. A., Aldrich, J., Byron, S., et al. (2017). Enrichment of PI3K-AKT-mTOR pathway activation in hepatic metastases from breast cancer. Clinical cancer research : An official journal of the American Association for Cancer Research, 23(16), 4919–4928. https://doi.org/10.1158/1078-0432.CCR-16-2656
Tehranian, C., Fankhauser, L., Harter, P. N., Ratcliffe, C. D. H., Zeiner, P. S., Messmer, J. M., et al. (2021). The PI3K/Akt/mTOR pathway as a preventive target in melanoma brain metastasis. Neuro-Oncology. https://doi.org/10.1093/neuonc/noab159
Ippen, F. M., Grosch, J. K., Subramanian, M., Kuter, B. M., Liederer, B. M., Plise, E. G., et al. (2019). Targeting the PI3K/Akt/mTOR pathway with the pan-Akt inhibitor GDC-0068 in PIK3CA-mutant breast cancer brain metastases. Neuro-Oncology, 21(11), 1401–1411. https://doi.org/10.1093/neuonc/noz105
Djukom, C., Porro, L. J., Mrazek, A., Townsend, C. M., Jr., Hellmich, M. R., & Chao, C. (2014). Dual inhibition of PI3K and mTOR signaling pathways decreases human pancreatic neuroendocrine tumor metastatic progression. Pancreas, 43(1), 88–92. https://doi.org/10.1097/MPA.0b013e3182a44ab4
Guimarães, I. S., Tessarollo, N. G., Lyra-Junior, P., dos Santos, D. Z., Zampier, R. C., de Oliveira, L., et al. (2015). Targeting the PI3K/AKT/mTOR pathway in cancer cells. Updates on Cancer Treatment.
Toda, K., Kawada, K., Iwamoto, M., Inamoto, S., Sasazuki, T., Shirasawa, S., et al. (2016). Metabolic alterations caused by KRAS mutations in colorectal cancer contribute to cell adaptation to glutamine depletion by upregulation of asparagine synthetase. Neoplasia, 18(11), 654–665. https://doi.org/10.1016/j.neo.2016.09.004
Yang, C. S., Matsuura, K., Huang, N. J., Robeson, A. C., Huang, B., Zhang, L., et al. (2015). Fatty acid synthase inhibition engages a novel caspase-2 regulatory mechanism to induce ovarian cancer cell death. Oncogene, 34(25), 3264–3272. https://doi.org/10.1038/onc.2014.271
Khawaja, M. R., Nick, A. M., Madhusudanannair, V., Fu, S., Hong, D., McQuinn, L. M., et al. (2016). Phase I dose escalation study of temsirolimus in combination with metformin in patients with advanced/refractory cancers. Cancer Chemotherapy and Pharmacology, 77(5), 973–977. https://doi.org/10.1007/s00280-016-3009-7
Bang, H., Ahn, S., Ji Kim, E., Kim, S. T., Park, H. Y., Lee, J., et al. (2020). Correlation between RICTOR overexpression and amplification in advanced solid tumors. Pathology, Research and Practice, 216(1), 152734. https://doi.org/10.1016/j.prp.2019.152734
Ross, J. S., Wang, K., Elkadi, O. R., Tarasen, A., Foulke, L., Sheehan, C. E., et al. (2014). Next-generation sequencing reveals frequent consistent genomic alterations in small cell undifferentiated lung cancer. Journal of Clinical Pathology, 67(9), 772–776. https://doi.org/10.1136/jclinpath-2014-202447
Umemura, S., Mimaki, S., Makinoshima, H., Tada, S., Ishii, G., Ohmatsu, H., et al. (2014). Therapeutic priority of the PI3K/AKT/mTOR pathway in small cell lung cancers as revealed by a comprehensive genomic analysis. Journal of Thoracic Oncology, 9(9), 1324–1331. https://doi.org/10.1097/jto.0000000000000250
Sakre, N., Wildey, G., Behtaj, M., Kresak, A., Yang, M., Fu, P., et al. (2017). RICTOR amplification identifies a subgroup in small cell lung cancer and predicts response to drugs targeting mTOR. Oncotarget, 8(4), 5992–6002, https://doi.org/10.18632/oncotarget.13362.
