
Abstract
The oriental fruit moth, Grapholita molesta (Busck) (Lepidoptera: Tortricidae) currently is one of the economically most destructive pest species of stone and pome fruits worldwide. Here we sequenced the complete mitochondrial genome of this pest. This genome is 15,776 bp long, with an A + T content of 81.24%, containing 37 typical animal mitochondrial genes and an A + T-rich region. All gene are arranged as hypothesized ancestral gene order of insects except for trnM, which was shuffled from 3′ downstream of trnQ to 5′ upstream of trnI. cox1 gene uses unusual CGA start codon, as that in all other sequenced lepidopteran mitochondrial genome. The secondary structures for the two rRNA genes were predicted. All helices typically present in insect mitochondrial rRNA genes are generated. A microsatellite sequence was inserted into the region of H2347 in rrnL in G. molesta and two other sequenced tortricid mitochondrial genomes, indicating that the insertion event in this helix might occurred anciently in family Tortricidae. All of the 22 typical animal tRNA genes have a typical cloverleaf structure except for trnS2, in which the D-stem pairings in the DHU arm are absent. An intergenic sequence is present between trnQ and nad2 as well as in other sequenced lepidopteran mitochondrial genomes, which was presumed to be a remnant of trnM gene and its boundary sequences after the duplication of trnM to the upstream of trnI in Lepidoptera. The A + T-rich region is 836 bp, containing six repeat sequences of “TTATTATTATTATTAAATA(G)TTT.”
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The oriental fruit moth, Grapholita molesta (Busck) (Lepidoptera: Tortricidae), originated from East Asia, currently is one of the economically most destructive pest species of stone and pome fruits worldwide [1, 2]. G. molesta larvae bore in fruits, causing direct damage, or feed on twigs, causing shoot dieback. Management of this pest is mainly based on the use of the insecticides and pheromone-based mating disruption [3]. In addition to controlling methods, recently, ecological strategies and evolutionary patterns were studied, that might facilitate the managements of this pest. Molecular markers, i.e. amplified fragment length polymorphism (AFLP) and microsatellite (SSR) have been used to investigate the population genetic structure of G. molesta [4, 5], however, both are length-based markers from nuclear genome.
Insect mitochondrial genomes are about 16 Kb in size with 37 genes, including 13 protein-coding genes, two ribosomal RNA genes (large and small ribosomal RNAs), and 22 tRNA genes [6]. Additionally, an A + T-rich region is present, functioning on the regulation of transcription and replication [7]. Mitochondrial genomes contain abundant molecular markers, such as sequences, gene arrangement patterns and RNA secondary structures, which were frequently used for studies of population genetics, species identification, and phylogeny at different hierarchical levels [8, 9].
Presently, twenty-six complete or nearly complete mitochondrial genomes sequences are available in GenBank for lepidopteran species. However, the number of sequenced lepidopteran mitochondrial genomes is very limited relative to the species-richness of Lepidoptera.
In this study, we describe the complete mitochondrial genome sequence of the oriental fruit moth, G. molesta, and compare its features with other available lepidopteran mitochondrial genomes.
Materials and methods
Insects and DNA extraction
Grapholita molesta larvae were collected on the peach trees and kept in absolute alcohol at −80°. Total genomic DNA was extracted from individual larva using a DNeasy tissue kit (Qiagen, Hilden, Germany) following manufacturer protocols.
PCR amplification and sequencing
The G. molesta mitochondrial genome was amplified through nine overlapping fragments by PCR amplification using modified universal primers [10, 11] according to the determined lepidopteran mitochondrial genome sequences and specific primers designed in this study.
PCRs were done using Takara LA Taq (Takara Biomedical, Japan) under the following conditions: initial denaturation for 2 min at 94° followed by 35 cycles of 10 s at 96°, 15 s at 45–55°, and 1–4 min at 60° and a subsequent final extension for 8 min at 60°. PCR components were added as recommended by Takara LA Taq, the manufacturer. PCR products were sequenced directly by primer walking from both directions after purification. Sequencing reactions were performed using a BigDye Terminator v3.1 Cycle Sequencing Kit (Applied Biosystems, USA) and run on an ABI 3730 capillary sequencer.
