
Abstract
Distant metastasis is the major contributor to the high mortality rate of colorectal cancer (CRC). To overcome the poor prognosis caused by distant metastasis, the mechanisms of CRC metastasis should be further explored. Epigenetic events are the main mediators of gene regulation and further affect tumor progression. Recent studies have found that some epigenetic enzymes are often dysregulated or mutated in multiple tumor types, which prompted us to study the roles of these enzymes in CRC metastasis. In this review, we summarized the alteration of enzymes related to various modifications, including histone modification, nonhistone modification, DNA methylation, and RNA methylation, and their epigenetic mechanisms during the progression of CRC metastasis. Existing data suggest that targeting epigenetic enzymes is a promising strategy for the treatment of CRC metastasis.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Colorectal cancer (CRC) is the world’s fourth most deadly cancer, and the disease-specific mortality rate is nearly 33% in the developed world[1, 2]. According to statistics provided by the National Cancer Center of China, it accounted for approximately 319,486 new cancer cases and 164,959 cancer-related deaths in males and 235,991 new cases and 121,203 deaths in females in 2020[3]. Many risk factors contribute to CRC, including increasing age, male sex, previous colonic polyps or previous CRC, unhealthy lifestyle or eating habits, inflammatory bowel disease and hereditary syndromes such as Lynch syndrome and familial adenomatous polyposis[1]. Although the outcomes of CRC patients have been improved with advanced technologies and therapies, the estimated death rate of CRC patients is still increasing year by year[3, 4].
The major contributor to the high mortality rate of CRC is distant metastasis. Approximately 25% of patients present with metastases at initial diagnosis, and almost 50% of patients with CRC ultimately develop metastases[5], which drove us to assess the mechanism contributing to CRC metastasis to prevent it from occurring. Tumor metastasis is a multistep process that presents as an invasion-metastasis cascade, including local invasion, intravasation and survival in the circulation, arrest at distant organs and extravasation into the parenchyma of distant tissues, formation of micrometastases and restart of the proliferative program[6]. Several biological processes contribute to CRC metastasis. Epithelial-to-mesenchymal transition (EMT), a major metastatic contributor, is a biological process in which epithelial cells transform into cells with a mesenchymal phenotype through specific procedures. EMT increases the metastatic potential of carcinoma cells by increasing their migratory and invasive capacities, which facilitates their movement out of primary tumor sites and into the circulation[7]. Cancer stem cells (CSCs), a small subset of cells with self-renewal and tumor-initiating ability within tumors, have also been proven to play an important role in CRC metastasis. Compared to non-CSCs, CSCs exhibit enhanced metastasis-related traits, such as motility, invasiveness and resistance to apoptosis[8]. Researchers found that colon cancer stem cells (CCSCs) are critical for the formation and maintenance of liver metastasis[9]. However, there is also a different view that CCSCs are indispensable for the outgrowth, but not the establishment, of metastases[10]. In addition to the above processes, the roles of the activation of multiple signaling pathways, metabolic reprogramming and immune escape in CRC metastasis cannot be ignored.
Wisniewski and coworkers compared the proteomes of formalin-fixed, paraffin-embedded (FFPE) samples containing colonic mucosa, primary colon tumors and nodal metastatic compartments. Although dramatic changes in the proteome were noted in the primary tumors compared to their matched normal tissues, the differences between the tumors and the metastases were much subtler[11]. Dynamic epigenetic modifications may be able to account for the mechanisms of CRC metastasis that cannot be explained simply by proteomic changes. At present, studies related to epigenetic modifications in the context of metastasis mainly focus on the following aspects: (a) epigenetic modification of EMT-inducing transcription factors (EMT-TFs), such as Snail and Slug, and their downstream effectors; (b) epigenetic modification of components of key CSC pathways that can also regulate the EMT process, such as the Wnt/β-catenin, Hedgehog, and Notch signaling pathways. (c) epigenetic modification of key effectors or metabolic enzymes in metabolic pathways and epigenetic modification of molecules that are crucial to recruit immunosuppressive cells. A large number of studies have shown that epigenetic enzymes are often dysregulated or mutated in cancer, and the subsequent changes in epigenetic modifications can further change gene transcription and protein stability to promote metastasis[12, 13]. In this review, we summarized several major epigenetic enzymes and their modifications and discussed their molecular mechanisms and clinical applications in the process of CRC metastasis.
Posttranslational modification in CRC metastasis
Posttranslational modifications include histone modifications and nonhistone modifications, which play a pivotal role in the development and progression of CRC. According to the different functional groups added, posttranslational modifications can be classified as acetylation, methylation, phosphorylation, ubiquitination, sumoylation, succinylation, ADP ribosylation, lactylation, isonicotinylation and so forth. In the next part of this review, more attention will be given to enzymes that mediate methylation and acetylation and their roles in CRC metastasis (Table 1). Lysine and arginine are the most common modification sites, and these modifications are catalyzed by acetylases and methylases. Simply, lysine and arginine residues on the N-terminal tails of histones are subject to methylation and acetylation, and these alternations further regulate the chromatin structure, thus affecting gene transcription. For nonhistone proteins, acetylation and methylation may regulate protein stability and further affect protein activity and function.
Acetylation and Deacetylation
CREB-binding protein (CBP)/p300
CBP and its homolog p300 are two acetyltransferase enzymes in humans and most higher eukaryotes[14]. CPB/p300 are multifunctional proteins that can acetylate diverse signaling effectors, enhancer-associated regulators and all four core histones[15]. Approximately 21,000 acetylation sites on 5,300 proteins can be acetylated by CBP/p300, and CBP/p300-regulated sites are significantly enriched for transcription and chromatin regulation[15].
To date, several studies have reported that CBP/p300 participates in the mechanism of CRC metastasis. LncRNA SATB2-AS1 recruits p300 and accelerates the p300-mediated acetylation of H3K27 and H3K9 at the SATB2 promoter to upregulate SATB2, which inhibits the invasion and migration of CRC cells[16]. CREPT, a tumor-related gene significantly associates with the poor overall survival (OS) of CRC, promotes the CRC metastasis by recruiting p300 and enhancing the acetylation of H3K27 and H4 in the c-myc promoter region to directly control its transcriptional efficacy[17, 18]. In addition to promoting histone acetylation, CBP/p300 also promotes the acetylation of nonhistone proteins to participate in the process of CRC metastasis. CREPT, as described above, can also enhance the interaction between p300 and β-catenin to promote p300-mediated acetylation and stability of β-catenin[18]. DOT1L is a new substrate of CBP, whose acetylation at K358 protects DOT1L from degradation to promote CRC metastasis[19]. ArhGAP30, a Rho GTPase-activating protein, promotes CRC cell migration in a p53-dependent manner, which upregulates p53 activity by enhancing the acetylation of p53 at the Lys382 site in the presence of p300[20]. A recent study found that CRC tumor-initiating cells (TICs) expressing CD110, a thrombopoietin (TPO)-responsive homodimeric receptor, mediated liver metastasis[21]. TPO expression strengthens the interaction between p300 and LRP6, which accelerates acetylation of LRP6 at the K802 site to activate LRP6 and further stimulate self-renewal of CD110 + TICs, thus promoting CRC metastasis[22]. Although these results indicate that CBP/p300 has an essential role in the mechanism of CRC metastasis, the relationship between CBP/p300 and some clinical features, such as tumor-node-metastasis (TNM) stage, survival rate and metastasis rate, is inconsistent[23,24,25]. These results indicate that CBP/p300 prefers to act as a downstream effector to assist the function of some important proteins rather than as a metastasis-initiating protein.
P300/CBP associated factor (PCAF)
PCAF, an acetyltransferase of the GNAT family, was first described by its competition with adenoviral oncoprotein E1A for binding to CBP/p300[26]. PCAF controls gene transcription by promoting the acetylation of H3K9[27], H3K14[28], and H4[29]; moreover, PCAF accelerates the acetylation of some nonhistone proteins, such as EZH2[30], PGK1[31], p53[32], and PTEN[33], to take part in the process of tumor progression. Previously, several studies reported that CXCL12 was involved in the metastasis of CRC[34, 35]. Romain and coworkers identified that both PCAF and CXCL12 were downregulated in colorectal tumor samples and found that a PCAF expression plasmid could significantly increase the CXCL12 gene expression level, which drove them to speculate that acetylation of the CXCL12 promoter may explain this phenomenon[36]. β-Catenin also plays a key role in CRC metastasis[37]. PCAF increases the transcriptional activity and nuclear accumulation of β-catenin, and this process greatly depends on the HAT2 domain of PCAF[38]. A pull-down assay revealed that PCAF not only coimmunoprecipitated with β-catenin but also promoted the acetylation of the K19 and K49 sites of β-catenin[38]. Encouraged by this result, researchers manipulated PCAF expression in CRC cells. The results were consistent with what they expected: knockdown of PCAF promoted cell differentiation and inhibited cell migration and tumor growth in vivo[38].
The histone deacetylase (HDAC) family
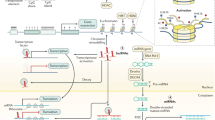
HDACs are a family of proteases that can be divided into four classes. HDACs deacetylate histones and lead to chromatin condensation, which might cause decreased or increased gene transcription. Several researchers have reported the clinical significance of HDAC family in CRC. They found that the positive rates of HDAC1, HDAC2, and HDAC3 expression were 36.4%, 57.9%, and 72.9%, respectively, in CRC tissues, and elevated HDAC levels significantly correlated with reduced patient survival[39, 40]. Furthermore, they also reported that the expression of all three HDAC isoforms was higher in the tumors with distant metastasis (HDAC1, P = 0.037; HDAC2, P = 0.045; HDAC3, P = 0.062), suggesting that they can play a prometastatic role in CRC[39]. An in vivo tumor xenograft assay showed that the HDAC inhibitor JNJ-26,481,585 strongly induced pan-H3 acetylation in tumor tissues and fully inhibited the growth of C170HM2 colorectal liver metastases[41]. These results indicated the prognostic impact of these HDACs and the treatment value of HDAC inhibitors. HDACs modulate CRC metastasis through various mechanisms. MMP3 and Claudin-1, proteins mainly associated with the invasion potential of cancer cells, can be epigenetically or nonepigenetically regulated by HDACs[42, 43]. It has been reported that HDACs can also reprogram the tumor immune microenvironment to modulate the metastatic process (Fig. 1). HDAC3 inhibitor treatment increases the Ac-H3 level at B7x promoter and promotes the interaction of C/EBP-α with the B7x promoter to upregulate the expression of B7x, an immune checkpoint molecule crucial to the immune escape of tumors, which contributes to HDAC inhibitor resistance in CRC[44]. Combined treatment with an HDAC inhibitor and B7x neutralizing antibody increased the infiltration of CD8 + and CD4 + T cells in CRC tissue from metastatic tumor xenografts and reduced the lung metastasis of the CRC model[44]. Nair and coworkers compared the RNA-sequencing data between CRC tissue-derived and normal tissue-derived immature myeloid-derived suppressor cells (I-MDSCs), a class of immune suppressive cells crucial to tumor metastasis, and found that 148 of the upregulated genes in tumor-infiltrating I-MDSCs were involved in HDAC activation and that HDAC inhibitors significantly reduced MDSC function and the expression of recruitment-associated genes ARG1, CCR2, and ITGAL[45]. All these results suggested that HDAC-mediated deacetylation might be involved in the mechanism of CRC metastasis and indicated the possibility of HDACs as targets of immune environment regulators in CRC.

HDAC inhibition represses tumor metastasis by reprogramming the tumor immune microenvironment
HDAC3 inhibition enhances B7x expression, and combined treatment with an HDAC inhibitor and a B7x neutralizing antibody increases the infiltration of CD8 + and CD4 + T cells. HDAC2 and HDAC3 inhibition can decrease the activity and recruitment of I-MDSCs in the tumor microenvironment, which further suppresses the metastatic ability of CRC cells.
The mammalian sirtuin (SIRT) family
The SIRT family contains seven members (SIRT1–7), which are evolutionarily conserved NAD+-dependent histone deacetylases or ADP-ribosyltransferases belonging to the class III HDAC family. Members of the SIRT family localize to different sites of the cell and are involved in many cellular processes, such as DNA repair, cell cycle regulation and cell metabolism, and the protection of cells from oxidative stress[46].
Many assays performed on patient tissues have shown that the SIRT family members are differentially expressed in CRC cancer tissues compared to adjacent normal tissues. For example, SIRT1, SIRT6 and SIRT7 are overexpressed in CRC tissues, and their upregulation is significantly associated with advanced tumor-node-metastasis (TNM) stage, a poor prognosis, decreased overall survival and disease-free survival in CRC patients[47,48,49,50]. Furthermore, the overexpression of SIRT1, SIRT6 and SIRT7 is positively correlated with lymph node or liver metastases[47, 48, 50]. Many articles have confirmed the prometastatic role of SIRT1. It was reported that Fra-1 is a component of AP-1 complex that can promote tumor-associtated EMT by directly regulating EMT-TFs, and SIRT1 enhances the EMT process in a Fra-1-dependent manner[49, 51]. However, the acetylation level of Fra-1 was not discussed in this article. Because of the crucial role of CSCs in CRC metastasis, Wang et al. found that SIRT1 could decrease H3K9ac enrichment on the promoter of miR-1185-1 and thus cause chromatin remodeling to repress miR-1185-1 expression, which further upregulated the CD24 level to promote the stemness and migration of CRC cells[52]. Some noncoding RNAs have been found to be associated with SIRT1 expression and to further contribute to the metastasis process of CRC. An in vitro assay proved that SIRT1 was a direct target of microRNA-199b and that SIRT1 further affects the acetylation level of CREB to upregulate the transcriptional activity of CREB[53]. In addition, SIRT6 promotes the EMT process of CRC cells in two different ways: it acts as a reader of snail and also suppresses TET1 transcription by modulating H3K9 deacetylation[50]. Controversially, many researchers have pointed out that SIRT6 and SIRT7 play a tumor inhibitory role via a variety of mechanisms, such as antagonizing the c-myc oncogene, regulating the DNA damage response, and inhibiting JAK2/STAT3 and PTEN/AKT signaling[54,55,56,57], but their roles in CRC metastasis were not discussed in these articles.
