
Abstract
Background
L-2-hydroxyglutaric aciduria (L2HGA) is a rare neurometabolic disorder that occurs due to accumulation of L-2-hydroxyglutaric acid in the cerebrospinal fluid (CSF), plasma and urine. The clinical manifestation of L2HGA includes intellectual disability, cerebellar ataxia, epilepsy, speech problems and macrocephaly.
Methods
In the present study, we ascertained a multigenerational consanguineous Pakistani family with 5 affected individuals. Clinical studies were performed through biochemical tests and brain CT scan. Locus mapping was carried out through genome-wide SNP genotyping, whole exome sequencing and Sanger sequencing. For in silico studies protein structural modeling and docking was done using I-TASSER, Cluspro and AutoDock VINA tools.
Results
Affected individuals presented with cognitive impairment, gait disturbance, speech difficulties and psychomotor delay. Radiologic analysis of a male patient revealed leukoaraiosis with hypoattenuation of cerebral white matter, suggestive of hypomyelination. Homozygosity mapping in this family revealed a linkage region on chromosome 14 between markers rs2039791 and rs781354. Subsequent whole exome analysis identified a novel frameshift mutation NM_024884.3:c.180delG, p.(Ala62Profs*24) in the second exon of L2HGDH. Sanger sequencing confirmed segregation of this mutation with the disease phenotype. The identification of the most N-terminal loss of function mutation published thus far further expands the mutational spectrum of L2HGDH.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
L-2-hydroxyglutaric aciduria [L2HGA (OMIM #236,792)] is a rare autosomal recessive neurodegenerative metabolic disorder, that occurs due to accumulation of L-2-hydroxyglutaric acid in the cerebrospinal fluid (CSF), plasma and urine (Duran et al. 1980; Chen et al. 1996). Phenotypic features of the affected individuals are variable and may include developmental delay, moderate to severe intellectual disability, epilepsy, behavioral problems, spasticity, macrocephaly, speech disorders and cerebellar ataxia (Barth et al.1992; Barth et al. 1998; Hanefeld et al.1994). Age of onset of L2HGA (OMIM #236,792) is variable and may occur at an early age with severe epilepsy and intellectual disability or in adulthood with moderate to mild symptoms. Different studies have documented that an elevated level of L2HGA (OMIM #236,792) in the brain may also lead to brain tumors (Steenweg et al. 2010; Haliloglu et al. 2008). Diagnosis of L2HGA can be established by means of radiological, biochemical and genetic testing. Metabolic screening includes plasma amino acid and urine organic acid analysis. Radiological examinations such as MRI and CT are necessary for the detection of brain abnormalities, especially with regard to subcortical cerebral white matter, globus pallidus, putamen, caudatus and dentatus, which are specifically affected by L2HGA (Moroni et al.2004; Seijo-Martínez et al. 2005; Topcu et al. 2005).
L2HGDH is expressed in various tissues with highest expression in brain, followed by muscles and testis (Vilarinho et al. 2005). The corresponding protein consists of 463 amino acids, which contains two domains i.e. a mitochondrial targeting sequence and a FAD dependent oxidoreductase domain (UniProtKB: Q9H9P8) (Goffette et al. 2006). L2HGDH acts as a mitochondrial enzyme which is involved in glutamate and glutamine metabolism pathways. Its prime function is to catalyze the oxidation of L-2-hydroxyglutarate (L2HG) to α2-ketoglutarate (α2KG) (Topçu et al. 2004; Vilarinho et al. 2009). Exact prevalence of L2HGA (OMIM #236,792) is unknown, but approximately 140 cases have been reported to date (Goffette et al. 2006; Topçu et al. 2004; Vilarinho et al. 2009; Jellouli et al. 2014; Larnaout et al. 2008; O'Connor et al.2009). Although there is no established treatment of L2HGA, Samuraki et al. reported effective treatment of a late onset patient with flavin adenine dinucleotide sodium (FAD) and levocarnitine chloride (Samuraki et al. 2008).
In the present study, we report on a consanguineous Pakistani family displaying mild intellectual disability. Genome-wide homozygosity mapping coupled with whole exome sequencing revealed a novel frameshift mutation NM_024884.3:c.180delG, p.(Ala62Profs*24) in the 2nd exon of L2HGDH. The identified mutation presumably creates a premature stop codon ether leading to nonsense mediated mRNA decay or truncation of the protein, which would distort the local folding of the polypeptide chain and lead to loss of its interacting sites.