Genome annotation and secondary structure prediction
tRNA genes were initially identified using the tRNAscan-SE search server with default parameters [12]. Sequences longer than 100 bp between the identified tRNA genes were used as queries in Blast searches in GenBank for identification of protein-coding and rRNA genes. Nucleotide sequences of protein-coding gene were translated using the invertebrate mitochondrial genetic code. The exact initiation and termination codons were identified in ClustalX version 2.0 [13] using reference sequences from other insects. The stop codon of these genes was inferred to be the first in-frame stop codon or, when necessary to avoid overlap with the downstream gene, an abbreviated stop codon corresponding well to the stop codon of other insect genes.
The secondary structure of large and small rRNAs (rrnL and rrnS) were derived from Drosophila melanogaster [14] and Drosophila virilis [15] with modifications made based on other predicted mitochondrial rRNA secondary structure [16]. Helix numbering follows the convention established at the CRW site [14] and Apis mellifera rRNA secondary structure [16] with minor modification. All structures were drawn in XRNA (developed by B. Weiser and available at http://rna.ucsc.edu/rnacenter/xrna/xrna.html).
All tRNA secondary structures were predicted using the tRNAscan-SE search server except for trnS2, which was predicted manually according to that predicted in other insects.
Results and discussions
Genome structure and base composition
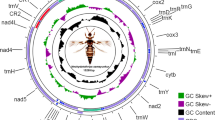
The complete mitochondrial genome of G. molesta is 15,776 bp long, containing 37 typical animal mitochondrial genes and an A + T-rich region (GenBank accession No. HQ116416). All gene are arranged as hypothesized ancestral gene order of insects except for trnM [17], which was shuffled from 3′ downstream of trnQ to 5′ upstream of trnI (Fig. 1). This arrangement pattern of trnM was presumed be an synapomorphic character in lepidopteran mitochondrial genomes [18, 19], however, the presently sequenced lepidopteran mitochondrial genomes used for comparative studies are all from the lineage of Ditrysia, representing limited group of Lepidoptera. Thus, more mitochondrial genomes are needed from other groups to prove the universality of trnM shuffling in this Order.

Structure of Grapholita molesta mitochondrial genome. cox1, cox2, and cox3 refer to the cytochrome oxidase subunits, cob refers to cytochrome b, nad1–nad6 refer to NADH dehydrogenase components, and rrnL and rrnS refer to ribosomal RNAs. Transfer RNA genes are denoted by one letter symbol according to the IPUC-IUB single-letter amino acid codes. L1, L2, S1 and S2 denote tRNALeu(CUN), tRNALeu(UUR), tRNASer(AGY) and tRNASer(UCN), respectively. AT indicates A + T-rich region. Gene names with lines indicate that the genes are coded on the minority strand while those without lines are on the majority strand
The entire mitochondrial genome of G. molesta is biased to use A and T, with an A + T content of 81.24%, as that of other insects. The AT skew for the majority strand is −0.064, while GC skew is −0.175, referring to the occurrence of more Ts than As and more Cs than Gs.
Protein-coding genes
All protein-coding genes start with ATN codons (one with ATA, three with ATT, one with ATC, and seven with ATG) except for cox1 (Table S1). In G. molesta mitochondrial genome, cox1 gene uses unusual CGA start codon, as that in all other sequenced lepidopteran mitochondrial genome. Annotated cox1 gene in G. molesta could be aligned well with its orthologous genes in other lepidopteran mitochondrial genome, confirmed the atypical start site of cox1 in Lepidoptera.
Seven of 13 protein-coding genes in G. molesta harbor the usual termination codon TAA, but cox1, cox2, cob, nad2, nad4 and nad5 use the incomplete termination codon T (Table S1). The assignment of incomplete stop codon on these genes could avoid overlapping nucleotides between their adjacent genes. These incomplete stop codons are commonly found in metazoan mitochondrial genes [18, 20].
The relative synonymous codon usage was analyzed, indicating a biased usage of A and T nucleotides (Table S2). UUA(Leu), AUU(Ile), UUU(Phe), AUA(Met) were the most frequently used codons as in other insects [18, 21]. All protein-coding genes show more T than A, while genes coded on the majority strand show more C than G and genes coded on the minority strand show less C than G (Table S3). This is congruent with the observation of skew values in insect mitochondrial genomes [22].
rRNA structure
Both rRNA genes are present in G. molesta mitochondrial genome, located between trnL1 and trnV for rrnL and between trnV and the A + T-rich region for rrnS. The length of the rrnL is 1382 bp, and the length of rrnS is 775 bp.