SIRT2 and SIRT4 seem to play metastasis-inhibiting roles in CRC. Recently, IDH1 has received great attention because of its effect on cancer metabolism[58]. Researchers found that IDH1 k224 acetylation tightly controls enzymatic activity through the HIF1a-SRC axis and promotes the liver metastasis of CRC and that SIRT2 can be a potential strategy for preventing CRC liver metastasis because of its role in inhibiting the acetylation and enzymatic activity of IDH[59]. Through the immunohistochemical analysis of SIRT2 protein expression in colorectal tissue specimens, they also confirmed that SIRT2 expression was significantly decreased in CRC tissues or liver metastases compared with corresponding colorectal normal tissues[59]. Similarly, SIRT4 expression was found to be negatively associated with lymph node metastasis, lymphatic invasion, and distant metastasis[60]. Consistent with its associated clinicopathological features, SIRT4 overexpression decreased the invasion and migration ability of CRC cells by suppressing miR-200c to further affect E-cadherin expression[60]. From these articles, we can easily conclude that the roles of the SIRT family in CRC metastasis are diverse. In addition to using different kinds of CRC models, integrated analysis of multiple SIRT members and assessment of the role of the same SIRT member in different stages of CRC development, such as pre- and post-metastasis, also need to be performed.
Methylation and demethylation
Enhancer of zeste homolog 2 (EZH2)
The EZH2 gene encodes histone lysine N-methyltransferase, a catalytic member of the PRC2 complex, which methylates H3K9 and H3K27 at the posttranslational level, leading to the transcriptional repression of target genes. EED and SUZ12, two other subunits of PRC2, are indispensable for the enzymatic activity of EZH2[61]. Additionally, it was reported that EZH2 can also methylate H3K4 in a PRC2-independent manner to play a gene activation role[62].
Many studies have demonstrated that the mRNA and protein expression of EZH2 is significantly increased in CRC tissues compared with adjacent noncancerous tissues, and that EZH2 overexpression is closely associated with reduced OS and disease-free survival (DFS)[63, 64]. In addition, the high level of EZH2 expression has been strongly linked to both regional lymph node (p < 0.001) and distant metastasis (p = 0.004) [63, 64]. Moreover, one allelic variant (rs3757441 C/C) of EZH2 is significantly associated with shorter progression-free survival (PFS) and OS in metastatic CRC patients[65]. Such findings indicated the prognostic and therapeutic value of EZH2 for advanced CRC patients.
Many studies have verified that EZH2 plays a prometastatic role in various cancer types, and that the pharmacological inhibition of EZH2 can significantly repress the migration and invasion of the cancer cells[66, 67]. Similarly, in CRC, EZH2 can act as a downstream regulator of some important signaling pathways and noncoding RNAs, thus promoting CRC metastasis. TGF-β-MTA1-SOX4 signaling drives the EMT process to promote CRC metastasis. In this signaling axis, EZH2 acts as a downstream regulator of EMT-associated factors such as E-cadherin, ZO-1, snail, and slug[68]. Activation of the Erk/Akt signaling pathway induces EZH2 overexpression to repress the transcription of ITGα2 and E-cadherin by affecting the enrichment level of H3K27me3 on their promoters[69]. In addition, EZH2 acts as a downstream regulator of lncRNAs/miRNAs to promote CRC metastasis[70,71,72,73,74,75]. For example, lncRNA MALAT1and lncRNA SNHG14 can regulate EZH2 to further affect H3K27me3 recruitment on the E-cadherin and EPHA7 promoters, respectively[71, 74]. MiR-101 inhibits the invasion and migration of CRC cells[76]. The downregulation of miR-101 enhances the stability of EZH2 by regulating O-GlcNAcylation on EZH2, which, in turn, further reduces miR-101 expression by recruiting H3K27me3 to its promoter[76]. Although many other articles have proven that EZH2 is a downstream target of many lncRNAs/miRNAs, it has not been discussed whether EZH2 exerts its prometastatic function through histone modification[70, 72, 73, 75]. Therefore, intensive studies of the mechanism are urgently needed.
Suppressor of variegation 3–9 homolog 1 and suppressor of variegation 3–9 homolog 2 (SUV39H1 and SUV39H2)
Suv39h1 and Suv39h2 are H3K9 selective histone methyltransferases that were first isolated and characterized in mice and identified as modulating chromatin dynamics in somatic cells[77, 78]. They epigenetically modulate functional proteins to control telomere length[79], heterochromatin organization, chromosome segregation, and mitotic progression[78, 80]. In recent years, many studies have proven that SUV39H1 and SUV39H2 play an important role in various cancer types, such as CRC[81, 82], melanoma[83], breast cancer[84], cervical cancer[85] and hematologic malignancies[86, 87].
In CRC, SUV39H1 is significantly upregulated in tumor tissues compared to normal colon tissues[88]. Overexpression of wild-type SUV39H1 increased H3K9me3 levels as well as the migration and invasion abilities of CRC cells, and this effect was eliminated when the SUV39H1-C326A mutant, which lacks enzymatic activity, was introduced[89]. Although the functional genes enriched by H3K9me3 were not discussed in this article, these results indicated the potential relation of SUV39H1 with the metastasis process of CRC. Similar to SUV39H1, it was reported that high SUV39H2 expression is strongly associated with distant metastasis and TNM stage and predicts shorter OS and PFS for CRC patients[81]. SUV39H2 enhances CRC metastasis by directly binding to the SLIT1 promoter and catalyzes H3K9me3 to suppress SLIT1 expression[81]. A small molecule inhibitor of SUV39H1 has been developed, and it can suppress human colon tumor xenograft growth in vivo[88], which confirmed the therapeutic value of SUV39H1 in CRC. Therefore, the metastasis inhibition ability of this SUV39H1 inhibitor needs to be further tested.
Protein arginine methyltransferases (PRMTs)
PRMT1 and PRMT5 are the major members of type I and type II PRMT families that catalyze asymmetric dimethylarginine (ADMA) and symmetric dimethylarginine (SDMA) deposition on proteins. They have been reported to regulate multiple cellular processes, including transcriptional activation and repression, RNA splicing, protein synthesis, DNA damage response, signal transduction and liquid–liquid phase separation[90]. A type I PRMT inhibitor (GSK3368715) has already been shown to inhibit proliferation in patient-derived DLBCL models and several cell lines that represent the majority of tumor types. A synergistic cancer cell growth inhibition effect was observed when PRMT5 was inhibited with GSK3368715, which further indicated the therapeutic value of type I and type II PRMTs[91].
In CRC, PRMT1 promotes cell migration and invasion through histone arginine methylation and nonhistone arginine methylation. PRMT1-mediated H4R3me2a directly recruits SMARCA4 to promote the migration of CRC cells by further activating TNS4 and EGFR[92]. For nonhistone arginine methylation, PRMT1 induces asymmetric demethylation of the R251 site of NONO, and compared to NONO WT cells, NONO R251K mutant-expressing CRC cells show reduced migration and invasion. Pharmacological inhibition of PRMT1 significantly reduces the ADMA level of NONO and abrogates the malignant phenotype associated with NONO R251 ADMA in both KRAS WT and KRAS mutant CRC cells[93].
PRMT5 modulates CRC cell migration ability through multiple pathways, including NF-kB/p65 signaling and EGFR/Akt/GSK3β signaling[94, 95]. However, the modification sites mediated by PRMT5 are not discussed in these articles. PRMT5 also has a synergistic effect with different kinds of modifications to promote CRC metastasis. SIRT7-mediated K3 and K243 deacetylation of WDR77 reduces WDR77 interaction with PRMT5 and further affects the transmethylase activity of the WDR77/PRMT5 complex, resulting in a reduction in H4R3me2 modification, which is related to the migration ability of CRC cells[96]. Although PRMT5 has been reported to methylate H3R8, H3R2, and H4R3 to facilitate transcriptional activation or regression in cancer[97], there are few studies related to the exact site methylated by PRMT5 in the process of CRC metastasis. Therefore, further studies are urgently needed.
Lysine-specific demethylase 1 (LSD1)
LSD1 was the first-discovered histone demethylase, and it mainly demethylates mono- or dimethylated H3K4 and K3K9[98]. In addition, it has also been proven to demethylate some nonhistone functional proteins, such as p53, E2F1, and DNMT1[98]. The catalytic activity of LSD1 resides in the AO domain and is dependent on its cofactor flavin-adenine dinucleotide (FAD)[99].
Several studies have shown that LSD1 is aberrantly expressed in multiple cancer types and has great significance in the process of cancer metastasis[100,101,102]. In CRC, the role of the LSD1 is controversial. Some studies reported that a higher level of LSD1 predicted a better outcome, and a lack of LSD1 significantly correlated with lymph node metastasis or advanced tumor stage[103, 104]. However, many studies have shown that LSD1 plays a prometastatic role in CRC. LSD1 induces demethylation of H3K4me2 at the CDH1 promoter, downregulates CDH1 expression, and consequently accelerates the EMT process[100]. TSPAN8 is a metastasis-promoting tetraspanin that coordinates with CD151 to promote cancer metastasis by recruiting MMP9 and MMP13[105]. LSD1 upregulates TSPAN8 expression by reducing H3K9me2 enrichment at the TSPAN8 promoter and further promotes the EMT process[106]. In addition, it has been reported that LSD1 interacts with Slug (EMT-related transcription factor) and represses the promoter activity of E-cadherin to promote invasion and migration of CRC cells, but whether this function depends on LSD1-mediated histone demethylation was not discussed in this paper[107]. LSD1 not only increases histone demethylation but also enhances the demethylation of nonhistone proteins to promote CRC metastasis. RIOK1 is an atypical serine/threonine kinase. RIOK1 increases the invasion and migration of CRC cells and promotes lung metastasis in vivo[108]. LSD1 demethylates RIOK1 to reverse SETD7-mediated RIOK1 methylation-dependent degradation, thus increasing its stability[108].
Jumonji domain-containing proteins (JMJD)
JMJD proteins are a family of histone demethylases containing the JMJC catalytic domain. Most of these family members are identified to mainly demethylate H3K4, H3K9, H3K27, H3K36 and H4K20, and a small group of JMJD proteins can also demethylate H3R2 and H4R3[98]. Several studies have shown that JMJD proteins affect the development of many cancer types[98, 109]. In CRC, the JMJD-driven mechanisms of metastasis have also been described in many articles.
A previous study reported that the levels of KDM4C mRNA (encoding JMJD2C protein) were significantly correlated with TNM stage, distant metastasis, OS and tumor recurrence in CRC[110]. In this study, JMJD2C increased the migration rate of CRC cells in vitro and promoted lung metastasis in vivo. JMJD2C is mainly located in the nuclei of CRC cell lines and decreases the levels of H3K9me3 and H3K36me3 on the MALAT1 promoter to enhance the transcriptional level of MALAT1, which promotes tumor growth and metastasis in CRC[110]. JMJD2D, another member of the JMJD family, is significantly upregulated in human colorectal tumor tissues versus control normal tissues and correlates with the level of proliferating cell nuclear antigen[111]. JMJD2D promotes CRC metastasis via multiple pathways, such as the Wnt/β-catenin signaling pathway, hedgehog signaling pathway and glycolysis[111,112,113]. In these pathways, JMJD2D demethylates the methyl groups of H3K9me3 at the promoters of β-catenin and its’ target genes (Myc, MMP9), Gli2, mTOR, HIF1 β, and PGK1 to increase their transcription and exert its prometastatic function[111,112,113]. JMJD1A was discovered to be an independent prognostic marker of CRC, and its expression levels were also significantly associated with lymph node metastasis, lymphatic invasion, venous invasion, and the depth of tumor invasion[114]. These signatures prompted researchers to explore the mechanism by which JMJD1A promotes CRC metastasis. They found that the demethylase activity of JMJD1A was required for CRC metastasis that it decreased H3K9me2 levels at the promoters of β-catenin, c-myc and MMP9 genes to activate Wnt/β-catenin signaling [115].
DNA modification
DNA methyltransferases (DNMTs)
DNA methylation is an epigenetic process in which a methyl group transfers onto the C5 position of the cytosine to form 5-methylcytosine (5mC) under the action of DNMTs. DNA methylation mainly occurs on CpG islands. In mammalian genomes, it has been reported that approximately 60 − 70% of CpG islands can be methylated[116]. The function of DNA methylation is closely related to maintaining the stability of genetic information, transcriptional inhibition and activation, X chromosome inactivation, reprogramming mammalian development and some diseases, such as neurological disorders and immunodeficiency[117]. Aberrant DNA methylation frequently occurs in cancer and is associated with tumor suppressor gene silencing and prevents their activation.