Methods
The current study was approved by the institutional ethical review board of Gomal University, Dera Ismail Khan, Pakistan, and patients were enrolled after obtaining written informed consent. The family was ascertained from Dera Ismail Khan, City in Khyber Pakhtunkhwa province of Pakistan. Blood samples were taken from available affected (V:4, V:5 and V:8) and unaffected (IV:2 and V:6) family members. DNA was extracted using standard laboratory protocols.
Clinical assessment
The clinical assessment of patients was carried out through biochemical tests e.g. liver functioning test (LFTs), renal function tests (RFTs), urine organic acid analysis and plasma amino acid analysis. Radiologic analysis was performed through CT scan of affected individual V:8.
Genome-wide SNP genotyping
Whole genome SNP genotyping was performed through microarray analysis using the Infinium Global Screening Array (Illumina, USA) kit. Raw data analysis was performed at the Life and Brain GmbH, Bonn, Germany. Homozygosity mapping to identify the disease associated locus, was carried out using GenomeStudio 2.0 Software (Illumina, USA).
Whole exome sequencing (WES)
For genetic analysis whole exome sequencing (WES) was performed for a single affected individual (V:4) via Agilent SureSelect V6 human All Exon library preparation, sequencing was conducted using a NovaSeq 6000 with 2 × 150 bp and 100 × coverage (50 × on-target coverage). Sequence alignment of raw fastq files to the human reference sequence (GRCh37/hg19 assembly) and variant calling was performed with the DRAGEN Germline Pipeline 3.2.8 on Illumina BaseSpace (https://basespace.illumina.com/). Variant annotation, analysis and homozygosity mapping was performed using VarSeq™ v2.2 (Golden Helix, Inc., Bozeman, MT, www.goldenhelix.com).
Segregation analysis
For Sanger DNA sequencing, primers were design through Primer3web tool (version 4.1.0) (Untergasser et al. 2012). Sanger sequencing was performed for available study participants (IV:2, V4-V8). Sequence analysis was performed by online BLAT tool package available in UCSC Genome Browser (Sanger et al.1977; Kent 2002) and offline BioEdit tool (version 7.0.5).
In silico studies
Computational analysis of mutant L2HGDH involved protein structural modeling and protein interaction capability.
Protein structure prediction
Structural modeling was done on I-TASSER tool (Yang and Zhang 2015) and models with highest confidence score (C- score) were selected for onward interaction studies. To further confirm the efficiency of predicted structure, I-TASSER (Yang and Zhang 2015) results were also crossed checked by SWISS MODEL (Bienert et al. 2016).
Molecular Docking and visualization
For interaction studies, ClusPro (Kozakov et al. 2017) tool was used for protein–protein docking between L2HGDH (wildtype and mutated protein) with its close interacting protein, which was predicted through STRING database (Szklarczyk et al. 2018). Protein-substrate docking was performed through AutoDock VINA tool (vina.scripps.edu) (Trott and Olson 2009). and molecular visualization through different offline tools e.g. LigPlot + (Version 2.1)( Laskowski et al.2011), PyMOL 2.3 (Schiffrin et al. 2020), Chimera 1.13.1(Pettersen et al. 2004) and Discovery Studio 2020(https://www.3dsbiovia.com/products/datasheets/discovery-studio-visualizer).
Results
The current study describes a consanguineous Pakistani family displaying intellectual disability with gait and speech problems recruited from Dera Ismail Khan City in Khyber Pakhtunkhwa province of Pakistan. The family pedigree and clinical history was assessed for five generations, with 4 affected individuals in the 5th generation and a single affected individual in the 4th generation (Fig. 1a).

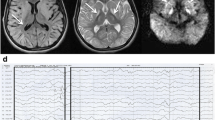
(a) Pedigree analysis illustrate autosomal recessive mode of disease segregation, and the genotype status of each analyzed individuals are represented as -/- (homozygous deletion) and -/G (heterozygous carrier). (b) Patient's photographs and (c) multiple panels of brain CT images of patients V:8 show white-matter atrophy
Molecular genetic analysis revealed a novel frameshift mutation in L2HGDH associated with L-2-hydroxyglutaric aciduria.
