
Abstract
An easy and effective regeneration system from leaf- and hypocotyl-derived protoplasts was established for carrot. The protoplast isolation efficiency after preplasmolytic treatment and digestion of source material in enzyme mixture consisted of 1% cellulase Onozuka R-10 and 0.1% pectolyase Y-23 reached on average 3 × 106 and 106 protoplasts per g of leaf and hypocotyl tissue, respectively. A modified thin alginate layer technique was applied for the protoplast culture. Direct somatic embryogenesis on a simplified Kao and Michayluk medium in the presence of 2,4-D and zeatin occurred during cultivation of both leaf- and hypocotyl-derived protoplasts for all accessions used. Morphologically normal plants were produced at very high efficiency within two months after initiation of the protoplast culture. Ninety three percent of in vitro derived plants were diploids. Pollen viability and seed set after self-pollination were similar to those of plants obtained from seeds.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Carrot (Daucus carota L. subsp. sativus Hoffm., 2n = 2x = 18) is one of the first plants used for successful growth in vitro. Early reports indicated the induction of callus tissue from root discs or embryoids formation from free-floating somatic carrot cells (Gautheret 1939; Steward 1958). Since then, carrot has become a model species for plant tissue culture systems. Research on protoplast culture in carrot began when successful protoplast isolation from sliced, 50-day-old, field-cultivated roots was reported (Kameya and Uchimiya 1972). At the same time, both cell division and plant development via direct somatic embryogenesis for protoplasts isolated from carrot cell suspension cultures were described (Grambow et al. 1972). To date, cell suspension cultures have been usually used as a protoplast donor material in carrot studies, especially in research on intra- and inter-specific as well intergeneric somatic hybridization (Dudits et al. 1977; Dudits et al. 1987; Han et al. 2009). Despite the fact, that suspension cultures are an excellent source of protoplasts due to their high embryogenic ability, the establishment and maintenance of suspension culture is laborious and time consuming, usually requiring several weeks in addition to the time required for callus induction. In contrast, leaf tissue can serve as an easy and direct source of protoplasts. Several types of plant material like leaves, cotyledons or hypocotyls are used as protoplast sources in many species. In carrot, petiole-derived protoplasts were successfully used for callus development and then plant regeneration by indirect somatic embryogenesis (Dirks et al. 1996). To our knowledge, other types of carrot green tissue have not been used as donor material for protoplast isolation. Thus, an efficient, fast, and easy system of plant regeneration from leaf- and hypocotyl-derived protoplasts is desired and would be valuable, e.g. in carrot research on protoplast fusion, in vitro selection or genetic transformation.
In this paper we propose an improved, highly efficient plant regeneration system via direct somatic embryogenesis from leaf and, for the first time, from hypocotyl carrot protoplasts.
Materials and methods
Protoplast donors
Seven carrot accessions were used including one open pollinated carrot cultivar Dolanka and three pairs of male-sterile (usually marked out as line A) and male-fertile breeding lines (line B) (Table 1). Protoplasts were isolated from leaves or hypocotyls of in vitro grown plants or seedlings, respectively (Fig. 1a, c). Aseptic material was derived from seeds sterilized using a three-step procedure. First, seeds were incubated in a water bath at 40°C, then transferred to 0.2% (v/v) solution of fungicide ‘Bravo’ (Syngenta) and placed on a gyratory shaker (160 rpm) and finally immersed in 20% (w/v) solution of chloramin T (sodium N-chlorotoluene-4-sulphonamide) (30 min each). After each step, the seeds were dipped in 70% ethanol for 30–60 s. After three washes with sterile water for 5 min each, the seeds were air-dried on a sterile filter paper and germinated in 9 cm Petri dishes on solid Murashige and Skoog (MS) medium with vitamins (Murashige and Skoog 1962) supplemented with 30 g l−1 sucrose and 6.5 g l−1 plant agar (Biocorp) and maintained at 26 ± 2°C in the dark. In case of protoplast isolation from leaves, about 20 seeds per dish were placed on the germination medium, and after about 7 days of culture seedlings were transferred to glass jars containing regeneration medium (R) composed of MS macro- and micro-elements, 0.1 mg l−1 thiamine HCl, 0.1 mg l−1 piridoxine HCl, 0.5 mg l−1 nicotinic acid, 3.0 mg l−1 glycine, 100 mg l−1 myo-inozytol, 20 g l−1 sucrose, and 2.5 g l−1 phytagel (Sigma). Cultures were kept in a climate room at 26 ± 2°C under 16 h photoperiod and light intensity of 55 μmol m−2s−1. In order to obtain elongated and etiolated hypocotyls, about 30–40 seeds were placed on the MS medium (the same as for seed germination) and kept in the dark for about 14 days.