Wang, S., Song, X., Li, X., Zhao, X., Chen, H., Wang, J., et al. (2016). RICTOR polymorphisms affect efficiency of platinum-based chemotherapy in Chinese non-small-cell lung cancer patients. Pharmacogenomics, 17(15), 1637–1647. https://doi.org/10.2217/pgs-2016-0070
Kim, S. T., Kim, S. Y., Klempner, S. J., Yoon, J., Kim, N., Ahn, S., et al. (2017). Rapamycin-insensitive companion of mTOR (RICTOR) amplification defines a subset of advanced gastric cancer and is sensitive to AZD2014-mediated mTORC1/2 inhibition. Annals of Oncology, 28(3), 547–554. https://doi.org/10.1093/annonc/mdw669
Park, S., Shim, J., Mortimer, P. G. S., Smith, S. A., Godin, R. E., Hollingsworth, S. J., et al. (2020). Biomarker-driven phase 2 umbrella trial study for patients with recurrent small cell lung cancer failing platinum-based chemotherapy. [Clinical Trial, Phase II; Journal Article; Randomized Controlled Trial; Research Support, Non‐U.S. Gov't]. Cancer, 126(17), 4002‐4012, https://doi.org/10.1002/cncr.33048.
Zhang, F., Zhang, X., Li, M., Chen, P., Zhang, B., Guo, H., et al. (2010). mTOR complex component Rictor interacts with PKCzeta and regulates cancer cell metastasis. Cancer Research, 70(22), 9360–9370. https://doi.org/10.1158/0008-5472.Can-10-0207
Krieger, K. L., Hu, W. F., Ripperger, T., & Woods, N. T. (2019). Functional impacts of the BRCA1-mTORC2 interaction in breast cancer. Int J Mol Sci, 20(23), https://doi.org/10.3390/ijms20235876.
Beck, C., Rodriguez-Vargas, J. M., Boehler, C., Robert, I., Heyer, V., Hanini, N., et al. (2019). PARP3, a new therapeutic target to alter Rictor/mTORC2 signaling and tumor progression in BRCA1-associated cancers. Cell Death and Differentiation, 26(9), 1615–1630. https://doi.org/10.1038/s41418-018-0233-1
Bian, Y. H., Xu, J., Zhao, W. Y., Zhang, Z. Z., Tu, L., Cao, H., et al. (2017). Targeting mTORC2 component rictor inhibits cell proliferation and promotes apoptosis in gastric cancer. Am J Transl Res, 9(9), 4317–4330.
Gulhati, P., Bowen, K. A., Liu, J., Stevens, P. D., Rychahou, P. G., Chen, M., et al. (2011). mTORC1 and mTORC2 regulate EMT, motility, and metastasis of colorectal cancer via RhoA and Rac1 signaling pathways. Cancer Research, 71(9), 3246–3256. https://doi.org/10.1158/0008-5472.Can-10-4058
Cao, R.-z., Min, L., Liu, S., Tian, R.-y., Jiang, H.-y., Liu, J., et al. (2021). Rictor activates Cav 1 through the Akt signaling pathway to inhibit the apoptosis of gastric cancer cells. [Original Research]. Frontiers in Oncology, 11(915), https://doi.org/10.3389/fonc.2021.641453.
Wang, L., Qi, J., Yu, J., Chen, H., Zou, Z., Lin, X., et al. (2017). Overexpression of Rictor protein in colorectal cancer is correlated with tumor progression and prognosis. Oncology letters, 14(5), 6198–6202. https://doi.org/10.3892/ol.2017.6936
Wen, F. F., Li, X. Y., Li, Y. Y., He, S., Xu, X. Y., Liu, Y. H., et al. (2020). Expression of Raptor and Rictor and their relationships with angiogenesis in colorectal cancer. Neoplasma, 67(3), 501–508. https://doi.org/10.4149/neo_2020_190705N597
Sticz, T., Molnár, A., Dankó, T., Hujber, Z., Petővári, G., Nagy, N., et al. (2019). The effects of different mTOR inhibitors in EGFR inhibitor resistant colon carcinoma cells. Pathology Oncology Research, 25(4), 1379–1386. https://doi.org/10.1007/s12253-018-0434-4
Jiang, W. J., Feng, R. X., Liu, J. T., Fan, L. L., Wang, H., & Sun, G. P. (2017). RICTOR expression in esophageal squamous cell carcinoma and its clinical significance. Medical Oncology, 34(3), 32. https://doi.org/10.1007/s12032-017-0894-5
Xu, Z., Hu, J., Cao, H., Pilo, M. G., Cigliano, A., Shao, Z., et al. (2018). Loss of Pten synergizes with c-Met to promote hepatocellular carcinoma development via mTORC2 pathway. Experimental & Molecular Medicine, 50(1), e417–e417. https://doi.org/10.1038/emm.2017.158
Kaibori, M., Shikata, N., Sakaguchi, T., Ishizaki, M., Matsui, K., Iida, H., et al. (2015). Influence of Rictor and raptor expression of mTOR signaling on long-term outcomes of patients with hepatocellular carcinoma. Digestive Diseases and Sciences, 60(4), 919–928. https://doi.org/10.1007/s10620-014-3417-7
Villanueva, A., Chiang, D. Y., Newell, P., Peix, J., Thung, S., Alsinet, C., et al. (2008). Pivotal role of mTOR signaling in hepatocellular carcinoma. Gastroenterology, 135(6), 1972–1983, 1983.e1971–1911, https://doi.org/10.1053/j.gastro.2008.08.008.