Clinical Findings
Phenotype
All affected family members showed mild intellectual disability and developing speech with weak communication skills. The patients had gait disturbance, however, no muscular dystrophy or skeletal anomalies were observed. No digital anomalies were determined, except syndactyly of feet in patient V:8. The affected individuals did not have a feeling of satiety even after excessive eating. Patient V:5 and V:8 had a history of epilepsy during the childhood. The head circumference in all patients was within normal range. Nonetheless, some degree of facial dysmorphism was observed due to drooping mouth (Fig. 1b). The general physique of the patients was normal. Examination of hearing, vision, visceral organs and skin was found normal (a summary of the phenotypic data is given in Table 1).
Radiological Findings
A CT scan was performed for a male patient (V:8). Plain CT demonstrates leukoaraiosis with hypoattenuation of cerebral white matter, particularly evident in frontal lobes. Also, a left-hemispheric preponderance becomes apparent (also involving capsula externa). The gyration appears normal, there is slight widening of the lateral ventricles, but no evident atrophy pattern in this 18-year-old male (Fig. 1c).
Biochemical Findings
Serum biochemistry reports showed high serum creatinine levels, however, blood urea, bilirubin, alanine transaminase (ALT), and alkaline phosphatase levels were normal. Urine organic acid analysis revealed marked excretion of 2-hydroxglutaric acid with a small peak for glutaric acid. A peak for 2-hydroxyglutaric lactones was not identified. Additionally, plasma amino acid analysis exhibited nonspecific variations in the level of different amino acids. For example, level of glutamate, glycine, alanine, leucine, ornithine and lysine were abnormally high, while, value of the cysteine was below the reference range. Patient’s biochemistry profile is illustrated in Table 2.
Molecular Findings
Whole genome homozygosity scan revealed a common linkage region on the q arm of chromosome 14 between SNP markers rs2039791 to rs781354 (45,171,670 bp – 52,879,326 bp). The size of the identified linkage interval spans over 7.7 Mb, which harbors 65 protein-coding genes (Fig. 2a).

Panel (a) shows SNP genotyping based HBD region and list of candidate genes between markers rs2039791 and rs781354. The causative gene L2HGDH is enclosed in red box (b) The structure of L2HGDH gene in which mutation harboring exon is encircled in red (c) Sanger sequencing chromatogram shows homozygous deletion in affected (V:8), which normal individuals (IV:2 & V:6) exhibited characteristic heterozygous/ carrier chromatogram. The position of deletion is framed in red box on the chromatogram
Whole exome data analysis identified a novel homozygous frameshift mutation NM_024884.3:c.180delG, p.(Ala62Profs*24) in the second exon of L2HGDH. Co-segregation of the identified mutation with the disease was confirmed by Sanger sequencing (Fig. 2c). This novel frameshift mutation was not listed in ClinVar, HGMD and gnomAD databases.
Structural Findings
Molecular modeling of Normal and Mutated L2HGDH proteins
After doing molecular modeling, the 3D-structures of both wild type and mutated L2HGDH were superimposed, which failed to overlap due to misfolding. It confirms that identified frameshift mutation results in structural distortion of L2HGDH (Fig. 3).

(a)Normal L2HGDH protein model (b)Mutant L2HGDH protein model (c)Superimposed structure (d)Normal L2HGDH protein docked to its substrate “L-2-hydroxyglutrate” molecule (e)Mutant L2HGDH protein docked to its substrate “L-2-hydroxyglutrate” molecule (f)Normal L2HGDH protein docked to its close interactor D2HGDH protein (g) Mutant L2HGDH protein docked to its close interactor D2HGDH protein
Protein–Protein Docking
Interaction studies of L2HGDH with D2HGDH have revealed remarkable alteration in docking sites (see Fig. 3). In addition to this, docking of L2HGDH with other close interactors i.e. ALDH4A1 and GLS2 proteins have exhibited significant alteration in the interacting sites (supplementary figure).
Enzyme–Substrate Docking
Interaction studies were also performed for L2HGDH proteins and its substrate i.e. L-2-hydroxyglutarate, which predicted five amino acids i.e. Gln-89, Tyr-195, Val-404, Ala-402 and Gly-403 of wild-type L2HGDH to be involved in interaction with its substrate via conventional hydrogen bonding. All these binding sites are within the FAD dependent enzyme domain. In mutated L2HGDH interacting sites within the FAD domain are lost due to frameshift and protein truncation. However, the mutant enzyme predictably showed interaction with its substrate on different positions i.e. Arg-42, Cys-38, Gly-40 and Cys-27 through conventional hydrogen bond, and Gly-28 through carbon hydrogen bond (see Fig. 3).