Both rrnL and rrnS conform to the secondary structure models proposed for these genes from other insects [20, 23, 24]. Forty-nine helices are present in G. molesta rrnL as in M. sexta [23], D. melanogaster [15] and A. mellifera [16], belonging to six domains (Fig. S1). The stem region of H991 was difficult to fold under the criteria of Watson–Crick pairs, and the structure of H991 with a large internal loop among H991, H1057 and H1087 is different from that of M. sexta. A 23 bp insertion was present in the loop region between H1664 and H1764 in M. sexta. In G. molesta, a microsatellite sequence of (TA)12 was inserted into the loop region of H2347. Alignment of the homologous regions in other sequenced lepidopteran mitochondrial genomes showed that similar microsatellite sequence of (TA)14 was also present in the stem region of H2347 in Spilonota lechriaspis [25] and Adoxophyes honmai (Lepidoptera: Tortricidae) [26] (Fig. 2), indicating that the insertion event in the region of H2347 in rrnL might be an synapomorphic character in family Tortricidae.

Alignment of the homologous regions including Helix 2347 of rrnL in all sequenced lepidopteran mitochondrial genomes Sequences were downloaded from GenBank, and the accession numbers are as in Table 1
The secondary structure of rrnS contains 29 helices present in Manduca sexta [23] and A. mellifera [16], belonging to three domains (Fig. 3). The structures of Helix H47, H673, H1047, H1241 and H1303 are different from those in M. sexta. H47 has a small loop in G. molesta compared to that in M. sexta. This region was variable within species [16, 23, 24], and has been used to predict the phylogenetic relationships among subfamilies of Braconidae (Insecta: Hymenoptera) combined with H39 and H367 [9]. H673 in G. molesta was more similar to that in some species of Hymenoptera [20, 24] and Diptera [15] than in species of Lepidoptera [23, 27]. The region of H673 is long, which could yields multiple possible secondary structures. The presently predicted structures of rRNA are mainly based on sequence comparison and mathematical methods, so it is not clear which structures are utilized in situ. The region composed of H1047, H1068, H1074 and H1113 in G. molesta was different in length especially in loop regions from that in M. sexta, indicating it is another variable region in rrnS within species [16, 23].

Predicted rrnS secondary structure in Grapholita molesta mitochondrial genome. Tertiary interactions and base triples are shown connected by continuous lines. A 5′ half of rrnL; B 3′ half of rrnL. Base-pairing is indicated as follows: Watson–Crick pairs by lines, wobble GU pairs by dots and other noncanonical pairs by circles
tRNA structure
All of the 22 typical animal tRNA genes were present in G. molesta mitochondrial genome, ranging from 65 to 71 bp. All tRNA genes have a typical cloverleaf structure except for trnS2 (Fig. S2). The D-stem pairings in the DHU arm are absent in trnS2, which has also been reported in other insects [24], and is common in Coleoptera [28]. The structure of trnS2 could not be identified and folded using conventional tRNA search methods such as tRNAscan-SE. We found the location of trnS2 by comparisons with those identified in other insects and then determined the exact boundaries according to the secondary structure folded manually. The anticodons for all tRNA genes are identical to their counterparts in most other published insect mitochondrial genomes.
In mitochondrial tRNA genes, noncanonical pairs were common in secondary structures. There are 16 wobble G–U pairs and four U–U pairs present in tRNA secondary structures in G. molesta.
Non-coding region
There are 14 non-coding regions ranging from 1 to 62 bp except for the A + T-rich region in G. molesta mitochondrial genome. A 62 bp intergenic sequence is present between trnQ and nad2, from where trnM was translocated to the upstream of trnI. In all other sequenced lepidopteran mitochondrial genomes, the same trnM rearrangement event occurred with a similar intergenic sequence ranging from 47 to 87 bp left in this region (Table 1). Additionally, the length of this intergenic sequence covers that of typical tRNA genes, thus, we presume that this region might be a remnant of trnM gene and its boundary sequences after the duplication of trnM to the upstream of trnI.