DNMT1, DNMT3A and DNMT3B are the most frequently studied DNMTs in CRC, and they contribute to CRC metastasis through various mechanisms (Table 2). To date, several studies have focused on the interaction between noncoding RNA and DNA methylation in the development of CRC. The human 14q32 locus encodes metastasis-suppressive miRNAs that suppress the adhesion, invasion, and migration properties of tumor cells and metastatic colonization of distant sites[118]. Oshima and his coworkers found that 14q32 locus-encoded miRNAs were overexpressed in a DNMT1/DNMT3B−/− DKO cell model and that the hypomethylation of MEG3-DMR, which acts as a cis-regulatory element for 14q32 miRNA expression, exhibited constitutive expression of 14q32 miRNAs[119]. Pharmacologic inhibition of DNA methylation by 5-Aza-dC, an inhibitor of DNA methyltransferases, induces 14q32 miRNA expression and restricts CRC liver metastasis[119]. In addition, DNMT1 and DNMT3B can be targeted by some miRNAs, such as miR-342, miR-124, and miR-506, thereby reducing the global DNA methylation level to restore the expression of tumor suppressive genes, such as E-cadherin, MGMT, P16, ADAM23, Hint1, RASSF1A, and RECK, thus further inhibiting the metastatic potential of CRC[120, 121]. Some cytokines, such as interleukins and chemokines, have also been reported to be involved in CRC metastasis. It has been reported that IL-23 selectively promotes the migration and invasion ability of SW620 cells compared with SW480, HT29, and HCT116 cells[122]. Socs3, an inhibitor of IL-23/stat signaling, was found to be differentially methylated in these cell lines, and this effect was DNMT1 dependent[122]. Wendt el al[123] found that CXCL12 was silenced by DNA hypermethylation in primary colorectal carcinomas as well as colorectal carcinoma-derived cell lines. Double knockout of DNMT1 and DNMT3b restored CXCL12 expression. Finally, the authors confirmed that stable expression of CXCL12 in CRC cell lines can significantly reduce metastatic tumor formation. These results further indicated the importance of DNA methylation in the process of CRC metastasis.
Ten-eleven translocation (TET) family
TET family contains TET1, TET2 and TET3 proteins, which catalyze the successive oxidation of 5mC to 5-hydroxymethylcytosine (5hmC), 5-formylcytosine (5fC), and 5-carboxylcytosine (5caC)[124]. TET proteins have been widely studied in hematological malignancies. Somatic alteration of TET2, TET1/TET2 deficiency (TET1/2 double knockout), and TET2/TET3 disruption (TET2/3 double knockout) can lead to a wide range of myeloid and lymphoid malignancies, late-onset B-cell lymphoma, and rapid and fully penetrant myeloid leukemia[124].
In contrast to the frequent mutation of TET proteins in blood cancers, TET proteins are always downregulated in many cancer types, such as melanoma[125], glioblastoma[126], breast cancer[127] and prostate cancer[128]. In CRC, loss of TET3 expression can coexist with TET3 frameshift mutation, which may be related to the development of CRC with high microsatellite instability (MSI-H)[129]. In addition, TET1 and TET2 are downregulated in BRAFV600E-mutated colon cancers. Overexpression of BRAFV600E in BRAF wild-type CRC cell lines can significantly repress TET1/TET2 expression, which leads to the hypermethylation of CIMP genes to promote the development of CpG island methylator phenotype colon cancer (CIMP-CC)[130]. Ma et al[131] found that the downregulation of TET1 inhibited the migration of CRC cells; they further found that TET1 regulates hypoxia-responsive genes such as VEGF, Glut1, and EPO by mediating the binding of HIF-1α to hypoxia-response element (HREs) of these genes by changing their CpG methylation levels (Table 2).
N6-methyladenosine (m6A) modification
m6A is the most prevalent internal modification in mRNAs and noncoding RNAs (ncRNAs) in higher eukaryotes and is highly conserved in eukaryotes[132]. Several lines of evidence indicate that m6A modification is present on lncRNAs, circRNAs[133], and pre-miRNAs[134] and suggest the importance and potential therapeutic value of m6A modification. In CRC, it was reported that m6A modification accelerates CRC progression by facilitating the glycolytic process of cancer cells[135, 136], inhibiting the immune response of the tumor microenvironment[137], maintaining tumor stem cells and promoting chemoresistance[138,139,140].
Methyltransferase-like3 (METTL3) and methyltransferase-like 14 (METTL14) are the most widely studied m6A writers in recent years. In CRC, METTL14 is remarkably downregulated in cancerous compared to paired normal samples. Decreased expression of METTL14 is positively correlated with larger tumor size, lymphatic invasion, and remote metastasis and implicates worse RFS[133, 141]. Furthermore, data from The Cancer Genome Atlas (TCGA) showed that METTL14 was positively correlated with OS and was an independent risk factor[141]. These results suggest that METTL14 is a reliable prognostic marker of CRC patients. METTL14 inhibits CRC metastasis by regulating multiple targets (Fig. 2). SOX4 is a downstream target of METTL14. METTL14 knockdown enhances SOX4 mRNA stability in an m6A-YTHDF2-dependent manner and further promotes the EMT process and PI3K/Akt signaling[142]. In addition, METTL14 inhibits CRC growth and metastasis by downregulating lncRNA XIST. METTL14 forms a complex with WTAP and induces the m6A process to suppress XIST expression through YTHDF2-dependent RNA degradation[141]. METTL14 also inhibits CRC metastasis via the miR-375/SP1 pathway[143]. METTL14-dependent m6A methylation enhances pre-miR-375 binding to DGCR8, thereby promoting the DGCR8-mediated maturation of pre-miR-375, which further targets SP1 to accelerate cell invasion and migration[143].
METTL3 is likely to be upregulated in CRC. High METTL3 expression correlates with lymph node invasion and distant metastasis. METTL3 enhances the metastatic potential of CRC cells by promoting m6A modification on pri-miR-1246 to upregulate the level of mature miR-1246, thereby affecting the function of its target gene SPRED2 and the activity of the MAPK pathway[133, 144]. Zhou et al[145] found that METTL3/YTHDF2-mediated m6A modification suppressed YPEL5 expression, which further regulated the PCNA and CCNB1 levels. LINC00460 enhances the interactions between DHX9 or IGF2BP2 and HMGA1, which leads to the upregulation of HMGA1 and promotes CRC growth and metastasis. Interestingly, this process depends on METTL3-mediated m6A modification of HMGA1 mRNA[146]. Chen and his coworkers identified circNSUN2 for the first time and found that it was positively associated with lymph node metastasis and liver metastasis in a cohort of clinical samples and that knockdown of circNSUN2 in patient-derived xenograft (PDX) CRC models significantly inhibited liver and lung metastasis[133]. YTHDC1, an m6A reader that identifies m6A-methylated circNSUN2, facilitates circNSUN2 export from the nucleus to the cytoplasm in an m6A-dependent manner. Consequently, circNSUN2 interacts with IGF2BP2 to stabilize HMGA2 mRNA. They also found that METTL3 plays an important role in this process, affecting the activity of circNSUN2. Once they mutated the m6A modification site (GAACU) in the circNUSN2-overexpressing construct, the m6A modification level of circNUSN2 was downregulated, and the invasion ability of CRC cells was attenuated. Additionally, METTL3 has also been found to mediate the m6A modification of lncRNA RP11 to trigger the dissemination of CRC cells via upregulation of Zeb1[147]. Taken together, these results reveal the diversity of m6A modification targets and suggest that interfering with the interaction between m6A modification and target RNAs is an important way to inhibit colon cancer metastasis.

METTL14- and METTL3-mediated m6A modification promotes CRC cell metastasis via various targets and mechanisms
METTL14 and METTL3 participate in CRC metastasis by inducing the m6A modification of multiple targets, including mRNAs, pre-miRNAs, miRNAs, lncRNAs, and circRNAs.
Potential clinical application and targeted therapy for CRC metastasis
Due to the lethality of CRC distant metastasis, some predictive markers with low-cost, rapid, high-accuracy and noninvasive characteristics need to be urgently discovered. Alterations in DNA methylation is a hallmark of CRC. A recent study from the Mayo clinic compared 14 methylated DNA markers (MDMs) in primary and metastatic CRC for feasibility in the detection of distantly recurrent/metastatic CRC in plasma[148]. They found that the levels of 14 selected MDMs (VAV3, CHST2, OPLAH, QKI, PPP2R5C, ARHGEF4, PDGFD, ZNF625, SFMBT2, LRRC4, DOCK10, IKZF1, NDRG4, BMP3) were remarkably similar between paired primary and metastatic CRC samples. Thirteen of the MDMs had high accuracy in detecting primary CRC at all stages, and the sensitivity of the 13 MDMs increased with increasing CRC stage (64% for stage I CRC; 62% and 65% for stage II CRC; 79% and 71% for stage III CRC, and 100% for stage IV CRC)[148]. Moreover, they observed that the trained model of MDMs with or without CEA distinguished patients with recurrent CRC from patients with no radiographic evidence of disease (NED) at the immediate previous follow-up with 80% (44–97%) sensitivity and detected metastatic CRC in patients actively undergoing palliative treatment with 93% (78–99%) sensitivity. The panel of MDMs with or without CEA detected recurrent CRC liver metastases with 100% (86–100%) sensitivity, lung metastases with 89% (52–100%) sensitivity, and peritoneal/nodal metastases with 57% (18–90%) sensitivity. Lesions with RECIST sum > 4 cm and ≤ 4 cm were detected with 100% (81–100%) and 83% (59–96%) sensitivity, respectively. The panel of MDMs with or without CEA detected recurrent rectal cancer with 92% (62–100%) sensitivity, left-sided colon cancer with 95% (75–100%) sensitivity, and right-sided colon cancer with 75% (35–97%) sensitivity[148]. This study indicated that the MDMs model was a highly sensitive, reliable, and stable model for detecting early- and late-stage CRC and a promising model for detecting CRC recurrence and metastasis, which strongly proved the clinical application of MDMs in CRC.
Some clinical trials related to epigenetic modifiers for advanced CRC have been tested to observe their efficacy (Table 3). In this review, we mainly focus on DNMT inhibitors, HDAC inhibitors and EZH2 inhibitors. Azacitidine, decitabine and guadecitabine are inhibitors of DNMT that have shown clinical efficacy in the treatment of hematologic malignancies[149, 150]. In CRC, the combined use of DNMT inhibitors and other therapies can be more effective. In a phase I/II study in refractory CIMP-high metastatic colorectal cancer (mCRC), azacitidine combined with capecitabine and oxaliplatin was well tolerated with high rates of stable disease (SD), although no objective responses were reported[151]. Another phase I study combining guadecitabine (SGI-110) with irinotecan in mCRC patients previously exposed to irinotecan reported that treatment with guadecitabine 45 mg/m2 and irinotecan 125 mg/m2 with growth factor support (GFS) was safe and tolerable in patients with mCRC, and 12/17 evaluable patients had SD as the best response, while one had a partial response (PR)[152]. DNMT inhibitors also showed efficacy when combined with targeted therapy. A phase I/II study of decitabine in combination with panitumumab showed good tolerance and activity (10% patients had PR and 50% had SD) in patients with KRAS wild-type mCRC previously treated with cetuximab[153]. Additionally, some researchers have tried to modulate the CRC immune microenvironment through epigenetic treatment. Unfortunately, the combination of guadecitabine with the GVAX colon vaccine was tolerable but showed no significant immunologic activity in mCRC[154].
HDAC inhibitors have shown clinical efficacy and have been approved for the treatment of hematologic malignancies[155]. Among various HDAC inhibitors, varinostat (a small molecule inhibitor of class I and II HDAC enzymes) has been the most studied in advanced solid tumors and mCRC. A phase I study evaluated the safety and efficacy of varinostat in gastrointestinal (GI) cancer. A total of 16 patients received either vorinostat 300 mg bid for 3 consecutive days followed by 4 rest days per cycle (n = 10) or vorinostat 400 mg qd for 21 consecutive days per cycle (n = 6). They reported that vorinostat 300 mg bid for 3 consecutive days followed by 4 days of rest was better tolerated, and 50% of patients achieved SD[156]. This study indicated that vorinostat may be an active agent in the treatment of GI cancer. However, the efficacy of vorinostat combined with 5-fluorouracil (5-FU)-based chemotherapy seems to be limited[157, 158]. This may be due to 5-FU resistance and dose-limiting toxicity (DLT) in selected patients. Fu and coworkers conducted two phase I studies in solid tumors and found that combined treatment with vorinostat and pazopanib yielded significantly longer PFS and OS in patients with metastatic TP53 mutant solid tumors, especially in those with metastatic sarcoma or mCRC[159, 160]. This finding supports the combined use of vorinostat and antiangiogenic targeted therapy in TP53-mutant mCRC.
EZH2 inhibitors have shown great therapeutic potency in preclinical models of several cancer types. In recent years, many EZH2 inhibitors have been developed and are undergoing clinical trials[161]. Among them, GSK126 (GSK2816126) and tazemetostat have been clinically tested in advanced CRC. GSK126 is a highly selective, S-adenosyl-methionine-competitive inhibitor of EZH2 that can decrease global H3K27me3 levels and reactivate silenced PRC2 target genes[162]. A phase I study reported that the maximum-tolerated dose (MTD) of GSK126 was 2,400 mg, and modest anticancer activity was observed at tolerable doses in patients with advanced solid tumors (including CRC) or B-cell lymphomas[163]. This finding supports the potential use of EZH2 inhibitors in advanced CRC patients. In January 2020, tazemetostat was approved by the FDA for the treatment of adults and adolescents aged ≥ 16 years with locally advanced or metastatic epithelioid sarcoma not eligible for complete resection[164]. Subsequently, several clinical trials conducted in non-Hodgkin lymphoma showed clinically meaningful and durable responses[165, 166]. However, there are few clinical studies of tazemetostat in solid tumors. A phase I study conducted in B-cell non-Hodgkin lymphoma and advanced solid tumors showed a 38% durable objective response rate in B-cell non-Hodgkin lymphoma but only a 5% durable objective response rate in solid tumors with tazemetostat treatment[167]. This result indicates the limited efficacy of monotherapy with EZH2 inhibitors in advanced solid tumors, and more clinical trials on the combined use of chemotherapy or other targeted therapies with EZH2 inhibitors should be carried out in mCRC.