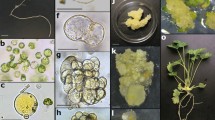
Plant regeneration from leaf- and hypocotyl-derived carrot protoplasts. a, c Donor material; b, d leaf and hypocotyl protoplasts, respectively, arrow indicates leaf protoplast with anthocyanins; e fluorescence of viable protoplasts after FDA staining; f enlarged leaf-derived protoplast with visible cytoplasmic strands, before mitotic division; g–i first symmetric and asymmetric division and cell colony formation, respectively; j early torpedo-shaped somatic embryo under light microscopy; k somatic embryos in cotyledonary stage formed in thin alginate layer; l thin alginate layer overgrown with somatic embryos after 2 months of culture; m protoplast-derived somatic embryos—start of germination in the light, on filter paper placed on the solid regeneration medium, 1 week after releasing from the aginate matrix; n protoplast-derived plants acclimatized to ex vitro conditions; o flowering plants; p viable (dark or magenta-red in colour picture) pollen grains after Alexander staining; q seed set obtained after self-pollination (bars in b, d–j, p 20 μm; k–l 0.5 cm)
Protoplast isolation and culture
Protoplasts were isolated from two types of organs, leaves with petioles from 3 to 4 week-old carrot plantlets and hypocotyls from 2 week-old seedlings using the protocol of Baranski et al. (2007), but with some modifications. In details, about 1 g of tissue was placed in a glass Petri dish with 8 ml of preplasmolysis solution (0.5 M mannitol), cut into fine pieces and then incubated for 1 h in the dark at 26 ± 2°C. Tissue digestion was performed overnight at 26 ± 2°C with gently shaking (30 rpm) in enzyme mixture consisting of 1% (w/v) cellulase Onozuka R-10 (Duchefa), 0.1% (w/v) pectolyase Y-23 (Duchefa), 20 mM 2-(N-Morpholino)ethanesulfonic acid (MES, Sigma), 5 mM CaCl2, and 0.6 M mannitol (Sigma), pH 5.6, filter-sterilized (0.22 μm, Millipore). The released protoplasts were separated from undigested tissue by filtration through a 80 μm nylon sieve (Millipore) and centrifuged at 100 g for 5 min. The pellet was resuspended in 8 ml of 0.5 M sucrose with 1 mM MES and overlaid with 2 ml of W5 medium (Menczel et al. 1981). After centrifugation at 145 g for 10 min the purified protoplasts were localized in the interphase between the two solutions. The collected protoplasts were washed in W5 solution and then in the culture medium, and centrifuged at 100 g for 5 min after each washing.
After purification, protoplast yield was determined by counting, using Fuchs Rosenthal hemocytometer chamber. The working density of protoplasts was adjusted to 8 × 105 protoplasts per ml. Culture system with protoplast embedding in a thin calcium alginate layer (Ca-alginate) was used, based on a slightly modified protocol of Damm et al. (1989). Equal volumes of protoplast suspension in the culture medium and 2.8% (w/v) alginic acid sodium salt (Sigma) in 0.4 M mannitol solution were mixed carefully. Aliquots (app. 300 μl) of protoplasts/alginate mixture were spread onto 1% (w/v) agar (Biocorp) containing 20 mM CaCl2, 0.4 M mannitol in 6 cm Petri dishes. By circular movement of the Petri dish protoplast suspension was forced to form a thin layer. After 1 h incubation at room temperature polymerization occurred and alginate layers with embedded protoplasts as a solid sheets were carefully transferred with the fine forceps from the agar plates to 4 ml of the liquid culture medium in 6 cm Petri dishes. The final density of embedded protoplasts was 4 × 105 per ml. Protoplast culture medium was based on the CPP medium (Dirks et al. 1996) and consisted of macro-, micro-elements and organic acids according to Kao and Mychayluk (1975), vitamins according to B5 medium (Gamborg et al. 1968), 74 g l−1 glucose, 250 mg l−1 casein enzymatic hydrolysate (Sigma), 0.1 mg l−1 2,4-dichlorophenoxyacetic acid (2,4-D), and 0.2 mg l−1 zeatin (pH 5.6, filter-sterilized). Protoplast cultures were incubated in the dark at 26 ± 2°C. After 10 days of culture, the medium was replaced by a fresh one.