Intergenic spacer region between trnS1 and nad1 may correspond to the binding site of mtTERM, a transcription attenuation factor [29], which was evidenced by a 7 bp motif (ATACTAA) conserved across Lepidoptera [23], 5 bp (TACTA) motif conserved across Coleoptera [28] and a 6 bp conserved motif (THACWW) in Hymenoptera [24]. The ATACTA motif is also present in G. molesta mitochondrial genome between trnS1 and nad1.
The longest intergenic region in G. molesta is the A + T-rich region, between rrnS and trnM. The length of A + T-rich region is 836 bp, and the A + T content is 95.9%. This region usually contains replication origins in both vertebrates and invertebrates [7, 30]. The sequence of “TTATTATTATTATTAAATA(G)TTT” was repeated six times in the A + T-rich region in G. molesta. However, the set of elements that may function in the initiation of genome replication could not be identified [7].
References
Borchert DM, Stinner RE, Walgenbach JF, Kennedy GG (2004) Oriental fruit moth (Lepidoptera : Tortricidae) phenology and management with methoxyfenozide in North Carolina apples. J Econ Entomol 97:1353–1364
Roelofs WL, Comeau A, Selle R (1969) Sex pheromone of the oriental fruit moth. Nature 224:723
Il’ichev AL, Williams DG, Gut LJ (2007) Dual pheromone dispenser for combined control of codling moth Cydia pomonella L and oriental fruit moth Grapholita molesta (Busck) (Lep., Tortricidae) in pears. J Appl Entomol 131:368–376
Timm AE, Geertsema H, Warnich L (2008) Population genetic structure of Grapholita molesta (Lepidoptera : Tortricidae) in South Africa. Ann Entomol Soc Am 101:197–203
Torriani MVG, Mazzi D, Hein S, Dorn S (2010) Structured populations of the oriental fruit moth in an agricultural ecosystem. Mol Ecol 19:2651–2660
Wolstenholme DR (1992) Animal mitochondrial DNA: structure and evolution. Int Rev of Cytol 141:173–216
Zhang DX, Hewitt GM (1997) Insect mitochondrial control region: a review of its structure, evolution and usefulness in evolutionary studies. Biochem Syst Ecol 25:99–120
Foottit RG, Maw HEL, Von Dohlen CD, Hebert PDN (2008) Species identification of aphids (Insecta: Hemiptera: Aphididae) through DNA barcodes. Mol Ecol Resour 8:1189–1201
Wei SJ, Min S, Sharkey MJ, Achterberg C, Chen XX (2010) Comparative mitogenomics of Braconidae (Insecta: Hymenoptera) and the phylogenetic utility of mitochondrial genomes with special reference to Holometabolous insects. BMC Genomics 11:371
Simon C, Frati F, Beckenbach A, Crespi B, Liu H, Flook P (1994) Evolution, weighting, and phylogenetic utility of mitochondrial gene sequences and a compilation of conserved polymerase chain reaction primers. Ann Entomol Soc Am 87:651–701
Simon C, Buckley TR, Frati F, Stewart JB, Beckenbach AT (2006) Incorporating molecular evolution into phylogenetic analysis, and a new compilation of conserved polymerase chain reaction primers for animal mitochondrial DNA. Annu Rev Ecol Evol Syst 37:545
Lowe TM, Eddy SR (1997) tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res 25:955–964
Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R (2007) Clustal W and Clustal X version 2.0. Bioinformatics 23:2947–2948
Cannone JJ, Subramanian S, Schnare MN, Collett JR, D’Souza LM, Du Y, Feng B, Lin N, Madabusi LV, Muller KM (2002) The comparative RNA web (CRW) site: an online database of comparative sequence and structure information for ribosomal, intron, and other RNAs. BMC Bioinformatics 3:1471–2105