Naruse, T., Yanamoto, S., Okuyama, K., Yamashita, K., Omori, K., Nakao, Y., et al. (2017). Therapeutic implication of mTORC2 in oral squamous cell carcinoma. Oral Oncology, 65, 23–32. https://doi.org/10.1016/j.oraloncology.2016.12.012
Ruicci, K. M., Plantinga, P., Pinto, N., Khan, M. I., Stecho, W., Dhaliwal, S. S., et al. (2019). Disruption of the RICTOR/mTORC2 complex enhances the response of head and neck squamous cell carcinoma cells to PI3K inhibition. Molecular oncology, 13(10), 2160–2177. https://doi.org/10.1002/1878-0261.12558
Kawasaki, G., Naruse, T., Furukawa, K., & Umeda, M. (2018). mTORC1 and mTORC2 expression levels in oral squamous cell carcinoma: An immunohistochemical and clinicopathological study. Anticancer Res, 38(3), 1623–1628, https://doi.org/10.21873/anticanres.12393.
Kondo, S., Hirakawa, H., Ikegami, T., Uehara, T., Agena, S., Uezato, J., et al. (2021). Raptor and rictor expression in patients with human papillomavirus-related oropharyngeal squamous cell carcinoma. BMC Cancer, 21(1), 87–87. https://doi.org/10.1186/s12885-021-07794-9
Liu, Y., Lu, Y., Li, A., Celiku, O., Han, S., Qian, M., et al. (2020). mTORC2/Rac1 pathway predisposes cancer aggressiveness in IDH1-mutated glioma. Cancers, 12(4), 787. https://doi.org/10.3390/cancers12040787
Alvarenga, A. W., Machado, L. E., Rodrigues, B. R., Lupinacci, F. C., Sanemastu, P., Matta, E., et al. (2017). Evaluation of Akt and RICTOR expression levels in astrocytomas of All grades. Journal of Histochemistry and Cytochemistry, 65(2), 93–103. https://doi.org/10.1369/0022155416675850
Petővári, G., Hujber, Z., Krencz, I., Dankó, T., Nagy, N., Tóth, F., et al. (2018). Targeting cellular metabolism using rapamycin and/or doxycycline enhances anti-tumour effects in human glioma cells. Cancer Cell International, 18, 211. https://doi.org/10.1186/s12935-018-0710-0
Akgül, S., Li, Y., Zheng, S., Kool, M., Treisman, D. M., Li, C., et al. (2018). Opposing tumor-promoting and -suppressive functions of Rictor/mTORC2 signaling in adult glioma and pediatric SHH medulloblastoma. Cell reports, 24(2), 463-478.e465. https://doi.org/10.1016/j.celrep.2018.06.050
Schmidt, K. M., Hellerbrand, C., Ruemmele, P., Michalski, C. W., Kong, B., Kroemer, A., et al. (2017). Inhibition of mTORC2 component RICTOR impairs tumor growth in pancreatic cancer models. Oncotarget, 8(15).
Driscoll, D. R., Karim, S. A., Sano, M., Gay, D. M., Jacob, W., Yu, J., et al. (2016). mTORC2 signaling drives the development and progression of pancreatic cancer. Cancer Research, 76(23), 6911–6923. https://doi.org/10.1158/0008-5472.Can-16-0810
Garcia, J. A., & Danielpour, D. (2008). Mammalian target of rapamycin inhibition as a therapeutic strategy in the management of urologic malignancies. Molecular Cancer Therapeutics, 7(6), 1347–1354. https://doi.org/10.1158/1535-7163.Mct-07-2408
Juengel, E., Kim, D., Makarević, J., Reiter, M., Tsaur, I., Bartsch, G., et al. (2015). Molecular analysis of sunitinib resistant renal cell carcinoma cells after sequential treatment with RAD001 (everolimus) or sorafenib. Journal of cellular and molecular medicine, 19(2), 430–441. https://doi.org/10.1111/jcmm.12471
Wen, S.-Y., Li, C.-H., Zhang, Y.-L., Bian, Y.-H., Ma, L., Ge, Q.-L., et al. (2014). Rictor is an independent prognostic factor for endometrial carcinoma. International journal of clinical and experimental pathology, 7(5), 2068–2078.