Conclusion and outlook
CRC metastasis is an important factor affecting the prognosis of CRC patients, and epigenetic modifications play a pivotal role in this process. In this review, we described the roles of several epigenetic enzymes in CRC and summarized their epigenetic mechanisms in the process of CRC metastasis. However, cancer metastasis is a highly complicated process that cannot simply be explained by the alteration of a single modification. Some studies have pointed out that the epigenome is widely altered in cancer[168, 169], which indicates that multiple epigenetic modifications may dynamically and synergistically affect gene function and promote tumor progression in a time- and space-dependent manner. Unfortunately, there are few studies on the synergistic role of multiple modifications in CRC metastasis. Therefore, more studies are needed to explore these potential synergistic effects to better understand the metastatic process of CRC.
In recent years, epigenetic changes have been gradually developed as clinical biomarkers for diagnosis, prognosis and treatment. Epigenetic modifiers have achieved good results in the clinical trials of hematologic malignancies and have received FDA approval for clinical application, suggesting that targeting epigenetic pathways is a promising strategy for cancer treatment. However, in solid tumors, the clinical effect of monotherapy with epigenetic modifiers seems to be limited. This may be due to epigenetic events that cooperate with the driver gene mutations and subsequently result in extensive changes in the tumor microenvironment. The combined use of multiple epigenetic modifiers and/or the combination of epigenetic modifiers with other therapies, such as traditional chemotherapy, targeted therapy and immunotherapy, may improve clinical outcomes. In addition, multiomics analysis of the tumor microenvironment in patients with mCRC, such as epigenomics, genomics, transcriptomics, proteomics and metabolomics analysis, may provide strong scientific evidence for the diagnosis, detection, prevention and epigenetic modifier-based combined treatment of mCRC in the future (Fig. 3).

Future direction for the development of epigenetic modifiers based mCRC therapy
Multiomics analysis of the tumor microenvironment in patients with mCRC, may provide strong scientific evidence for the diagnosis, detection, prevention and epigenetic modifier-based combined treatment of mCRC.
Data availability Statement
No data, models, or code were generated or used during the study.
References
Cunningham D, Atkin W, Lenz H-J, Lynch HT, Minsky B, Nordlinger B, Starling N (2010) Colorectal cancer. The Lancet 375(9719):1030–1047. doi:https://doi.org/10.1016/s0140-6736(10)60353-4
Brody H (2015) Colorectal cancer. Nature 521(7551):S1. doi:https://doi.org/10.1038/521S1a
Cao W, Chen HD, Yu YW, Li N, Chen WQ (2021) Changing profiles of cancer burden worldwide and in China: a secondary analysis of the global cancer statistics 2020. Chin Med J (Engl) 134(7):783–791. doi:https://doi.org/10.1097/CM9.0000000000001474
Chen W, Zheng R, Baade PD, Zhang S, Zeng H, Bray F, Jemal A, Yu XQ, He J (2016) Cancer statistics in China, 2015. CA Cancer J Clin 66(2):115–132. doi:https://doi.org/10.3322/caac.21338
Van Cutsem E, Cervantes A, Nordlinger B, Arnold D, Group EGW (2014) Metastatic colorectal cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol 25 Suppl 3iii1–9. doi:https://doi.org/10.1093/annonc/mdu260
Valastyan S, Weinberg RA (2011) Tumor metastasis: molecular insights and evolving paradigms. Cell 147(2):275–292. doi:https://doi.org/10.1016/j.cell.2011.09.024
Chaffer CL, San Juan BP, Lim E, Weinberg RA (2016) EMT, cell plasticity and metastasis. Cancer Metastasis Rev 35(4):645–654. doi:https://doi.org/10.1007/s10555-016-9648-7
Chaffer CL, Weinberg RA (2011) A perspective on cancer cell metastasis. Science 331(6024):1559–1564. doi:https://doi.org/10.1126/science.1203543
de Sousa e Melo F, Kurtova AV, Harnoss JM, Kljavin N, Hoeck JD, Hung J, Anderson JE, Storm EE, Modrusan Z, Koeppen H, Dijkgraaf GJ, Piskol R, de Sauvage FJ (2017) A distinct role for Lgr5(+) stem cells in primary and metastatic colon cancer. Nature 543(7647):676–680. doi:https://doi.org/10.1038/nature21713
Fumagalli A, Oost KC, Kester L, Morgner J, Bornes L, Bruens L, Spaargaren L, Azkanaz M, Schelfhorst T, Beerling E, Heinz MC, Postrach D, Seinstra D, Sieuwerts AM, Martens JWM, van der Elst S, van Baalen M, Bhowmick D, Vrisekoop N, Ellenbroek SIJ, Suijkerbuijk SJE, Snippert HJ, van Rheenen J (2020) Plasticity of Lgr5-Negative Cancer Cells Drives Metastasis in Colorectal Cancer. Cell Stem Cell 26(4):569–578e567. doi:https://doi.org/10.1016/j.stem.2020.02.008
Wisniewski JR, Ostasiewicz P, Dus K, Zielinska DF, Gnad F, Mann M (2012) Extensive quantitative remodeling of the proteome between normal colon tissue and adenocarcinoma. Mol Syst Biol 8:611. doi:https://doi.org/10.1038/msb.2012.44
Cruz-Rodriguez N, Combita AL, Zabaleta J (2018) Epigenetics in Hematological Malignancies. Methods Mol Biol 1856:87–101. doi:https://doi.org/10.1007/978-1-4939-8751-1_5
Han TS, Ban HS, Hur K, Cho HS (2018) The Epigenetic Regulation of HCC Metastasis. Int J Mol Sci 19(12). doi:https://doi.org/10.3390/ijms19123978
Dancy BM, Cole PA (2015) Protein lysine acetylation by p300/CBP. Chem Rev 115(6):2419–2452. doi:https://doi.org/10.1021/cr500452k
Weinert BT, Narita T, Satpathy S, Srinivasan B, Hansen BK, Scholz C, Hamilton WB, Zucconi BE, Wang WW, Liu WR, Brickman JM, Kesicki EA, Lai A, Bromberg KD, Cole PA, Choudhary C (2018) Time-Resolved Analysis Reveals Rapid Dynamics and Broad Scope of the CBP/p300 Acetylome. Cell 174(1):231–244e212. doi:https://doi.org/10.1016/j.cell.2018.04.033
Wang YQ, Jiang DM, Hu SS, Zhao L, Wang L, Yang MH, Ai ML, Jiang HJ, Han Y, Ding YQ, Wang S (2019) SATB2-AS1 Suppresses Colorectal Carcinoma Aggressiveness by Inhibiting SATB2-Dependent Snail Transcription and Epithelial-Mesenchymal Transition. Cancer Res 79(14):3542–3556. doi:https://doi.org/10.1158/0008-5472.CAN-18-2900
Zheng G, Li W, Zuo B, Guo Z, Xi W, Wei M, Chen P, Wen W, Yang AG (2016) High expression of CREPT promotes tumor growth and is correlated with poor prognosis in colorectal cancer. Biochem Biophys Res Commun 480(3):436–442. doi:https://doi.org/10.1016/j.bbrc.2016.10.067
Zhang Y, Wang S, Kang W, Liu C, Dong Y, Ren F, Wang Y, Zhang J, Wang G, To KF, Zhang X, Sung JJ, Chang Z, Yu J (2018) CREPT facilitates colorectal cancer growth through inducing Wnt/beta-catenin pathway by enhancing p300-mediated beta-catenin acetylation. Oncogene 37(26):3485–3500. doi:https://doi.org/10.1038/s41388-018-0161-z
Liu C, Yang Q, Zhu Q, Lu X, Li M, Hou T, Li Z, Tang M, Li Y, Wang H, Yang Y, Wang H, Zhao Y, Wen H, Liu X, Mao Z, Zhu WG (2020) CBP mediated DOT1L acetylation confers DOT1L stability and promotes cancer metastasis. Theranostics 10(4):1758–1776. doi:https://doi.org/10.7150/thno.39013
Wang J, Qian J, Hu Y, Kong X, Chen H, Shi Q, Jiang L, Wu C, Zou W, Chen Y, Xu J, Fang JY (2014) ArhGAP30 promotes p53 acetylation and function in colorectal cancer. Nat Commun 5:4735. doi:https://doi.org/10.1038/ncomms5735
Gao W, Chen L, Ma Z, Du Z, Zhao Z, Hu Z, Li Q (2013) Isolation and Phenotypic Characterization of Colorectal Cancer Stem Cells With Organ-Specific Metastatic Potential. Gastroenterology 145(3):636–646e635. doi:https://doi.org/10.1053/j.gastro.2013.05.049
Wu Z, Wei D, Gao W, Xu Y, Hu Z, Ma Z, Gao C, Zhu X, Li Q (2015) TPO-Induced Metabolic Reprogramming Drives Liver Metastasis of Colorectal Cancer CD110 + Tumor-Initiating Cells. Cell Stem Cell 17(1):47–59. doi:https://doi.org/10.1016/j.stem.2015.05.016
Ishihama K, Yamakawa M, Semba S, Takeda H, Kawata S, Kimura S, Kimura W (2007) Expression of HDAC1 and CBP/p300 in human colorectal carcinomas. J Clin Pathol 60(11):1205–1210. doi:https://doi.org/10.1136/jcp.2005.029165
Huh JW, Kim HC, Kim SH, Park YA, Cho YB, Yun SH, Lee WY, Chun HK (2013) Prognostic impact of p300 expression in patients with colorectal cancer. J Surg Oncol 108(6):374–377. doi:https://doi.org/10.1002/jso.23405
Kowalczyk AE, Krazinski BE, Godlewski J, Kiewisz J, Kwiatkowski P, Sliwinska-Jewsiewicka A, Kiezun J, Sulik M, Kmiec Z (2017) Expression of the EP300, TP53 and BAX genes in colorectal cancer: Correlations with clinicopathological parameters and survival. Oncol Rep 38(1):201–210. doi:https://doi.org/10.3892/or.2017.5687
Yang XJ, Ogryzko VV, Nishikawa J, Howard BH, Nakatani Y (1996) A p300/CBP-associated factor that competes with the adenoviral oncoprotein E1A. Nature 382(6589):319–324. doi:https://doi.org/10.1038/382319a0
Jin Q, Zhuang L, Lai B, Wang C, Li W, Dolan B, Lu Y, Wang Z, Zhao K, Peng W, Dent SY, Ge K (2014) Gcn5 and PCAF negatively regulate interferon-beta production through HAT-independent inhibition of TBK1. EMBO Rep 15(11):1192–1201. doi:https://doi.org/10.15252/embr.201438990
Love IM, Sekaric P, Shi D, Grossman SR, Androphy EJ (2012) The histone acetyltransferase PCAF regulates p21 transcription through stress-induced acetylation of histone H3. Cell Cycle 11(13):2458–2466. doi:https://doi.org/10.4161/cc.20864
Zheng X, Gai X, Ding F, Lu Z, Tu K, Yao Y, Liu Q (2013) Histone acetyltransferase PCAF up-regulated cell apoptosis in hepatocellular carcinoma via acetylating histone H4 and inactivating AKT signaling. Mol Cancer 12(1):96. doi:https://doi.org/10.1186/1476-4598-12-96
Wan J, Zhan J, Li S, Ma J, Xu W, Liu C, Xue X, Xie Y, Fang W, Chin YE, Zhang H (2015) PCAF-primed EZH2 acetylation regulates its stability and promotes lung adenocarcinoma progression. Nucleic Acids Res 43(7):3591–3604. doi:https://doi.org/10.1093/nar/gkv238
Hu H, Zhu W, Qin J, Chen M, Gong L, Li L, Liu X, Tao Y, Yin H, Zhou H, Zhou L, Ye D, Ye Q, Gao D (2017) Acetylation of PGK1 promotes liver cancer cell proliferation and tumorigenesis. Hepatology 65(2):515–528. doi:https://doi.org/10.1002/hep.28887
Xenaki G, Ontikatze T, Rajendran R, Stratford IJ, Dive C, Krstic-Demonacos M, Demonacos C (2008) PCAF is an HIF-1alpha cofactor that regulates p53 transcriptional activity in hypoxia. Oncogene 27(44):5785–5796. doi:https://doi.org/10.1038/onc.2008.192
Okumura K, Mendoza M, Bachoo RM, DePinho RA, Cavenee WK, Furnari FB (2006) PCAF modulates PTEN activity. J Biol Chem 281(36):26562–26568. doi:https://doi.org/10.1074/jbc.M605391200