Schnare MN, Damberger SH, Gray MW, Gutell RR (1996) Comprehensive Comparison of structural characteristics in eukaryotic cytoplasmic large subunit (23S-like) ribosomal RNA. J Mol Biol 256:701–719
Gillespie JJ, Johnston JS, Cannone JJ, Gutell RR (2006) Characteristics of the nuclear (18S, 5.8S, 28S and 5S) and mitochondrial (12S and 16S) rRNA genes of Apis mellifera (Insecta: Hymenoptera): structure, organization, and retrotransposable elements. Insect Mol Biol 15:657–686
Boore JL, Lavrov DV, Brown WM (1998) Gene translocation links insects and crustaceans. Nature 392:667–668
Liao F, Wang L, Wu S, Li YP, Zhao L, Huang GM, Niu CJ, Liu YQ, Li MG (2010) The complete mitochondrial genome of the fall webworm, Hyphantria cunea (Lepidoptera: Arctiidae). Int J Biol Sci 6:172–186
Kim I, Lee EM, Seol KY, Yun EY, Lee YB, Hwang JS, Jin BR (2006) The mitochondrial genome of the Korean hairstreak, Coreana raphaelis (Lepidoptera : Lycaenidae). Insect Mol Biol 15:217–225
Wei SJ, Shi M, He JH, Sharkey MJ, Chen XX (2009) The complete mitochondrial genome of Diadegma semiclausum (Hymenoptera: Ichneumonidae) indicates extensive independent evolutionary events. Genome 52:308–319
Lessinger AC, Martins Junqueira AC, Lemos TA, Kemper EL, Da Silva FR, Vettore AL, Arruda P (2000) The mitochondrial genome of the primary screwworm fly Cochliomyia hominivorax (Diptera: Calliphoridae). Insect Mol Biol 9:521–529
Wei SJ, Shi M, Chen XX, Sharkey MJ, van Achterberg C, Ye GY, He JH (2010) New views on strand asymmetry in insect mitochondrial genomes. PloS ONE 5:1767–1780
Cameron SL, Whiting MF (2008) The complete mitochondrial genome of the tobacco hornworm, Manduca sexta, (Insecta: Lepidoptera: Sphingidae), and an examination of mitochondrial gene variability within butterflies and moths. Gene 408:112–123
Wei S, Tang P, Zheng L, Shi M, Chen X (2010) The complete mitochondrial genome of Evania appendigaster (Hymenoptera: Evaniidae) has low A + T content and a long intergenic spacer between atp8 and atp6. Mol Biol Rep 37:1931–1942
Zhao JL, Zhang YY, Luo AR, Jiang GF, Cameron SL, Zhu CD (2011) The complete mitochondrial genome of Spilonota lechriaspis Meyrick (Lepidoptera: Tortricidae). Mol Biol Rep. doi:10.1007/s11033-010-0491-6
Lee ES, Shin KS, Kim MS, Park H, Cho S, Kim CB (2006) The mitochondrial genome of the smaller tea tortrix Adoxophyes honmai (Lepidoptera: Tortricidae). Gene 373:52–57
Niehuis O, Naumann CM, Misof B (2006) Identification of evolutionary conserved structural elements in the mt SSU rRNA of Zygaenoidea (Lepidoptera): a comparative sequence analysis. Org Divers Evol 6:17–32
Sheffield NC, Song H, Cameron SL, Whiting MF (2008) A comparative analysis of mitochondrial genomes in coleoptera (Arthropoda: Insecta) and genome descriptions of six new beetles. Mol Biol Evol 25:2499–2509
Cornuet JM, Garnery L, Solignac M (1991) Putative origin and function of the intergenic region between COI and COII of Apis mellifera L mitochondrial DNA. Genetics 128:393–403
Boore JL (1999) Animal mitochondrial genomes. Nucleic Acids Res 27:1767–1780
Hong MY, Lee EM, Jo YH, Park HC, Kim SR, Hwang JS, Jin BR, Kang PD, Kim K-G, Han YS (2008) Complete nucleotide sequence and organization of the mitogenome of the silk moth Caligula boisduvalii (Lepidoptera: Saturniidae) and comparison with other lepidopteran insects. Gene 413:49–57
Yang L, Wei ZJ, Hong GY, Jiang ST, Wen LP (2008) The complete nucleotide sequence of the mitochondrial genome of Phthonandria atrilineata (Lepidoptera: Geometridae). Mol Biol Rep 36:1441–1449
Coates BS, Sumerford DV, Hellmich RL, Lewis LC (2005) Partial mitochondrial genome sequences of Ostrinia nubilalis and Ostrinia furnicalis. Int J Biol Sci 1:13–18