Discussion
L-2-hydroxyglutaric aciduria is a rare form of autosomal recessive neuro-metabolic disorder that is caused by mutations in L2HGDH. The corresponding protein acts as a mitochondrial enzyme which bio-oxidizes the L-2-hydroxyglutaric acid to α-ketoglutarate (Olgac et al. 2019), and is involved in butanoate metabolism, glutamate and glutamine metabolism pathways (Olgac et al. 2019; Ma et al. 2017). There are two defined features, the mitochondrial targeting sequence and a FAD dependent oxidoreductase domain (UniProtKB: Q9H9P8). Insufficient enzyme activity leads to toxic levels of L-2-hydroxyglutaric acid in the cerebrospinal fluid (CSF), plasma and urine. The main phenotypical features associated with L2HGA include leukodystrophy, intellectual disability, psycho-motor abnormalities, macrocephaly, intention tremors, abnormal gait, epilepsy and cerebellar atrophy (Haliloglu et al. 2008).
Penderis et al. (2007) described a spontaneous canine model of L-2-hydroxyglutaric aciduria in outbred bull terriers dogs. All affected dogs exhibited increased urinary excretion of L-2-hydroxyglutarate (L-2-HG), while 12 dogs in which MRI imaging was performed showed symmetric regions of hyper intensity comparable to that seen in humans (Penderis et al. 2007).
Similarly, Ma et al. (2017) developed an L2hgdh null mice and found range of phenotypes i.e. increased level of L-2-hydroxyglutarate (L-2-HG) levels in multiple tissues, especially in the brain and testis. L2hgdh null mice demonstrated white matter deterioration, extensive gliosis, microglia-mediated neuro-inflammation, and an expansion of oligodendrocyte progenitor cells. Additionally, L2HGDH deficiency in the later stages results in hippocampal neurogenesis and late-onset neurodegeneration (Ma et al. 2017). Oldham and coworkers identified L-2-hydroxyglutarate (L2HG) as an important factor for the hypoxia response. Earlier, L2HG was reported to be produced by the malate dehydrogenase via mitochondrial 2-oxoglutarate reduction. Elevated level of 2-oxoglutarate is considered responsible for accumulation of L2HG, which happens due to dysfunction of tricarboxylic acid cycle and increased mitochondrial reducing potential. These changes were associated with homeostasis of cellular redox, because elevated level of L2HG in cell prevents glycolysis as well as electron transport, in order to counterbalance the unfavorable consequences of mitochondrial reductive stress provoked by hypoxia. Therefore, L2HG combines cytoplasmic and mitochondrial based energy metabolism in a new cellular redox regulation model (Oldham et al. 2015). Qiu et al. (2020) reported that both mitochondrial enzyme i.e. L2HGDH and D2HGDH catalyzes the oxidation of L2HG and D2HG into α-ketoglutarate. The studies have shown that MYC is the essential factor that regulates the expression of both L2HGDH and D2HGDH. It basically regulates the TET DNA hydroxylases and RNA demethylases, and thereby controls the cellular epigenome and epitranscriptome (Qiu et al. 2020). In addition Ye et al. (2018) have demonstrated the role of 2-HG other than epigenetic control and linked the expression of 2-HG (D-and L-2-Hydroxyglutarates) to T cell regulation and suggest its presumable role in tumor immunity (Ye et al.2018).