Drury LJ, Ziarek JJ, Gravel S, Veldkamp CT, Takekoshi T, Hwang ST, Heveker N, Volkman BF, Dwinell MB (2011) Monomeric and dimeric CXCL12 inhibit metastasis through distinct CXCR4 interactions and signaling pathways. Proc Natl Acad Sci U S A 108(43):17655–17660. doi:https://doi.org/10.1073/pnas.1101133108
Wang D, Wang X, Si M, Yang J, Sun S, Wu H, Cui S, Qu X, Yu X (2020) Exosome-encapsulated miRNAs contribute to CXCL12/CXCR4-induced liver metastasis of colorectal cancer by enhancing M2 polarization of macrophages. Cancer Lett 474:36–52. doi:https://doi.org/10.1016/j.canlet.2020.01.005
Romain B, Benbrika-Nehmar R, Marisa L, Legrain M, Lobstein V, Oravecz A, Poidevin L, Bour C, Freund JN, Duluc I, Guenot D, Pencreach E (2017) Histone hypoacetylation contributes to CXCL12 downregulation in colon cancer: impact on tumor growth and cell migration. Oncotarget 8(24):38351–38366. doi:https://doi.org/10.18632/oncotarget.16323
Tenbaum SP, Ordonez-Moran P, Puig I, Chicote I, Arques O, Landolfi S, Fernandez Y, Herance JR, Gispert JD, Mendizabal L, Aguilar S, Ramon y Cajal S, Schwartz S Jr, Vivancos A, Espin E, Rojas S, Baselga J, Tabernero J, Munoz A, Palmer HG (2012) beta-catenin confers resistance to PI3K and AKT inhibitors and subverts FOXO3a to promote metastasis in colon cancer. Nat Med 18(6):892–901. doi:https://doi.org/10.1038/nm.2772
Ge X, Jin Q, Zhang F, Yan T, Zhai Q (2009) PCAF acetylates {beta}-catenin and improves its stability. Mol Biol Cell 20(1):419–427. doi:https://doi.org/10.1091/mbc.E08-08-0792
Weichert W, Roske A, Niesporek S, Noske A, Buckendahl AC, Dietel M, Gekeler V, Boehm M, Beckers T, Denkert C (2008) Class I histone deacetylase expression has independent prognostic impact in human colorectal cancer: specific role of class I histone deacetylases in vitro and in vivo. Clin Cancer Res 14(6):1669–1677. doi:https://doi.org/10.1158/1078-0432.CCR-07-0990
Nemati M, Ajami N, Estiar MA, Rezapour S, Gavgani RR, Hashemzadeh S, Kafil HS, Sakhinia E (2018) Deregulated expression of HDAC3 in colorectal cancer and its clinical significance. Adv Clin Exp Med 27(3):305–311. doi:https://doi.org/10.17219/acem/66207
Arts J, King P, Marien A, Floren W, Belien A, Janssen L, Pilatte I, Roux B, Decrane L, Gilissen R, Hickson I, Vreys V, Cox E, Bol K, Talloen W, Goris I, Andries L, Du Jardin M, Janicot M, Page M, van Emelen K, Angibaud P (2009) JNJ-26481585, a novel “second-generation” oral histone deacetylase inhibitor, shows broad-spectrum preclinical antitumoral activity. Clin Cancer Res 15(22):6841–6851. doi:https://doi.org/10.1158/1078-0432.CCR-09-0547
Kumamoto K, Fujita K, Kurotani R, Saito M, Unoki M, Hagiwara N, Shiga H, Bowman ED, Yanaihara N, Okamura S, Nagashima M, Miyamoto K, Takenoshita S, Yokota J, Harris CC (2009) ING2 is upregulated in colon cancer and increases invasion by enhanced MMP13 expression. Int J Cancer 125(6):1306–1315. doi:https://doi.org/10.1002/ijc.24437
Krishnan M, Singh AB, Smith JJ, Sharma A, Chen X, Eschrich S, Yeatman TJ, Beauchamp RD, Dhawan P (2010) HDAC inhibitors regulate claudin-1 expression in colon cancer cells through modulation of mRNA stability. Oncogene 29(2):305–312. doi:https://doi.org/10.1038/onc.2009.324
Li Y, Liu Y, Zhao N, Yang X, Li Y, Zhai F, Zang X, Cui W (2020) Checkpoint regulator B7x is epigenetically regulated by HDAC3 and mediates resistance to HDAC inhibitors by reprogramming the tumor immune environment in colorectal cancer. Cell Death Dis 11(9):753. doi:https://doi.org/10.1038/s41419-020-02968-y
Sasidharan Nair V, Saleh R, Toor SM, Taha RZ, Ahmed AA, Kurer MA, Murshed K, Alajez NM, Abu Nada M, Elkord E (2020) Transcriptomic profiling disclosed the role of DNA methylation and histone modifications in tumor-infiltrating myeloid-derived suppressor cell subsets in colorectal cancer. Clin Epigenetics 12(1):13. doi:https://doi.org/10.1186/s13148-020-0808-9
Carafa V, Rotili D, Forgione M, Cuomo F, Serretiello E, Hailu GS, Jarho E, Lahtela-Kakkonen M, Mai A, Altucci L (2016) Sirtuin functions and modulation: from chemistry to the clinic. Clin Epigenetics 8:61. doi:https://doi.org/10.1186/s13148-016-0224-3
Lv L, Shen Z, Zhang J, Zhang H, Dong J, Yan Y, Liu F, Jiang K, Ye Y, Wang S (2014) Clinicopathological significance of SIRT1 expression in colorectal adenocarcinoma. Med Oncol 31(6):965. doi:https://doi.org/10.1007/s12032-014-0965-9
Yu H, Ye W, Wu J, Meng X, Liu RY, Ying X, Zhou Y, Wang H, Pan C, Huang W (2014) Overexpression of sirt7 exhibits oncogenic property and serves as a prognostic factor in colorectal cancer. Clin Cancer Res 20(13):3434–3445. doi:https://doi.org/10.1158/1078-0432.CCR-13-2952
Cheng F, Su L, Yao C, Liu L, Shen J, Liu C, Chen X, Luo Y, Jiang L, Shan J, Chen J, Zhu W, Shao J, Qian C (2016) SIRT1 promotes epithelial-mesenchymal transition and metastasis in colorectal cancer by regulating Fra-1 expression. Cancer Lett 375(2):274–283. doi:https://doi.org/10.1016/j.canlet.2016.03.010
Geng CH, Zhang CL, Zhang JY, Gao P, He M, Li YL (2018) Overexpression of Sirt6 is a novel biomarker of malignant human colon carcinoma. J Cell Biochem 119(5):3957–3967. doi:https://doi.org/10.1002/jcb.26539
Bakiri L, Macho-Maschler S, Custic I, Niemiec J, Guio-Carrion A, Hasenfuss SC, Eger A, Muller M, Beug H, Wagner EF (2015) Fra-1/AP-1 induces EMT in mammary epithelial cells by modulating Zeb1/2 and TGFbeta expression. Cell Death Differ 22(2):336–350. doi:https://doi.org/10.1038/cdd.2014.157
Wang TW, Chern E, Hsu CW, Tseng KC, Chao HM (2020) SIRT1-Mediated Expression of CD24 and Epigenetic Suppression of Novel Tumor Suppressor miR-1185-1 Increases Colorectal Cancer Stemness. Cancer Res 80(23):5257–5269. doi:https://doi.org/10.1158/0008-5472.CAN-19-3188
Zhan-long shen BW, Ke-wei, Jiang (2016) Downregulation of miR-199b is associated with distant metastasis in colorectal cancer via activation of SIRT1 and inhibition of CREB/KISS1 signaling. Oncotarget 7(23):35092–35105
Lin Z, Yang H, Tan C, Li J, Liu Z, Quan Q, Kong S, Ye J, Gao B, Fang D (2013) USP10 antagonizes c-Myc transcriptional activation through SIRT6 stabilization to suppress tumor formation. Cell Rep 5(6):1639–1649. doi:https://doi.org/10.1016/j.celrep.2013.11.029
Rizzo A, Iachettini S, Salvati E, Zizza P, Maresca C, D’Angelo C, Benarroch-Popivker D, Capolupo A, Del Gaudio F, Cosconati S, Di Maro S, Merlino F, Novellino E, Amoreo CA, Mottolese M, Sperduti I, Gilson E, Biroccio A (2017) SIRT6 interacts with TRF2 and promotes its degradation in response to DNA damage. Nucleic Acids Res 45(4):1820–1834. doi:https://doi.org/10.1093/nar/gkw1202
Li N, Mao D, Cao Y, Li H, Ren F, Li K (2018) Downregulation of SIRT6 by miR-34c-5p is associated with poor prognosis and promotes colon cancer proliferation through inhibiting apoptosis via the JAK2/STAT3 signaling pathway. Int J Oncol 52(5):1515–1527. doi:https://doi.org/10.3892/ijo.2018.4304
Tian J, Yuan L (2018) Sirtuin 6 inhibits colon cancer progression by modulating PTEN/AKT signaling. Biomed Pharmacother 106:109–116. doi:https://doi.org/10.1016/j.biopha.2018.06.070
Dang L, White DW, Gross S, Bennett BD, Bittinger MA, Driggers EM, Fantin VR, Jang HG, Jin S, Keenan MC, Marks KM, Prins RM, Ward PS, Yen KE, Liau LM, Rabinowitz JD, Cantley LC, Thompson CB, Vander Heiden MG, Su SM (2009) Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature 462(7274):739–744. doi:https://doi.org/10.1038/nature08617
Wang B, Ye Y, Yang X, Liu B, Wang Z, Chen S, Jiang K, Zhang W, Jiang H, Mustonen H, Puolakkainen P, Wang S, Luo J, Shen Z (2020) SIRT2-dependent IDH1 deacetylation inhibits colorectal cancer and liver metastases. EMBO Rep 21(4):e48183. doi:https://doi.org/10.15252/embr.201948183
Miyo M, Yamamoto H, Konno M, Colvin H, Nishida N, Koseki J, Kawamoto K, Ogawa H, Hamabe A, Uemura M, Nishimura J, Hata T, Takemasa I, Mizushima T, Doki Y, Mori M, Ishii H (2015) Tumour-suppressive function of SIRT4 in human colorectal cancer. Br J Cancer 113(3):492–499. doi:https://doi.org/10.1038/bjc.2015.226
Cao R, Zhang Y (2004) SUZ12 is required for both the histone methyltransferase activity and the silencing function of the EED-EZH2 complex. Mol Cell 15(1):57–67. doi:https://doi.org/10.1016/j.molcel.2004.06.020
Xu K, Wu ZJ, Groner AC, He HH, Cai C, Lis RT, Wu X, Stack EC, Loda M, Liu T, Xu H, Cato L, Thornton JE, Gregory RI, Morrissey C, Vessella RL, Montironi R, Magi-Galluzzi C, Kantoff PW, Balk SP, Liu XS, Brown M (2012) EZH2 oncogenic activity in castration-resistant prostate cancer cells is Polycomb-independent. Science 338(6113):1465–1469. doi:https://doi.org/10.1126/science.1227604
Liu YL, Gao X, Jiang Y, Zhang G, Sun ZC, Cui BB, Yang YM (2015) Expression and clinicopathological significance of EED, SUZ12 and EZH2 mRNA in colorectal cancer. J Cancer Res Clin Oncol 141(4):661–669. doi:https://doi.org/10.1007/s00432-014-1854-5
Chen Z, Yang P, Li W, He F, Wei J, Zhang T, Zhong J, Chen H, Cao J (2018) Expression of EZH2 is associated with poor outcome in colorectal cancer. Oncol Lett 15(3):2953–2961. doi:https://doi.org/10.3892/ol.2017.7647
Crea F, Fornaro L, Paolicchi E, Masi G, Frumento P, Loupakis F, Salvatore L, Cremolini C, Schirripa M, Graziano F, Ronzoni M, Ricci V, Farrar WL, Falcone A, Danesi R (2012) An EZH2 polymorphism is associated with clinical outcome in metastatic colorectal cancer patients. Ann Oncol 23(5):1207–1213. doi:https://doi.org/10.1093/annonc/mdr387
Liu Q, Wang G, Li Q, Jiang W, Kim JS, Wang R, Zhu S, Wang X, Yan L, Yi Y, Zhang L, Meng Q, Li C, Zhao D, Qiao Y, Li Y, Gursel DB, Chinnaiyan AM, Chen K, Cao Q (2019) Polycomb group proteins EZH2 and EED directly regulate androgen receptor in advanced prostate cancer. Int J Cancer 145(2):415–426. doi:https://doi.org/10.1002/ijc.32118
Jin B, Zhang P, Zou H, Ye H, Wang Y, Zhang J, Yang H, Pan J (2020) Verification of EZH2 as a druggable target in metastatic uveal melanoma. Mol Cancer 19(1):52. doi:https://doi.org/10.1186/s12943-020-01173-x
Li L, Liu J, Xue H, Li C, Liu Q, Zhou Y, Wang T, Wang H, Qian H, Wen T (2020) A TGF-beta-MTA1-SOX4-EZH2 signaling axis drives epithelial-mesenchymal transition in tumor metastasis. Oncogene 39(10):2125–2139. doi:https://doi.org/10.1038/s41388-019-1132-8
Ferraro A, Mourtzoukou D, Kosmidou V, Avlonitis S, Kontogeorgos G, Zografos G, Pintzas A (2013) EZH2 is regulated by ERK/AKT and targets integrin alpha2 gene to control Epithelial-Mesenchymal Transition and anoikis in colon cancer cells. Int J Biochem Cell Biol 45(2):243–254. doi:https://doi.org/10.1016/j.biocel.2012.10.009