Plant regeneration and acclimatization
After about 3–6 weeks of culture, visible protoplast-derived macrocolonies and somatic embryos were released from Ca-alginate layers by incubation for 1–2 h at room temperature in 20 mM sodium citrate solution containing 0.2 M mannitol. Then the alginate residues and citrate solution were removed by centrifugation (5 min at 100 g). The pellet was carefully washed in the CPPD medium containing 0.1 mg l−1 NAA and 0.2 mg l−1 zeatin, pH 5.6 (Dirks et al. 1996), and centrifuged for 5 min at 100 g. Finally, colonies and embryos derived from one alginate layer were resuspended in 4 ml of the CPPD medium and plated in 1 ml aliquots on filter paper placed on the solidified R medium. About 2–3 weeks later small rooted plantlets were transferred to a fresh R medium for further growth. During plant regeneration the cultures were maintained in a climate room at 26 ± 2°C under 16 h photoperiod and light intensity 55 μmol m−2s−1.
If not mentioned otherwise, all media and solutions were adjusted to pH 5.8 and autoclaved.
Rooted plants of cv. Dolanka with 3–4 leaves were transplanted to the moss-coconut fiber substrate (Ceres International) and acclimatized to ex vitro conditions for 2 weeks in a climate chamber at 20 ± 1°C under 16 h photoperiod and light intensity 30 μmol m−2s−1. In the first week the relative humidity was adjusted to 90% while in the second week humidity was reduced by 2% every day. During acclimatization, the plants were watered moderately.
Characterization of regenerants
Ploidy level of acclimatized plants was determined by flow cytometry according to Kielkowska and Adamus (2010). Potted plants were grown in the greenhouse for about 4 months and then they were vernalized for the next 3 months at 10 ± 2°C to induce flowering. Pollen viability was evaluated according to the Alexander’s procedure (1969) based on differential staining of aborted (colored as light green) and non-aborted grains (colored as magenta-red) (Fig. 1p). When the stigma was receptive the flowers were self-pollinated and seed set was scored.
Data collection and statistical analysis
Protoplast isolation efficiency was recorded using hemocytometer and was expressed as protoplast number per gram of fresh weight (fw) source tissue. Protoplast viability was assessed by staining with fluorescein diacetate (FDA) according to Anthony et al. (1999) on the first, second, third, and sixth day of culture for cv. Dolanka and only on the first day for the remaining accessions. Briefly, 60 μl of 0.3% filter-sterilized FDA acetone stock solution was dissolved in 4 ml of culture medium to prepare FDA working solution. 100 μl of that solution was added to the Petri dish with culture of immobilized protoplasts. Apple-green fluorescence of live cells (Fig. 1e) was examined under Zeiss Axiovert S100 microscope equipped with the appropriate filter set (λEx = 485 nm, λEm = 515 nm). The percentage of cell colonies formation (number of cell colonies/total number of observed protoplasts × 100) was determined and expressed as plating efficiency. Additionally, in the first experiments with Dolanka the number of two-, four-, and multi-cell colonies was determined at the same time points.
The treatments were set up in three replicates, 3–4 independent experiments were carried out. For protoplast viability and colony formation, counting was carried out in five microscopic fields on 100–300 cells per Petri dish. The mean values and standard errors were calculated. The overall effect of treatments was assessed using analysis of variance (ANOVA) and Tukey’s honestly significant difference (HSD) test in Statistica 8.0 (Stat Soft. Inc.).
Results
Protoplast isolation efficiency
Spherical protoplasts were released from the leaf and hypocotyl tissues after 14–16 h of incubation in the enzyme mixture. In leaf protoplasts, numerous green chloroplasts randomly distributed in the cytosol were visible whereas hypocotyl protoplasts were bright with densely organized cytoplasm (Fig. 1b, d). Some leaf protoplasts were pink probably due to the presence of anthocyanins in a vacuole. Both leaf and hypocotyl protoplasts were usually small and similar in size, reaching on average 28.36 ± 0.22 and 36.76 ± 0.50 μm in diameter, respectively. The yield of leaf protoplasts was on average 3.21 ± 0.47 × 106 per g fresh weight, and was over three times higher (P < 0.001) then that of hypocotyl protoplasts (0.96 ± 0.09 × 106, Table 1). This tendency was observed for all accessions. The efficiency varied from 1.92 to 5.03 × 106 for leaf protoplasts and from 0.64 to 1.38 × 106 for hypocotyl tissue depending on the accession, but differences between the accessions were not significant (P = 0.457 and P = 0.487, respectively).