To date, 83 mutations in L2HGDH have been published (according to HGMD, Feb. 2021), however, only two mutations i.e. c.1003C > T p.(Arg335*) (Sass et al. 2008) and c.178G > A p.(Gly60Arg) (Ullah et al.2018) have been described in Pakistani families. In this study, we are reporting on a multigenerational Pakistani family presenting with mild intellectual disability, psychomotor retardation, gait disturbance and epilepsy. Whole exome sequencing identified a frameshift mutation NM_024884.3: c.180delG, p.(Ala62Profs*24) in L2HGDH, the most N-terminal loss of function mutation in this gene published thus far. The synopsis of the molecular findings and the clinical presentation of patients, based on the biochemical profile and brain CT findings is in concordance with the diagnosis of L2HGA. Subsequent structural and interaction analysis were conducted to predict the functional impact of the mutation in case of protein truncation. Analysis revealed remarkable changes in the local folding of L2HGDH and interaction with its substrate (L-2-hydroxyglutarate) and close interactors (D2HGDH, ALDH4A1, GLS2). However, as the mutation is located in close proximity to the N-terminus, nonsense mediated mRNA decay cannot be ruled out as the underlying patho-mechanism in this family. Some biochemical studies have shown that 2-hydroxyglutaric aciduria may be associated with elevated levels of lysine (Samuraki et al. 2008). Interestingly, biochemical profiling of one of our patients showed additional abnormally high levels of glutamate, glycine, alanine, leucine and ornithine amino acids, but it remains unclear whether these findings can be attributed to the mutation in L2HGDH. The comparative clinical analysis of the present family with previously reported Pakistani L2HGA family determined partial overlap (Ullah et al. 2018). However, tonic–clonic seizure and macrocephaly was not present in the patients presented here. Further, Peng et al. (2018) have reported a few missense and frameshift mutations in Chinese patients, who exhibited mild phenotypes comparable to the patients included in the current study (Peng et al. 2018). Additionally, none of the patients do exhibit cerebral neoplasms thus far.
Based on the findings of the current study, it is suggested that pediatricians in developing countries (especially in Pakistan) should offer screening of metabolic disorders in children, because early diagnosis and therapeutic interventions may effectively reduce the progression of the disease.
Conclusion
Herein, we report on the most N-terminal loss-of-function mutation in L2HGDH [NM_024884.3: c.180delG p.(Ala62Profs*24)] in a consanguineous family causing L-2-hydroxyglutaric aciduria. This finding further expands the mutational spectrum of L2HGDH.
Data availability
The reference sequence data was obtained from UCSC genome browser (http://genome.ucsc.edu/). The patient’s data (sequence, photographs, pedigrees) is stored in the password protected computer of Lab of Medical Genetics at Gomal University, D.I.Khan and is available upon request.
References
Barth P, Hoffmann G, Jaeken J et al (1992) L-2-hydroxyglutaric acidemia: A novel inherited neurometabolic disease. Ann Neurol 32(1):66–71. https://doi.org/10.1002/ana.410320111
Barth PG, Wanders RJ, Scholte HR et al (1998) L-2-hydroxyglutaric aciduria and lactic acidosis. J Inherit Metab Dis 21:251–254
Bienert S, Waterhouse A, De Beer TAP, et al (2016) The SWISS-MODEL Repository—new features and functionality. Nucleic Acids Res 45(D1). https://doi.org/10.1093/nar/gkw1132.
BIOVIA DISCOVERY STUDIO VISUALIZER. https://www.3dsbiovia.com/products/datasheets/discovery-studio-visualizer. Accessed July 21, 2020.
Chen E, Nyhan W, Jakobs C et al (1996) l-2-Hydroxyglutaric aciduria: Neuropathological correlations and first report of severe neurodegenerative disease and neonatal death. J Inherit Metab Dis 19(3):335–343. https://doi.org/10.1007/bf01799264
Duran M, Kamerling JP, Bakker HD, van Gennip AH, Wadman SK (1980) L-2 Hydroxyglutaric aciduria: an inborn error of metabolism? J Inherit Metab Dis 3:109–112
Goffette S, Duprez T, Nassogne M, Vincent M, Jakobs C, Sindic C (2006) l-2-Hydroxyglutaric aciduria: clinical, genetic, and brain MRI characteristics in two adult sisters. Eur J Neurol 13(5):499–504. https://doi.org/10.1111/j.1468-1331.2006.01282.x
Haliloglu G, Jobard F, Oguz K et al (2008) L-2-Hydroxyglutaric Aciduria and Brain Tumors in Children with Mutations in theL2HGDHGene: Neuroimaging Findings. Neuropediatric 39(02):119–122. https://doi.org/10.1055/s-2008-1081217
Hanefeld F, Kruse B, Bruhn H, Frahm J (1994) In Vivo Proton Magnetic Resonance Spectroscopy of the Brain in a Patient with L-2-Hydroxyglutaric Acidemia. Pediatr Res 35(5):614–616. https://doi.org/10.1203/00006450-199405000-00015
Jellouli N, Hadj Salem I, Ellouz E et al (2014) Founder effect confirmation of c.241A>G mutation in the L2HGDH gene and characterization of oxidative stress parameters in six Tunisian families with L-2-hydroxyglutaric aciduria. J Hum Genet 59(4):216–222. https://doi.org/10.1038/jhg.2014.4
Kent WJ (2002) The Human Genome Browser at UCSC. Genome Res 12(6):996–1006. https://doi.org/10.1101/gr.229102
Kozakov D, Hall DR, Xia B et al (2017) The ClusPro web server for protein–protein docking. Nat Protoc 12(2):255–278. https://doi.org/10.1038/nprot.2016.169
Larnaout A, Amouri R, Kefi M, Hentati F (2008) L-2-hydroxyglutaric aciduria: clinical and molecular study in three Tunisian families. Identification of a new mutation and inter-familial phenotype variability. J Inherit Metab Dis 31:S375–S379
Laskowski RA, Swindells MB (2011) LigPlot : Multiple Ligand-Protein Interaction Diagrams for Drug Discovery. J Chem Inf Model 51(10):2778–2786. https://doi.org/10.1021/ci200227u
Ma S, Sun R, Jiang B, Gao, J., Deng, W., Liu, P, et al (2017) L2hgdh deficiency accumulates L-2-hydroxyglutarate with progressive leukoencephalopathy and neurodegeneration. Molec. Cell 37: e00492–16. Note: Electronic Article.