Zhang Y, Lin C, Liao G, Liu S, Ding J, Tang F, Wang Z, Liang X, Li B, Wei Y, Huang Q, Li X, Tang B (2015) MicroRNA-506 suppresses tumor proliferation and metastasis in colon cancer by directly targeting the oncogene EZH2. Oncotarget 6(32):32586–32601. doi:https://doi.org/10.18632/oncotarget.5309
Li P, Zhang X, Wang H, Wang L, Liu T, Du L, Yang Y, Wang C (2017) MALAT1 Is Associated with Poor Response to Oxaliplatin-Based Chemotherapy in Colorectal Cancer Patients and Promotes Chemoresistance through EZH2. Mol Cancer Ther 16(4):739–751. doi:https://doi.org/10.1158/1535-7163.MCT-16-0591
Shi L, Hong X, Ba L, He X, Xiong Y, Ding Q, Yang S, Peng G (2019) Long non-coding RNA ZNFX1-AS1 promotes the tumor progression and metastasis of colorectal cancer by acting as a competing endogenous RNA of miR-144 to regulate EZH2 expression. Cell Death Dis 10(3):150. doi:https://doi.org/10.1038/s41419-019-1332-8
Zhang M, Duan W, Sun W (2019) LncRNA SNHG6 promotes the migration, invasion, and epithelial-mesenchymal transition of colorectal cancer cells by miR-26a/EZH2 axis. Onco Targets Ther 12:3349–3360. doi:https://doi.org/10.2147/OTT.S197433
Di W, Weinan X, Xin L, Zhiwei Y, Xinyue G, Jinxue T, Mingqi L (2019) Long noncoding RNA SNHG14 facilitates colorectal cancer metastasis through targeting EZH2-regulated EPHA7. Cell Death Dis 10(7):514. doi:https://doi.org/10.1038/s41419-019-1707-x
Kong WQ, Liang JJ, Du J, Ye ZX, Gao P, Liang YL (2020) Long Noncoding RNA DLX6-AS1 Regulates the Growth and Aggressiveness of Colorectal Cancer Cells Via Mediating miR-26a/EZH2 Axis. Cancer Biother Radiopharm. doi:https://doi.org/10.1089/cbr.2020.3589
Jiang M, Xu B, Li X, Shang Y, Chu Y, Wang W, Chen D, Wu N, Hu S, Zhang S, Li M, Wu K, Yang X, Liang J, Nie Y, Fan D (2019) O-GlcNAcylation promotes colorectal cancer metastasis via the miR-101-O-GlcNAc/EZH2 regulatory feedback circuit. Oncogene 38(3):301–316. doi:https://doi.org/10.1038/s41388-018-0435-5
O’Carroll D, Scherthan H, Peters AH, Opravil S, Haynes AR, Laible G, Rea S, Schmid M, Lebersorger A, Jerratsch M, Sattler L, Mattei MG, Denny P, Brown SD, Schweizer D, Jenuwein T (2000) Isolation and characterization of Suv39h2, a second histone H3 methyltransferase gene that displays testis-specific expression. Mol Cell Biol 20(24):9423–9433. doi:https://doi.org/10.1128/mcb.20.24.9423-9433.2000
Melcher M, Schmid M, Aagaard L, Selenko P, Laible G, Jenuwein T (2000) Structure-function analysis of SUV39H1 reveals a dominant role in heterochromatin organization, chromosome segregation, and mitotic progression. Mol Cell Biol 20(10):3728–3741. doi:https://doi.org/10.1128/mcb.20.10.3728-3741.2000
Garcia-Cao M, O’Sullivan R, Peters AH, Jenuwein T, Blasco MA (2004) Epigenetic regulation of telomere length in mammalian cells by the Suv39h1 and Suv39h2 histone methyltransferases. Nat Genet 36(1):94–99. doi:https://doi.org/10.1038/ng1278
Velazquez Camacho O, Galan C, Swist-Rosowska K, Ching R, Gamalinda M, Karabiber F, De La Rosa-Velazquez I, Engist B, Koschorz B, Shukeir N, Onishi-Seebacher M, van de Nobelen S, Jenuwein T (2017) Major satellite repeat RNA stabilize heterochromatin retention of Suv39h enzymes by RNA-nucleosome association and RNA:DNA hybrid formation. Elife 6. doi:https://doi.org/10.7554/eLife.25293
Shuai W, Wu J, Chen S, Liu R, Ye Z, Kuang C, Fu X, Wang G, Li Y, Peng Q, Shi W, Li Y, Zhou Q, Huang W (2018) SUV39H2 promotes colorectal cancer proliferation and metastasis via tri-methylation of the SLIT1 promoter. Cancer Lett 422:56–69. doi:https://doi.org/10.1016/j.canlet.2018.02.023
Lu C, Yang D, Klement JD, Oh IK, Savage NM, Waller JL, Colby AH, Grinstaff MW, Oberlies NH, Pearce CJ, Xie Z, Kulp SK, Coss CC, Phelps MA, Albers T, Lebedyeva IO, Liu K (2019) SUV39H1 Represses the Expression of Cytotoxic T-Lymphocyte Effector Genes to Promote Colon Tumor Immune Evasion. Cancer Immunol Res 7(3):414–427. doi:https://doi.org/10.1158/2326-6066.CIR-18-0126
Kim G, Kim JY, Lim SC, Lee KY, Kim O, Choi HS (2018) SUV39H1/DNMT3A-dependent methylation of the RB1 promoter stimulates PIN1 expression and melanoma development. FASEB J 32(10):5647–5660. doi:https://doi.org/10.1096/fj.201700645RRRRR
Mo W, Liu Q, Lin CC, Dai H, Peng Y, Liang Y, Peng G, Meric-Bernstam F, Mills GB, Li K, Lin SY (2016) mTOR Inhibitors Suppress Homologous Recombination Repair and Synergize with PARP Inhibitors via Regulating SUV39H1 in BRCA-Proficient Triple-Negative Breast Cancer. Clin Cancer Res 22(7):1699–1712. doi:https://doi.org/10.1158/1078-0432.CCR-15-1772
Zhang L, Tian S, Zhao M, Yang T, Quan S, Yang Q, Song L, Yang X (2020) SUV39H1-DNMT3A-mediated epigenetic regulation of Tim-3 and galectin-9 in the cervical cancer. Cancer Cell Int 20:325. doi:https://doi.org/10.1186/s12935-020-01380-y
Carvalho Alves-Silva J, do Amaral Rabello D, Oliveira Bravo M, Lucena-Araujo A, Madureira de Oliveira D, Morato de Oliveira F, Magalhaes Rego E, Pittella-Silva F, Saldanha-Araujo F (2017) Aberrant levels of SUV39H1 and SUV39H2 methyltransferase are associated with genomic instability in chronic lymphocytic leukemia. Environ Mol Mutagen 58(9):654–661. doi:https://doi.org/10.1002/em.22128
Chu Y, Chen Y, Guo H, Li M, Wang B, Shi D, Cheng X, Guan J, Wang X, Xue C, Cheng T, Shi J, Yuan W (2020) SUV39H1 regulates the progression of MLL-AF9-induced acute myeloid leukemia. Oncogene 39(50):7239–7252. doi:https://doi.org/10.1038/s41388-020-01495-6
Lu C, Klement JD, Yang D, Albers T, Lebedyeva IO, Waller JL, Liu K (2020) SUV39H1 regulates human colon carcinoma apoptosis and cell cycle to promote tumor growth. Cancer Lett 476:87–96. doi:https://doi.org/10.1016/j.canlet.2020.02.004
Yokoyama Y, Hieda M, Nishioka Y, Matsumoto A, Higashi S, Kimura H, Yamamoto H, Mori M, Matsuura S, Matsuura N (2013) Cancer-associated upregulation of histone H3 lysine 9 trimethylation promotes cell motility in vitro and drives tumor formation in vivo. Cancer Sci 104(7):889–895. doi:https://doi.org/10.1111/cas.12166
Guccione E, Richard S (2019) The regulation, functions and clinical relevance of arginine methylation. Nat Rev Mol Cell Biol 20(10):642–657. doi:https://doi.org/10.1038/s41580-019-0155-x
Fedoriw A, Rajapurkar SR, O’Brien S, Gerhart SV, Mitchell LH, Adams ND, Rioux N, Lingaraj T, Ribich SA, Pappalardi MB, Shah N, Laraio J, Liu Y, Butticello M, Carpenter CL, Creasy C, Korenchuk S, McCabe MT, McHugh CF, Nagarajan R, Wagner C, Zappacosta F, Annan R, Concha NO, Thomas RA, Hart TK, Smith JJ, Copeland RA, Moyer MP, Campbell J, Stickland K, Mills J, Jacques-O’Hagan S, Allain C, Johnston D, Raimondi A, Porter Scott M, Waters N, Swinger K, Boriack-Sjodin A, Riera T, Shapiro G, Chesworth R, Prinjha RK, Kruger RG, Barbash O, Mohammad HP (2019) Anti-tumor Activity of the Type I PRMT Inhibitor, GSK3368715, Synergizes with PRMT5 Inhibition through MTAP Loss. Cancer Cell 36(1):100–114 e125. doi:https://doi.org/10.1016/j.ccell.2019.05.014
Yao B, Gui T, Zeng X, Deng Y, Wang Z, Wang Y, Yang D, Li Q, Xu P, Hu R, Li X, Chen B, Wang J, Zen K, Li H, Davis MJ, Herold MJ, Pan HF, Jiang ZW, Huang DCS, Liu M, Ju J, Zhao Q (2021) PRMT1-mediated H4R3me2a recruits SMARCA4 to promote colorectal cancer progression by enhancing EGFR signaling. Genome Med 13(1):58. doi:https://doi.org/10.1186/s13073-021-00871-5
Yin XK, Wang YL, Wang F, Feng WX, Bai SM, Zhao WW, Feng LL, Wei MB, Qin CL, Wang F, Chen ZL, Yi HJ, Huang Y, Xie PY, Kim T, Wang YN, Hou JW, Li CW, Liu Q, Fan XJ, Hung MC, Wan XB (2021) PRMT1 enhances oncogenic arginine methylation of NONO in colorectal cancer. Oncogene 40(7):1375–1389. doi:https://doi.org/10.1038/s41388-020-01617-0
Hartley AV, Wang B, Jiang G, Wei H, Sun M, Prabhu L, Martin M, Safa A, Sun S, Liu Y, Lu T (2020) Regulation of a PRMT5/NF-kappaB Axis by Phosphorylation of PRMT5 at Serine 15 in Colorectal Cancer. Int J Mol Sci 21(10). doi:https://doi.org/10.3390/ijms21103684
Yan Y, Zhao P, Wang Z, Liu Z, Wang Z, Zhang J, Ding Y, Hua X, Yu L (2021) PRMT5 regulates colorectal cancer cell growth and EMT via EGFR/Akt/GSK3beta signaling cascades. Aging 13(3):4468–4481. doi:https://doi.org/10.18632/aging.202407
Qi H, Shi X, Yu M, Liu B, Liu M, Song S, Chen S, Zou J, Zhu WG, Luo J (2018) Sirtuin 7-mediated deacetylation of WD repeat domain 77 (WDR77) suppresses cancer cell growth by reducing WDR77/PRMT5 transmethylase complex activity. J Biol Chem 293(46):17769–17779. doi:https://doi.org/10.1074/jbc.RA118.003629
Kim H, Ronai ZA (2020) PRMT5 function and targeting in cancer. Cell Stress 4(8):199–215. doi:https://doi.org/10.15698/cst2020.08.228
Kooistra SM, Helin K (2012) Molecular mechanisms and potential functions of histone demethylases. Nat Rev Mol Cell Biol 13(5):297–311. doi:https://doi.org/10.1038/nrm3327
Maiques-Diaz A, Somervaille TC (2016) LSD1: biologic roles and therapeutic targeting. Epigenomics 8(8):1103–1116. doi:https://doi.org/10.2217/epi-2016-0009
Ding J, Zhang ZM, Xia Y, Liao GQ, Pan Y, Liu S, Zhang Y, Yan ZS (2013) LSD1-mediated epigenetic modification contributes to proliferation and metastasis of colon cancer. Br J Cancer 109(4):994–1003. doi:https://doi.org/10.1038/bjc.2013.364
Alsaqer SF, Tashkandi MM, Kartha VK, Yang YT, Alkheriji Y, Salama A, Varelas X, Kukuruzinska M, Monti S, Bais MV (2017) Inhibition of LSD1 epigenetically attenuates oral cancer growth and metastasis. Oncotarget 8(43):73372–73386. doi:https://doi.org/10.18632/oncotarget.19637
Liu J, Feng J, Li L, Lin L, Ji J, Lin C, Liu L, Zhang N, Duan D, Li Z, Huang B, Zhang Y, Lu J (2020) Arginine methylation-dependent LSD1 stability promotes invasion and metastasis of breast cancer. EMBO Rep 21(2):e48597. doi:https://doi.org/10.15252/embr.201948597
Carvalho S, Freitas M, Antunes L, Monteiro-Reis S, Vieira-Coimbra M, Tavares A, Paulino S, Videira JF, Jeronimo C, Henrique R (2018) Prognostic value of histone marks H3K27me3 and H3K9me3 and modifying enzymes EZH2, SETDB1 and LSD-1 in colorectal cancer. J Cancer Res Clin Oncol 144(11):2127–2137. doi:https://doi.org/10.1007/s00432-018-2733-2
Ramirez-Ramirez R, Gutierrez-Angulo M, Peregrina-Sandoval J, Moreno-Ortiz JM, Franco-Topete RA, Cerda-Camacho FJ, Ayala-Madrigal ML (2020) Somatic deletion of KDM1A/LSD1 gene is associated to advanced colorectal cancer stages. J Clin Pathol 73(2):107–111. doi:https://doi.org/10.1136/jclinpath-2019-206128