Protoplast viability and cell colony formation
To assess protoplast viability and plating efficiency, leaf protoplasts of cv. Dolanka were used in preliminary experiments. In the first day of culture the proportion of viable protoplasts was high (74%), in the next 2 days it decreased to 52% and remained at this level afterwards (Fig. 2a). Three days after isolation most protoplasts enlarged and started to change their shape from spherical to oval indicating a reconstruction of the cell wall (Fig. 1f). Two kinds of the first cell division, i.e. symmetrical as well as asymmetrical were observed (Fig. 1g, h). In 5-days-old cultures two-, four-, and multi-cell colonies were formed (Fig. 2b). At that time about 15% of plated protoplasts underwent the first cell division while 10 days later the frequency of cell colony formation was over three times higher (53%, P < 0.01). In all examined periods of the culture similar numbers of two- and four-cell colonies were observed (8 and 5%, respectively—Fig. 2b) whereas the number of multi-cell colonies increased from 3 to 38%. Microscopic observations showed that cells in the colonies were tightly packed and had dense cytoplasm, suggesting that they might be embryogenic (Fig. 1i). Due to lack of synchrony in the development, the size of the colonies varied considerably.

Carrot leaf protoplast culture of cv. ‘Dolanka’ after embedding in alginate layers. a Viability [%] of protoplasts in the first days of the culture. b Developmental stage of cell colonies after 5, 10, and 15 days of the culture. Bars show standard error
Additionally, the reaction to the culture conditions of leaf and hypocotyl protoplasts isolated from cv. Dolanka and six breeding lines was examined (Fig. 3). Viability of hypocotyl protoplasts after embedding in alginate layers, as estimated using FDA staining, was higher than viability of leaf protoplasts (P = 0.01) and reached on average 67.21 ± 1.54% and 59.69 ± 2.41%, respectively. The survival rate for both protoplast types was genotype-independent (Fig. 3a). Both leaf and hypocotyl protoplasts of all accessions responded positive to immobilization in alginate and the applied culture medium, starting mitotic divisions by 4 days after isolation. Approximately after 14 days of culture the first cell colonies up to 250 μm in size were macroscopically visible. Plating efficiency of hypocotyl protoplasts was approximately twice as high as that of leaf protoplasts (P < 0.001). On tenth day of culture the number of leaf- and hypocotyl-derived cell colonies reached 21 and 52%, respectively and on the twentieth day it increased to 40 and 67%, respectively. Big differences in the rate of mitotic divisions between leaf and hypocotyl protoplasts were observed for several accessions, especially at the beginning of their culture. The proportion of colony formation highly depended on the accession (P = 0.01) and varied from 13% (line 2B) to 59% (line 2874A) for leaf protoplasts and from 54% (line 2B) to 75% (‘Dolanka’) for hypocotyl protoplasts (Fig. 3c). Such significant impact of the accession on the plating efficiency was revealed in both time points of evaluation (after 10 and 20 days) for both types of protoplasts.

Viability of leaf and hypocotyl protoplasts of various carrot accessions in the day of isolation (a) and the plating efficiency after 10 (b) and 20 (c) days of the culture. Bars show standard error
Plant regeneration
Independently on the protoplasts source, continuous growth of microcolonies was achieved following microcalli or proembryonic mass (PEM) development. On the 3–4th week of protoplast culture, PEM efficiently transformed into globular somatic embryos, that were morphologically distinct from microcalli. Globular embryos started to differentiate into torpedo-shape somatic embryos (Fig. 1j) and already 1–2 months after protoplast isolation embryos at cotyledonary stage were visible on the top of alginate layers or were floating in the culture medium (Fig. 1k-l). The rate of PEM and somatic embryos formation was high and similar in all accessions. Microcalli, PEMs, as well as somatic embryos of different sizes and stages of development, released from alginate matrix by its depolymerization, were transferred to a hormone free regeneration medium. In the first week of culture on solid medium the embryos started to grow and turned green (Fig. 1m), and small plantlets with one-two leaves and roots developed. After about 1 month, conversion of most somatic embryos into plants was completed. The frequency of germination of protoplast-derived somatic embryos was extremely high and thousands of plantlets were obtained that made estimation of their real number hardly possible. The plantlets showed no visible morphological abnormalities and they relatively easily acclimatized to ex vitro conditions with approximately 95% efficiency (Fig. 1n-o). Most of plants regenerated from leaf protoplasts of cv. Dolanka were diploids (93%, Table 2) and only a few were tetraploids. Pollen viability of diploid regenerants varied from 8 to 98%, reaching on average 57% and these values were similar with those obtained for seed-derived plants (Table 2). After self-pollination of flowers in the primary umbel on average 0.9 g of seeds was produced (Table 2, Fig. 1q).