Salvato P, Simonato M, Battisti A, Negrisolo E (2008) The complete mitochondrial genome of the bag-shelter moth Ochrogaster lunifer (Lepidoptera, Notodontidae). BMC Genomics 9:331
Jiang ST, Hong GY, Yu M, Li N, Yang Y, Liu YQ, Wei ZJ (2009) Characterization of the complete mitochondrial genome of the giant silkworm moth, Eriogyna pyretorum (Lepidoptera: Saturniidae). Int J Biol Sci 5:351–365
Li W, Zhang X, Fan Z, Yue B, Huang F, King E, Ran J (2010) Structural characteristics and phylogenetic analysis of the mitochondrial genome of the sugarcane borer, Diatraea saccharalis (Lepidoptera: Crambidae). DNA Cell Biol 30:3–8
Pan MH, Yu QY, Xia YL, Dai FY, Liu YQ, Lu C, Zhang Z, Xiang ZH (2008) Characterization of mitochondrial genome of Chinese wild mulberry silkworm, Bomyx mandarina (Lepidoptera: Bombycidae). Sci China Ser C 51:693–701
Yukuhiro K, Sezutsu H, Itoh M, Shimizu K, Banno Y (2002) Significant levels of sequence divergence and gene rearrangements have occurred between the mitochondrial genomes of the wild mulberry silkmoth, Bombyx mandarina, and its close relative, the domesticated silkmoth, Bombyx mori. Mol Biol Evol 19:1385–1389
Hong G, Jiang S, Yu M, Yang Y, Li F, Xue F, Wei Z (2009) The complete nucleotide sequence of the mitochondrial genome of the cabbage butterfly, Artogeia melete (Lepidoptera: Pieridae). Acta Biochim Biophys Sin 41:446–455
Kim SR, Kim MI, Hong MY, Kim KY, Kang PD, Hwang JS, Han YS, Jin BR, Kim I (2008) The complete mitogenome sequence of the Japanese oak silkmoth, Antheraea yamamai (Lepidoptera: Saturniidae). Mol Biol Rep 36:1871–1880
Hu XL, Cao GL, Xue RY, Zheng XJ, Zhang X, Duan HR, Gong CL (2010) The complete mitogenome and phylogenetic analysis of Bombyx mandarina strain Qingzhou. Mol Biol Rep 37:2599–2608
Hu J, Zhang D, Hao J, Huang D, Cameron S, Zhu C (2010) The complete mitochondrial genome of the yellow coaster, Acraea issoria (Lepidoptera: Nymphalidae: Heliconiinae: Acraeini): sequence, gene organization and a unique tRNA translocation event. Mol Biol Rep 37:3431–3438
Acknowledgments
We thank Jian-feng Wu and Long-long Weng for their help in molecular works and two anonymous reviewers for their valuable comments and suggestions. Funding for this study was provided jointly by the 973 Program (No. 2009CB119004), Special Fund for Agro-scientific Research in the Public Interest (No. 201103024), Modern Agro-industry Technology Research System (No. CARS-31) and Beijing New Star Program on Science and Technology (NO. 2010B027).
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
11033_2011_1049_MOESM1_ESM.pdf
Fig. S1 Predicted rrnL secondary structure in Grapholita molesta mitochondrial genome. Tertiary interactions and base triples are shown connected by continuous lines. a 5′ half of rrnL; b 3′ half of rrnL. Base-pairing is indicated as follows: Watson–Crick pairs by lines, wobble GU pairs by dots and other noncanonical pairs by circles. (PDF 394 kb)
11033_2011_1049_MOESM3_ESM.pdf
Fig. S2 Predicted secondary structures for the 22 typical tRNA genes of Grapholita molesta mitochondrial genome. Symbols are as in Fig. S1 (PDF 700 kb)
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Gong, Yj., Shi, Bc., Kang, Zj. et al. The complete mitochondrial genome of the oriental fruit moth Grapholita molesta (Busck) (Lepidoptera: Tortricidae). Mol Biol Rep 39, 2893–2900 (2012). https://doi.org/10.1007/s11033-011-1049-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11033-011-1049-y