Yue S, Mu W, Zoller M (2013) Tspan8 and CD151 promote metastasis by distinct mechanisms. Eur J Cancer 49(13):2934–2948. doi:https://doi.org/10.1016/j.ejca.2013.03.032
Zhang HS, Liu HY, Zhou Z, Sun HL, Liu MY (2020) TSPAN8 promotes colorectal cancer cell growth and migration in LSD1-dependent manner. Life Sci 241:117114. doi:https://doi.org/10.1016/j.lfs.2019.117114
Ferrari-Amorotti G, Fragliasso V, Esteki R, Prudente Z, Soliera AR, Cattelani S, Manzotti G, Grisendi G, Dominici M, Pieraccioli M, Raschella G, Chiodoni C, Colombo MP, Calabretta B (2013) Inhibiting interactions of lysine demethylase LSD1 with snail/slug blocks cancer cell invasion. Cancer Res 73(1):235–245. doi:https://doi.org/10.1158/0008-5472.CAN-12-1739
Hong X, Huang H, Qiu X, Ding Z, Feng X, Zhu Y, Zhuo H, Hou J, Zhao J, Cai W, Sha R, Hong X, Li Y, Song H, Zhang Z (2018) Targeting posttranslational modifications of RIOK1 inhibits the progression of colorectal and gastric cancers. Elife 7. doi:https://doi.org/10.7554/eLife.29511
Berry WL, Janknecht R (2013) KDM4/JMJD2 histone demethylases: epigenetic regulators in cancer cells. Cancer Res 73(10):2936–2942. doi:https://doi.org/10.1158/0008-5472.CAN-12-4300
Wu X, Li R, Song Q, Zhang C, Jia R, Han Z, Zhou L, Sui H, Liu X, Zhu H, Yang L, Wang Y, Ji Q, Li Q (2019) JMJD2C promotes colorectal cancer metastasis via regulating histone methylation of MALAT1 promoter and enhancing beta-catenin signaling pathway. J Exp Clin Cancer Res 38(1):435. doi:https://doi.org/10.1186/s13046-019-1439-x
Peng K, Kou L, Yu L, Bai C, Li M, Mo P, Li W, Yu C (2019) Histone Demethylase JMJD2D Interacts With beta-Catenin to Induce Transcription and Activate Colorectal Cancer Cell Proliferation and Tumor Growth in Mice. Gastroenterology 156(4):1112–1126. doi:https://doi.org/10.1053/j.gastro.2018.11.036
Zhuo M, Chen W, Shang S, Guo P, Peng K, Li M, Mo P, Zhang Y, Qiu X, Li W, Yu C (2020) Inflammation-induced JMJD2D promotes colitis recovery and colon tumorigenesis by activating Hedgehog signaling. Oncogene 39(16):3336–3353. doi:https://doi.org/10.1038/s41388-020-1219-2
Peng K, Zhuo M, Li M, Chen Q, Mo P, Yu C (2020) Histone demethylase JMJD2D activates HIF1 signaling pathway via multiple mechanisms to promote colorectal cancer glycolysis and progression. Oncogene 39(47):7076–7091. doi:https://doi.org/10.1038/s41388-020-01483-w
Uemura M, Yamamoto H, Takemasa I, Mimori K, Hemmi H, Mizushima T, Ikeda M, Sekimoto M, Matsuura N, Doki Y, Mori M (2010) Jumonji domain containing 1A is a novel prognostic marker for colorectal cancer: in vivo identification from hypoxic tumor cells. Clin Cancer Res 16(18):4636–4646. doi:https://doi.org/10.1158/1078-0432.CCR-10-0407
Peng K, Su G, Ji J, Yang X, Miao M, Mo P, Li M, Xu J, Li W, Yu C (2018) Histone demethylase JMJD1A promotes colorectal cancer growth and metastasis by enhancing Wnt/beta-catenin signaling. J Biol Chem 293(27):10606–10619. doi:https://doi.org/10.1074/jbc.RA118.001730
Li E, Zhang Y (2014) DNA methylation in mammals. Cold Spring Harb Perspect Biol 6(5):a019133. doi:https://doi.org/10.1101/cshperspect.a019133
Greenberg MVC, Bourc’his D (2019) The diverse roles of DNA methylation in mammalian development and disease. Nat Rev Mol Cell Biol 20(10):590–607. doi:https://doi.org/10.1038/s41580-019-0159-6
Uppal A, Wightman SC, Mallon S, Oshima G, Pitroda SP, Zhang Q, Huang X, Darga TE, Huang L, Andrade J, Liu H, Ferguson MK, Greene GL, Posner MC, Hellman S, Khodarev NN, Weichselbaum RR (2015) 14q32-encoded microRNAs mediate an oligometastatic phenotype. Oncotarget 6(6):3540–3552. doi:https://doi.org/10.18632/oncotarget.2920
Oshima G, Poli EC, Bolt MJ, Chlenski A, Forde M, Jutzy JMS, Biyani N, Posner MC, Pitroda SP, Weichselbaum RR, Khodarev NN (2019) DNA Methylation Controls Metastasis-Suppressive 14q32-Encoded miRNAs. Cancer Res 79(3):650–662. doi:https://doi.org/10.1158/0008-5472.CAN-18-0692
Wang H, Wu J, Meng X, Ying X, Zuo Y, Liu R, Pan Z, Kang T, Huang W (2011) MicroRNA-342 inhibits colorectal cancer cell proliferation and invasion by directly targeting DNA methyltransferase 1. Carcinogenesis 32(7):1033–1042. doi:https://doi.org/10.1093/carcin/bgr081
Chen Z, Liu S, Tian L, Wu M, Ai F, Tang W, Zhao L, Ding J, Zhang L, Tang A (2015) miR-124 and miR-506 inhibit colorectal cancer progression by targeting DNMT3B and DNMT1. Oncotarget 6(35):38139–38150. doi:https://doi.org/10.18632/oncotarget.5709
Zhang L, Li J, Li L, Zhang J, Wang X, Yang C, Li Y, Lan F, Lin P (2014) IL-23 selectively promotes the metastasis of colorectal carcinoma cells with impaired Socs3 expression via the STAT5 pathway. Carcinogenesis 35(6):1330–1340. doi:https://doi.org/10.1093/carcin/bgu017
Wendt MK, Johanesen PA, Kang-Decker N, Binion DG, Shah V, Dwinell MB (2006) Silencing of epithelial CXCL12 expression by DNA hypermethylation promotes colonic carcinoma metastasis. Oncogene 25(36):4986–4997. doi:https://doi.org/10.1038/sj.onc.1209505
Rasmussen KD, Helin K (2016) Role of TET enzymes in DNA methylation, development, and cancer. Genes Dev 30(7):733–750. doi:https://doi.org/10.1101/gad.276568.115
Lian CG, Xu Y, Ceol C, Wu F, Larson A, Dresser K, Xu W, Tan L, Hu Y, Zhan Q, Lee CW, Hu D, Lian BQ, Kleffel S, Yang Y, Neiswender J, Khorasani AJ, Fang R, Lezcano C, Duncan LM, Scolyer RA, Thompson JF, Kakavand H, Houvras Y, Zon LI, Mihm MC Jr, Kaiser UB, Schatton T, Woda BA, Murphy GF, Shi YG (2012) Loss of 5-hydroxymethylcytosine is an epigenetic hallmark of melanoma. Cell 150(6):1135–1146. doi:https://doi.org/10.1016/j.cell.2012.07.033
Carella A, Tejedor JR, Garcia MG, Urdinguio RG, Bayon GF, Sierra M, Lopez V, Garcia-Torano E, Santamarina-Ojeda P, Perez RF, Bigot T, Mangas C, Corte-Torres MD, Saenz-de-Santa-Maria I, Mollejo M, Melendez B, Astudillo A, Chiara MD, Fernandez AF, Fraga MF (2020) Epigenetic downregulation of TET3 reduces genome-wide 5hmC levels and promotes glioblastoma tumorigenesis. Int J Cancer 146(2):373–387. doi:https://doi.org/10.1002/ijc.32520
Evelyne Collignon AC, Clémence Al Wardi, Martin Bizet, Emilie Calonne, Sarah Dedeurwaerder, Soizic Garaud, Céline Naveaux 3, Whitney Barham 4, Andrew Wilson 4, Sophie Bouchat, Pascale Hubert, Carine Van Lint, Fiona Yull, Christos Sotiriou, Karen Willard-Gallo, Agnès Noel, François Fuks (2018) (Immunity drives TET1 regulation in cancer through NF-κB.pdf). Sci Adv 4(6):eaap7309
Nickerson ML, Das S, Im KM, Turan S, Berndt SI, Li H, Lou H, Brodie SA, Billaud JN, Zhang T, Bouk AJ, Butcher D, Wang Z, Sun L, Misner K, Tan W, Esnakula A, Esposito D, Huang WY, Hoover RN, Tucker MA, Keller JR, Boland J, Brown K, Anderson SK, Moore LE, Isaacs WB, Chanock SJ, Yeager M, Dean M, Andresson T (2017) TET2 binds the androgen receptor and loss is associated with prostate cancer. Oncogene 36(15):2172–2183. doi:https://doi.org/10.1038/onc.2016.376
Mo HY, An CH, Choi EJ, Yoo NJ, Lee SH (2020) Somatic mutation and loss of expression of a candidate tumor suppressor gene TET3 in gastric and colorectal cancers. Pathol Res Pract 216(3):152759. doi:https://doi.org/10.1016/j.prp.2019.152759
Noreen F, Kung T, Tornillo L, Parker H, Silva M, Weis S, Marra G, Rad R, Truninger K, Schar P (2019) DNA methylation instability by BRAF-mediated TET silencing and lifestyle-exposure divides colon cancer pathways. Clin Epigenetics 11(1):196. doi:https://doi.org/10.1186/s13148-019-0791-1
Ma L, Qi T, Wang S, Hao M, Sakhawat A, Liang T, Zhang L, Cong X, Huang Y (2019) Tet methylcytosine dioxygenase 1 promotes hypoxic gene induction and cell migration in colon cancer. J Cell Physiol 234(5):6286–6297. doi:https://doi.org/10.1002/jcp.27359
Fu Y, Dominissini D, Rechavi G, He C (2014) Gene expression regulation mediated through reversible m(6)A RNA methylation. Nat Rev Genet 15(5):293–306. doi:https://doi.org/10.1038/nrg3724
Chen RX, Chen X, Xia LP, Zhang JX, Pan ZZ, Ma XD, Han K, Chen JW, Judde JG, Deas O, Wang F, Ma NF, Guan X, Yun JP, Wang FW, Xu RH, Dan X (2019) N(6)-methyladenosine modification of circNSUN2 facilitates cytoplasmic export and stabilizes HMGA2 to promote colorectal liver metastasis. Nat Commun 10(1):4695. doi:https://doi.org/10.1038/s41467-019-12651-2
Han J, Wang JZ, Yang X, Yu H, Zhou R, Lu HC, Yuan WB, Lu JC, Zhou ZJ, Lu Q, Wei JF, Yang H (2019) METTL3 promote tumor proliferation of bladder cancer by accelerating pri-miR221/222 maturation in m6A-dependent manner. Mol Cancer 18(1):110. doi:https://doi.org/10.1186/s12943-019-1036-9
Wang Y, Lu JH, Wu QN, Jin Y, Wang DS, Chen YX, Liu J, Luo XJ, Meng Q, Pu HY, Wang YN, Hu PS, Liu ZX, Zeng ZL, Zhao Q, Deng R, Zhu XF, Ju HQ, Xu RH (2019) LncRNA LINRIS stabilizes IGF2BP2 and promotes the aerobic glycolysis in colorectal cancer. Mol Cancer 18(1):174. doi:https://doi.org/10.1186/s12943-019-1105-0
Shen C, Xuan B, Yan T, Ma Y, Xu P, Tian X, Zhang X, Cao Y, Ma D, Zhu X, Zhang Y, Fang JY, Chen H, Hong J (2020) m(6)A-dependent glycolysis enhances colorectal cancer progression. Mol Cancer 19(1):72. doi:https://doi.org/10.1186/s12943-020-01190-w
Wang L, Hui H, Agrawal K, Kang Y, Li N, Tang R, Yuan J, Rana TM (2020) m(6) A RNA methyltransferases METTL3/14 regulate immune responses to anti-PD-1 therapy. EMBO J 39(20):e104514. doi:https://doi.org/10.15252/embj.2020104514
Bai Y, Yang C, Wu R, Huang L, Song S, Li W, Yan P, Lin C, Li D, Zhang Y (2019) YTHDF1 Regulates Tumorigenicity and Cancer Stem Cell-Like Activity in Human Colorectal Carcinoma. Front Oncol 9:332. doi:https://doi.org/10.3389/fonc.2019.00332
Zhang Y, Kang M, Zhang B, Meng F, Song J, Kaneko H, Shimamoto F, Tang B (2019) m(6)A modification-mediated CBX8 induction regulates stemness and chemosensitivity of colon cancer via upregulation of LGR5. Mol Cancer 18(1):185. doi:https://doi.org/10.1186/s12943-019-1116-x
Relier S, Ripoll J, Guillorit H, Amalric A, Achour C, Boissiere F, Vialaret J, Attina A, Debart F, Choquet A, Macari F, Marchand V, Motorin Y, Samalin E, Vasseur JJ, Pannequin J, Aguilo F, Lopez-Crapez E, Hirtz C, Rivals E, Bastide A, David A (2021) FTO-mediated cytoplasmic m(6)Am demethylation adjusts stem-like properties in colorectal cancer cell. Nat Commun 12(1):1716. doi:https://doi.org/10.1038/s41467-021-21758-4
Yang X, Zhang S, He C, Xue P, Zhang L, He Z, Zang L, Feng B, Sun J, Zheng M (2020) METTL14 suppresses proliferation and metastasis of colorectal cancer by down-regulating oncogenic long non-coding RNA XIST. Mol Cancer 19(1):46. doi:https://doi.org/10.1186/s12943-020-1146-4