Moroni I, Bugiani M, D’Incerti L et al (2004) L-2-hydroxyglutaric aciduria and brain malignant tumors: A predisposing condition? Neurology 62(10):1882–1884. https://doi.org/10.1212/01.wnl.0000125335.21381.87
O’Connor G, King M, Salomons G, Jakobs C, Hardiman O (2009) A novel mutation as a cause of L-2-hydroxyglutaric aciduria. J Neurol 256:672–673
Oldham WM, Clish CB, Yang Y, Loscalzo J (2015) Hypoxia-mediated increases in L-2-hydroxyglutarate coordinate the metabolic response to reductive stress. Cell Metab 22(2):291–303. https://doi.org/10.1016/j.cmet.2015.06.021
Olgac A, TekinOrgun L, Ezgü FS, Biberoǧlu G, Tümer L (2019) A 7 year old Boy with Hand Tremors and a Novel Mutation for L 2 hydroxyglutaric Aciduria. Balkan J Med Genet 22(2):93–96. https://doi.org/10.2478/bjmg-2019-0015 (eCollection 2019 Dec)
Penderis J, Calvin J, Abramson C et al (2007) L-2-hydroxyglutaric aciduria: characterisation of the molecular defect in a spontaneous canine model. J Med Genet 44(5):334–340
Peng W, Ma XW, Yang X et al (2018) Two novel L2HGDH mutations identified in a rare Chinese family with L-2-hydroxyglutaric aciduria. BMC Med Genet 19(1):1–5
Pettersen EF, Goddard TD, Huang CC et al (2004) UCSF Chimera?A visualization system for exploratory research and analysis. J Comput Chem 25(13):1605–1612. https://doi.org/10.1002/jcc.20084
Qiu Z, Lin AP, Jiang S et al (2020) MYC Regulation of D2HGDH and L2HGDH Influences the Epigenome and Epitranscriptome. Cell Chem. Biol. 27(5):538–50. https://doi.org/10.1016/j.chembiol.2020.02.002
Samuraki M, Komai K, Hasegawa Y, Kimura M, Yamaguchi S, Terada N, Yamada M (2008) A successfully treated adult patient with L-2-hydroxyglutaric aciduria. Neurology 70(13):1051–2. https://doi.org/10.1212/01.wnl.0000287141.90944.95
Sanger F, Nicklen S, Coulson AR (1977) DNA sequencing with chain-terminating inhibitors. PNAS 74(12):5463–5467. https://doi.org/10.1073/pnas.74.12.5463
Sass JO, Jobard F, Topçu M, Mahfoud A, Werlé E, Cure S, et al. (2008) Fischer J. L-2-hydroxyglutaric aciduria: identification of ten novel mutations in the L2HGDH gene. J Inherit Metab Dis 275–279.