Sahu, D., Huan, J., Wang, H., Sahoo, D., Casteel, D. E., Klemke, R. L., et al. (2021). Bladder cancer invasion is mediated by mTORC2-driven regulation of nitric oxide and invadopodia formation. American Journal of Pathology. https://doi.org/10.1016/j.ajpath.2021.08.002
Gibault, L., Ferreira, C., Pérot, G., Audebourg, A., Chibon, F., Bonnin, S., et al. (2012). From PTEN loss of expression to RICTOR role in smooth muscle differentiation: Complex involvement of the mTOR pathway in leiomyosarcomas and pleomorphic sarcomas. Modern Pathology, 25(2), 197–211. https://doi.org/10.1038/modpathol.2011.163
Okada, T., Lee, A. Y., Qin, L. X., Agaram, N., Mimae, T., Shen, Y., et al. (2016). Integrin-α10 dependency identifies RAC and RICTOR as therapeutic targets in high-grade myxofibrosarcoma. Cancer Discovery, 6(10), 1148–1165. https://doi.org/10.1158/2159-8290.Cd-15-1481
Zeng, Z., dos Sarbassov, D., Samudio, I. J., Yee, K. W., Munsell, M. F., Ellen Jackson, C., et al. (2007). Rapamycin derivatives reduce mTORC2 signaling and inhibit AKT activation in AML. Blood, 109(8), 3509–3512. https://doi.org/10.1182/blood-2006-06-030833
Zhuang, J., Hawkins, S. F., Glenn, M. A., Lin, K., Johnson, G. G., Carter, A., et al. (2010). Akt is activated in chronic lymphocytic leukemia cells and delivers a pro-survival signal: The therapeutic potential of Akt inhibition. Haematologica, 95(1), 110–118. https://doi.org/10.3324/haematol.2009.010272
Simioni, C., Martelli, A. M., Zauli, G., Melloni, E., & Neri, L. M. (2019). Targeting mTOR in acute lymphoblastic leukemia. Cells, 8(2), https://doi.org/10.3390/cells8020190.
Watanabe, T., Sato, A., Kobayashi-Watanabe, N., Sueoka-Aragane, N., Kimura, S., & Sueoka, E. (2016). Torin2 potentiates anticancer effects on adult T-cell leukemia/lymphoma by inhibiting mammalian target of rapamycin. Anticancer Research, 36(1), 95–102.
Carayol, N., Vakana, E., Sassano, A., Kaur, S., Goussetis, D. J., Glaser, H., et al. (2010). Critical roles for mTORC2- and rapamycin-insensitive mTORC1-complexes in growth and survival of BCR-ABL-expressing leukemic cells. Proceedings of the National Academy of Sciences of the United States of America, 107(28), 12469–12474. https://doi.org/10.1073/pnas.1005114107
Müller, A., Zang, C., Chumduri, C., Dörken, B., Daniel, P. T., & Scholz, C. W. (2013). Concurrent inhibition of PI3K and mTORC1/mTORC2 overcomes resistance to rapamycin induced apoptosis by down-regulation of Mcl-1 in mantle cell lymphoma. International Journal of Cancer, 133(8), 1813–1824. https://doi.org/10.1002/ijc.28206
Smith, A. P., Hoek, K., & Becker, D. (2005). Whole-genome expression profiling of the melanoma progression pathway reveals marked molecular differences between nevi/melanoma in situ and advanced-stage melanomas. Cancer Biology & Therapy, 4(9), 1018–1029. https://doi.org/10.4161/cbt.4.9.2165
Haqq, C., Nosrati, M., Sudilovsky, D., Crothers, J., Khodabakhsh, D., Pulliam, B. L., et al. (2005). The gene expression signatures of melanoma progression. Proceedings of the National Academy of Sciences of the United States of America, 102(17), 6092–6097. https://doi.org/10.1073/pnas.0501564102
Zhang, X., Wang, X., Xu, T., Zhong, S., & Shen, Z. (2015). Targeting of mTORC2 may have advantages over selective targeting of mTORC1 in the treatment of malignant pheochromocytoma. Tumour Biology, 36(7), 5273–5281. https://doi.org/10.1007/s13277-015-3187-7
Zecchini, V., & Frezza, C. (2017). Metabolic synthetic lethality in cancer therapy. Biochimica et Biophysica Acta (BBA) - Bioenergetics, 1858(8), 723–731, https://doi.org/10.1016/j.bbabio.2016.12.003.