Chen X, Xu M, Xu X, Zeng K, Liu X, Pan B, Li C, Sun L, Qin J, Xu T, He B, Pan Y, Sun H, Wang S (2020) METTL14-mediated N6-methyladenosine modification of SOX4 mRNA inhibits tumor metastasis in colorectal cancer. Mol Cancer 19(1):106. doi:https://doi.org/10.1186/s12943-020-01220-7
Chen X, Xu M, Xu X, Zeng K, Liu X, Sun L, Pan B, He B, Pan Y, Sun H, Xia X, Wang S (2020) METTL14 Suppresses CRC Progression via Regulating N6-Methyladenosine-Dependent Primary miR-375 Processing. Mol Ther 28(2):599–612. doi:https://doi.org/10.1016/j.ymthe.2019.11.016
Peng W, Li J, Chen R, Gu Q, Yang P, Qian W, Ji D, Wang Q, Zhang Z, Tang J, Sun Y (2019) Upregulated METTL3 promotes metastasis of colorectal Cancer via miR-1246/SPRED2/MAPK signaling pathway. J Exp Clin Cancer Res 38(1):393. doi:https://doi.org/10.1186/s13046-019-1408-4
Zhou D, Tang W, Xu Y, Xu Y, Xu B, Fu S, Wang Y, Chen F, Chen Y, Han Y, Wang G (2021) METTL3/YTHDF2 m6A axis accelerates colorectal carcinogenesis through epigenetically suppressing YPEL5. Mol Oncol 15(8):2172–2184. doi:https://doi.org/10.1002/1878-0261.12898
Hou P, Meng S, Li M, Lin T, Chu S, Li Z, Zheng J, Gu Y, Bai J (2021) LINC00460/DHX9/IGF2BP2 complex promotes colorectal cancer proliferation and metastasis by mediating HMGA1 mRNA stability depending on m6A modification. J Exp Clin Cancer Res 40(1):52. doi:https://doi.org/10.1186/s13046-021-01857-2
Wu Y, Yang X, Chen Z, Tian L, Jiang G, Chen F, Li J, An P, Lu L, Luo N, Du J, Shan H, Liu H, Wang H (2019) m(6)A-induced lncRNA RP11 triggers the dissemination of colorectal cancer cells via upregulation of Zeb1. Mol Cancer 18(1):87. doi:https://doi.org/10.1186/s12943-019-1014-2
Xie H, Mahoney DW, Foote PH, Burger KN, Doering KA, Taylor WR, Then SS, Cao X, McGlinch M, Berger CK, Wu TT, Hubbard JM, Allawi HT, Kaiser MW, Lidgard GP, Ahlquist DA, Kisiel JB (2021) Novel Methylated DNA Markers in the Surveillance of Colorectal Cancer Recurrence. Clin Cancer Res 27(1):141–149. doi:https://doi.org/10.1158/1078-0432.CCR-20-2589
Dhillon S (2020) Decitabine/Cedazuridine: First Approval. Drugs 80(13):1373–1378. doi:https://doi.org/10.1007/s40265-020-01389-7
Fenaux P, Mufti GJ, Hellstrom-Lindberg E, Santini V, Finelli C, Giagounidis A, Schoch R, Gattermann N, Sanz G, List A, Gore SD, Seymour JF, Bennett JM, Byrd J, Backstrom J, Zimmerman L, McKenzie D, Beach CL, Silverman LR (2009) Efficacy of azacitidine compared with that of conventional care regimens in the treatment of higher-risk myelodysplastic syndromes: a randomised, open-label, phase III study. Lancet Oncol 10(3):223–232. doi:https://doi.org/10.1016/s1470-2045(09)70003-8
Overman MJ, Morris V, Moinova H, Manyam G, Ensor J, Lee MS, Eng C, Kee B, Fogelman D, Shroff RT, LaFramboise T, Mazard T, Feng T, Hamilton S, Broom B, Lutterbaugh J, Issa JP, Markowitz SD, Kopetz S (2016) Phase I/II study of azacitidine and capecitabine/oxaliplatin (CAPOX) in refractory CIMP-high metastatic colorectal cancer: evaluation of circulating methylated vimentin. Oncotarget 7(41):67495–67506. doi:https://doi.org/10.18632/oncotarget.11317
Lee V, Wang J, Zahurak M, Gootjes E, Verheul HM, Parkinson R, Kerner Z, Sharma A, Rosner G, De Jesus-Acosta A, Laheru D, Le DT, Oganesian A, Lilly E, Brown T, Jones P, Baylin S, Ahuja N, Azad N (2018) A Phase I Trial of a Guadecitabine (SGI-110) and Irinotecan in Metastatic Colorectal Cancer Patients Previously Exposed to Irinotecan. Clin Cancer Res 24(24):6160–6167. doi:https://doi.org/10.1158/1078-0432.CCR-18-0421
Garrido-Laguna I, McGregor KA, Wade M, Weis J, Gilcrease W, Burr L, Soldi R, Jakubowski L, Davidson C, Morrell G, Olpin JD, Boucher K, Jones D, Sharma S (2013) A phase I/II study of decitabine in combination with panitumumab in patients with wild-type (wt) KRAS metastatic colorectal cancer. Invest New Drugs 31(5):1257–1264. doi:https://doi.org/10.1007/s10637-013-9947-6
Bever KM, Thomas DL 2, Zhang J, Diaz Rivera EA, Rosner GL, Zhu Q, Nauroth JM, Christmas B, Thompson ED, Anders RA, Judkins C, Liu M, Jaffee EM, Ahuja N, Zheng L, Azad NS (2021) A feasibility study of combined epigenetic and vaccine therapy in advanced colorectal cancer with pharmacodynamic endpoint. Clin Epigenetics 13(1):25. doi:https://doi.org/10.1186/s13148-021-01014-8
Kim YH, Bagot M, Pinter-Brown L, Rook AH, Porcu P, Horwitz SM, Whittaker S, Tokura Y, Vermeer M, Zinzani PL, Sokol L, Morris S, Kim EJ, Ortiz-Romero PL, Eradat H, Scarisbrick J, Tsianakas A, Elmets C, Dalle S, Fisher DC, Halwani A, Poligone B, Greer J, Fierro MT, Khot A, Moskowitz AJ, Musiek A, Shustov A, Pro B, Geskin LJ, Dwyer K, Moriya J, Leoni M, Humphrey JS, Hudgens S, Grebennik DO, Tobinai K, Duvic M, Investigators M (2018) Mogamulizumab versus vorinostat in previously treated cutaneous T-cell lymphoma (MAVORIC): an international, open-label, randomised, controlled phase 3 trial. Lancet Oncol 19(9):1192–1204. doi:https://doi.org/10.1016/S1470-2045(18)30379-6
Doi T, Hamaguchi T, Shirao K, Chin K, Hatake K, Noguchi K, Otsuki T, Mehta A, Ohtsu A (2013) Evaluation of safety, pharmacokinetics, and efficacy of vorinostat, a histone deacetylase inhibitor, in the treatment of gastrointestinal (GI) cancer in a phase I clinical trial. Int J Clin Oncol 18(1):87–95. doi:https://doi.org/10.1007/s10147-011-0348-6
Wilson PM, El-Khoueiry A, Iqbal S, Fazzone W, LaBonte MJ, Groshen S, Yang D, Danenberg KD, Cole S, Kornacki M, Ladner RD, Lenz HJ (2010) A phase I/II trial of vorinostat in combination with 5-fluorouracil in patients with metastatic colorectal cancer who previously failed 5-FU-based chemotherapy. Cancer Chemother Pharmacol 65(5):979–988. doi:https://doi.org/10.1007/s00280-009-1236-x
Fakih MG, Groman A, McMahon J, Wilding G, Muindi JR (2012) A randomized phase II study of two doses of vorinostat in combination with 5-FU/LV in patients with refractory colorectal cancer. Cancer Chemother Pharmacol 69(3):743–751. doi:https://doi.org/10.1007/s00280-011-1762-1
Fu S, Hou MM, Naing A, Janku F, Hess K, Zinner R, Subbiah V, Hong D, Wheler J, Piha-Paul S, Tsimberidou A, Karp D, Araujo D, Kee B, Hwu P, Wolff R, Kurzrock R, Meric-Bernstam F (2015) Phase I study of pazopanib and vorinostat: a therapeutic approach for inhibiting mutant p53-mediated angiogenesis and facilitating mutant p53 degradation. Ann Oncol 26(5):1012–1018. doi:https://doi.org/10.1093/annonc/mdv066
Wang Y, Janku F, Piha-Paul S, Hess K, Broaddus R, Liu L, Shi N, Overman M, Kopetz S, Subbiah V, Naing A, Hong D, Tsimberidou AM, Karp D, Yao J, Fu S (2020) Phase I studies of vorinostat with ixazomib or pazopanib imply a role of antiangiogenesis-based therapy for TP53 mutant malignancies. Sci Rep 10(1):3080. doi:https://doi.org/10.1038/s41598-020-58366-z
Duan R, Du W, Guo W (2020) EZH2: a novel target for cancer treatment. J Hematol Oncol 13(1):104. doi:https://doi.org/10.1186/s13045-020-00937-8
McCabe MT, Ott HM, Ganji G, Korenchuk S, Thompson C, Van Aller GS, Liu Y, Graves AP, Della Pietra A 3, Diaz E, LaFrance LV, Mellinger M, Duquenne C, Tian X, Kruger RG, McHugh CF, Brandt M, Miller WH, Dhanak D, Verma SK, Tummino PJ, Creasy CL (2012) EZH2 inhibition as a therapeutic strategy for lymphoma with EZH2-activating mutations. Nature 492(7427):108–112. doi:https://doi.org/10.1038/nature11606
Yap TA, Winter JN, Giulino-Roth L, Longley J, Lopez J, Michot JM, Leonard JP, Ribrag V, McCabe MT, Creasy CL, Stern M, Pene Dumitrescu T, Wang X, Frey S, Carver J, Horner T, Oh C, Khaled A, Dhar A, Johnson PWM (2019) Phase I Study of the Novel Enhancer of Zeste Homolog 2 (EZH2) Inhibitor GSK2816126 in Patients with Advanced Hematologic and Solid Tumors. Clin Cancer Res 25(24):7331–7339. doi:https://doi.org/10.1158/1078-0432.CCR-18-4121
Hoy SM (2020) Tazemetostat: First Approval. Drugs 80(5):513–521. doi:https://doi.org/10.1007/s40265-020-01288-x
Morschhauser F, Tilly H, Chaidos A, McKay P, Phillips T, Assouline S, Batlevi CL, Campbell P, Ribrag V, Damaj GL, Dickinson M, Jurczak W, Kazmierczak M, Opat S, Radford J, Schmitt A, Yang J, Whalen J, Agarwal S, Adib D, Salles G (2020) Tazemetostat for patients with relapsed or refractory follicular lymphoma: an open-label, single-arm, multicentre, phase 2 trial. Lancet Oncol 21(11):1433–1442. doi:https://doi.org/10.1016/S1470-2045(20)30441-1
Izutsu K, Ando K, Nishikori M, Shibayama H, Teshima T, Kuroda J, Kato K, Imaizumi Y, Nosaka K, Sakai R, Hojo S, Nakanishi T, Rai S (2021) Phase II study of tazemetostat for relapsed or refractory B-cell non-Hodgkin lymphoma with EZH2 mutation in Japan. Cancer Sci 112(9):3627–3635. doi:https://doi.org/10.1111/cas.15040
Italiano A, Soria JC, Toulmonde M, Michot JM, Lucchesi C, Varga A, Coindre JM, Blakemore SJ, Clawson A, Suttle B, McDonald AA, Woodruff M, Ribich S, Hedrick E, Keilhack H, Thomson B, Owa T, Copeland RA, Ho PTC, Ribrag V (2018) Tazemetostat, an EZH2 inhibitor, in relapsed or refractory B-cell non-Hodgkin lymphoma and advanced solid tumours: a first-in-human, open-label, phase 1 study. Lancet Oncol 19(5):649–659. doi:https://doi.org/10.1016/S1470-2045(18)30145-1
Baylin SB, Jones PA (2016) Epigenetic Determinants of Cancer. Cold Spring Harb Perspect Biol 8(9). doi:https://doi.org/10.1101/cshperspect.a019505
Wang H, Li J, Huang R, Fang L, Yu S (2021) SIRT4 and SIRT6 Serve as Novel Prognostic Biomarkers With Competitive Functions in Serous Ovarian Cancer. Front Genet 12:666630. doi:https://doi.org/10.3389/fgene.2021.666630
Acknowledgements
This study was funded by Higher-Level Talents of Inner Mongolia University award (to Changshan Wang), and the National Natural Science Foundation of China (no. 81660024). and the Natural Science Foundation of Inner Mongolia Autonomous Region (No. 2020MS08096). Open Project of the State Key Laboratory of Reproductive Regulation & Breeding of Grassland Livestock.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
All authors declare no conflict of interest.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Riya Su and Xinlin Wu authors contributed equally to this work.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Su, R., Wu, X., Tao, L. et al. The role of epigenetic modifications in Colorectal Cancer Metastasis. Clin Exp Metastasis 39, 521–539 (2022). https://doi.org/10.1007/s10585-022-10163-w
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10585-022-10163-w