Schiffrin B, Radford SE, Brockwell DJ, Calabrese AN (2020) PyXlink Viewer: a flexible tool for visualisation of protein chemical crosslinking data within the PyMOL molecular graphics system. https://doi.org/10.1101/2020.06.16.154773
Seijo-Martínez M, Navarro C, Castro del Río M et al (2005) L-2-Hydroxyglutaric Aciduria. Arch Neurol 62(4):666. https://doi.org/10.1001/archneur.62.4.666
Steenweg M, Jakobs C, Errami A et al (2010) An overview of L-2-hydroxyglutarate dehydrogenase gene (L2HGDH) variants: a genotype-phenotype study. Hum Mutat 31(4):380–390. https://doi.org/10.1002/humu.21197
Steenweg M, Salomons G, Yapici Z et al (2009) l-2-Hydroxyglutaric Aciduria: Pattern of MR Imaging Abnormalities in 56 Patients. Radiolog 251(3):856–865. https://doi.org/10.1148/radiol.2513080647
Szklarczyk D, Gable AL, Lyon D, et al (2018) STRING v11: protein–protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res 47(D1). https://doi.org/10.1093/nar/gky1131.
Topcu M, Aydin OF, Yalcinkaya C et al (2005) L-2-hydroxyglutaric aciduria: a report of 29 patients. Turk J Pediatr 47:1–7
Topçu M, Jobard F, Halliez S et al (2004) l-2-Hydroxyglutaric aciduria identification of a mutant gene C14orf160 localized on chromosome 14q22 1. Hum Mol Genet 13(22):2803–2811. https://doi.org/10.1093/hmg/ddh300
Trott O, Olson AJ (2009) AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem. https://doi.org/10.1002/jcc.21334
Ullah MI, Nasir A, Ahmad A, et al (2018) Identification of novel L2HGDH mutation in a large consanguineous Pakistani family- a case report. BMC medical genetics 19(1). https://doi.org/10.1186/s12881-018-0532-x
Untergasser A, Cutcutache I, Koressaar T et al (2012) Primer3-new capabilities and interfaces. Nucleic Acids Res 40(15):e115. https://doi.org/10.1093/nar/gks596
Vilarinho L, Cardoso M, Gaspar P et al (2005) Novel L2HGDH mutations in 21 patients with L-2-hydroxyglutaric aciduria of Portuguese origin. Hum Mutant 26(4):395–396. https://doi.org/10.1002/humu.9373
Vilarinho L, Tafulo S, Sibilio M et al (2009) Identification of novel L2HGDH gene mutations and update of the pathological spectrum. J Hum Genet 55(1):55–58. https://doi.org/10.1038/jhg.2009.110
Yang J, Zhang Y (2015) I-TASSER server new development for protein structure and function predictions. Nucleic Acids Res 43(W1):W174–W181. https://doi.org/10.1093/nar/gkv342
Ye D, Guan KL, Xiong Y (2018) Metabolism activity and targeting of D-and L-2-hydroxyglutarates. Trends Cancer. 4(2):151–65. https://doi.org/10.1016/j.trecan.2017.12.005
Acknowledgment
We are grateful to the volunteer family for their valuable participation in the present biochemical genetics study. MAK is thankful to Higher Education Commission (HCE) of Pakistan for supporting the present study through an NPRU grant (5564/KPK/NRPU/R&D/HEC/2016). The current data has not been published anywhere, except presented in the M.Phil. and Ph.D. Thesis of few students that are already on-board in this manuscript. The doctoral studies of MM is supported by HEC Pakistan by granting Research studentship in the said project.
Funding
The current study was supported through an HEC Pakistan funded “NRPU grant (5564/KPK/NRPU/R&D/HEC/2016)”.
Author information
Authors and Affiliations
Contributions
MM, MZA, SK & SA have recruited the family and done clinical analysis. MM and ST has performed experiments and genome data analysis. BB, JB, EP & KW performed Sanger sequencing and data analysis. CE performed the radiologic analysis on CT scan. MAK and CW conceptualized and supervised the study and remained involved in data analysis, manuscript drafting & fund acquisition. All the authors have read, edited and approve the final version of manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
The present molecular study was approved by the institutional ethical review board of Gomal University D.I.Khan (IRB# 04/ ERB/GU), and Kohat University of Science and Technology, Kohat, Pakistan.
Consent to Publish
The patient’s guardians have given their consent to publish their clinical information and photographs.
Conflict of interest/ Competing interests
None declare by all authors.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Muzammil Ahmad Khan and Christian Windpassinger equally share the corresponding authorship
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Muzammal, M., Ali, M.Z., Brugger, B. et al. A novel protein truncating mutation in L2HGDH causes L-2-hydroxyglutaric aciduria in a consanguineous Pakistani family. Metab Brain Dis 37, 243–252 (2022). https://doi.org/10.1007/s11011-021-00832-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11011-021-00832-2