Bizjak, M., Malavašič, P., Dolinar, K., Pohar, J., Pirkmajer, S., & Pavlin, M. (2017). Combined treatment with metformin and 2-deoxy glucose induces detachment of viable MDA-MB-231 breast cancer cells in vitro. Scientific reports, 7(1), 1761–1761. https://doi.org/10.1038/s41598-017-01801-5
Cheong, J.-H., Park, E. S., Liang, J., Dennison, J. B., Tsavachidou, D., Nguyen-Charles, C., et al. (2011). Dual inhibition of tumor energy pathway by 2-deoxyglucose and metformin is effective against a broad spectrum of preclinical cancer models. Molecular Cancer Therapeutics, 10(12), 2350–2362. https://doi.org/10.1158/1535-7163.Mct-11-0497
Jones, A. T., Narov, K., Yang, J., Sampson, J. R., & Shen, M. H. (2019). Efficacy of dual inhibition of glycolysis and glutaminolysis for therapy of renal lesions in Tsc2(+/-) mice. Neoplasia (New York, N.Y.), 21(2), 230–238, https://doi.org/10.1016/j.neo.2018.12.003.
Li, J., Eu, J. Q., Kong, L. R., Wang, L., Lim, Y. C., Goh, B. C., et al. (2020). Targeting metabolism in cancer cells and the tumour microenvironment for cancer therapy. Molecules, 25(20), 4831.
Yuan, P., Ito, K., Perez-Lorenzo, R., Del Guzzo, C., Lee, J. H., Shen, C.-H., et al. (2013). Phenformin enhances the therapeutic benefit of BRAFV600E inhibition in melanoma. Proceedings of the National Academy of Sciences, 110(45), 18226–18231.
Vitiello, G. A., Medina, B. D., Zeng, S., Bowler, T. G., Zhang, J. Q., Loo, J. K., et al. (2018). Mitochondrial inhibition augments the efficacy of imatinib by resetting the metabolic phenotype of gastrointestinal stromal tumor. Clinical Cancer Research, 24(4), 972–984.
Yakes, F. M., Chen, J., Tan, J., Yamaguchi, K., Shi, Y., Yu, P., et al. (2011). Cabozantinib (XL184), a novel MET and VEGFR2 inhibitor, simultaneously suppresses metastasis, angiogenesis, and tumor growth. Molecular cancer therapeutics, 10(12), 2298–2308.
Leone, R. D., Zhao, L., Englert, J. M., Sun, I.-M., Oh, M.-H., Sun, I.-H., et al. (2019). Glutamine blockade induces divergent metabolic programs to overcome tumor immune evasion. Science, 366(6468), 1013–1021.
Kaushik, I., Ramachandran, S., Prasad, S., & Srivastava, S. K. (2021). Drug rechanneling: A novel paradigm for cancer treatment. Seminars in Cancer Biology, 68, 279–290. https://doi.org/10.1016/j.semcancer.2020.03.011
Scatena, C., Roncella, M., Di Paolo, A., Aretini, P., Menicagli, M., Fanelli, G., et al. (2018). Doxycycline, an inhibitor of mitochondrial biogenesis, effectively reduces cancer stem cells (CSCs) in early breast cancer patients: A clinical pilot study. Frontiers in Oncology, 8, 452. https://doi.org/10.3389/fonc.2018.00452
Ózsvári, B., Sotgia, F., & Lisanti, M. P. (2020). First-in-class candidate therapeutics that target mitochondria and effectively prevent cancer cell metastasis: Mitoriboscins and TPP compounds. Aging, 12(11), 10162–10179, https://doi.org/10.18632/aging.103336.
Dankó, T., Petővári, G., Sztankovics, D., Moldvai, D., Raffay, R., Lőrincz, P., et al. (2021). Rapamycin plus doxycycline combination affects growth arrest and selective autophagy-dependent cell death in breast cancer cells. Int J Mol Sci, 22(15), https://doi.org/10.3390/ijms22158019.
Funding
Open access funding provided by Semmelweis University. The studies and research projects of the authors were funded by the National Research, Development, and Innovation Office of Hungary (NKFI-FK-128404) and the Higher Education Excellence Program at Semmelweis University, Hungary (FIKP).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
The included figures were created with BioRender (https://biorender.com) covered by 1st Department of Pathology and Experimental Cancer Research institutional license.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Sebestyén, A., Dankó, T., Sztankovics, D. et al. The role of metabolic ecosystem in cancer progression — metabolic plasticity and mTOR hyperactivity in tumor tissues. Cancer Metastasis Rev 40, 989–1033 (2021). https://doi.org/10.1007/s10555-021-10006-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10555-021-10006-2