Discussion
To date, carrot suspension cultures were used as a source of protoplasts in research on intra-specific, inter-specific or inter-generic somatic hybridization with D. capilifolius, Hordeum vulgare, Oryza sativa, and Nicotiana tabacum (Ichikawa et al. 1987; Tanno-Suenaga et al. 1988; Kisaka and Kameya 1994; Kisaka et al. 1994; Kisaka et al., 1997; Yamamoto et al. 2000). Plating efficiencies of suspension-derived protoplasts was usually low reaching only up to 20% (Didits 1984). Additionally, three to 8 months were required for the establishment of suspension cultures. Hypocotyl and leaf tissues may be an alternative source of donor material. Protoplast isolation from hypocotyls and leaves can be successfully performed after 10–14 days of seed germination and after 3–4 weeks of axenic plant growth, respectively. Besides, both leaf and hypocotyl tissue are known as valuable donor materials releasing a large number of protoplasts (Meyer et al. 2009, Sheng et al. 2010). Isolation efficiency examined for seven carrot accessions in the present work was relatively high, reaching from 2 to 5 × 106 protoplasts per g of leaf tissue and around 106 protoplasts per g of hypocotyl tissue. These values are similar to those reported for other species (Wiszniewska and Pindel 2009; Davey et al. 2010). Lower protoplast yield obtained from hypocotyls than from leaves is still sufficiently high, even to perform protoplast fusion. Satisfactory release of protoplasts from the source tissues was achieved probably due to the combined effect of the optimized protocol implementing the plasmolysis treatment prior the enzymatic digestion, enzyme mixture consisted of cellulase and pectolyase (the more active pectinase), and gentle agitation of the mixture during digestion for long enough period of time. It is known, that each of these treatments can be applied alone or in various combinations to improve the protoplast isolation efficiency (Davey et al. 2005; Sonntag et al. 2009; Castelblanque et al. 2010; Prange et al. 2010; Sheng et al. 2010). The use of both leaf- and hypocotyl-derived protoplasts could be especially useful in somatic hybridization, where specific morphological markers, like chloroplasts in mesophyll protoplasts and cytoplasmic strands in hypocotyl protoplasts offer an easy system for recognition of the cell fusion under the light microscope. Additionally, fusion of morphologically different protoplasts offer possibility for heterokaryon selection using fluorescent microscopy. Leaf protoplasts have natural red autofluorescence coming from chloroplasts while chloroplast-free protoplasts can be live-stained with a fluorochrome emitting in another spectral range, like FDA, fluorescing yellow-green light. That dual fluorescence, observed in heterokaryons, can be then used for their individual selection with a glass capillary of a micromanipulator combined with the microscope (Orczyk et al. 2003; Przetakiewicz et al. 2007).
Several culture systems including liquid over agar, protoplast embedding in agar or agarose, feeder layers, nurse culture or filter paper overlaid on agar have been shown to improve protoplast plating efficiency (Pati et al. 2005). Immobilization of protoplasts in a semi-solid matrix as calcium alginate is one of the methods used to avoid cell agglutination and improve survival rate during the culture. It was proved for several species such as Arabidopsis thaliana, Beta vulgaris, Daucus carota, and recently Nicotiana tabacum, Lotus corniculatus, and Rosa sp. that embedding in alginate supports the cell wall regeneration and promotes mitotic divisions as a result of protoplast separation (Hall et al. 1993; Dirks et al. 1996; Dovzhenko et al. 2003; Pati et al. 2005; Borgato et al. 2007; Pati et al. 2008). Another advantage of that culture system, is easier handling of the cultures which permits replacement of the culture media without disturbing the development of the microcolonies and may occur essential in prevention against microbial contamination. Several techniques of protoplast embedding in alginate matrix are known, i.e. alginate beads, alginate discs, alginate layers, thin alginate layers (TAL), and extra thin alginate films (ETAF). According to many reports, thickness of the alginate matrix plays an important role in protoplast divisions, namely the thiner matrix the higher plating efficiency as the consequence of increased compound diffusion from the medium to the protoplasts (Dovzhenko et al. 1998, Dovzhenko et al. 2003, Pati et al. 2005). In these systems different volumes of alginate are required to embed protoplasts, resulting in different thickness of the gel, i.e. 1 ml, 625 or 100 μl of protoplast-alginate mixture can be used to form alginate disc and alginate layer, TAL or ETAF, respectively (Hall et al. 1993; Dirks et al. 1996; Dovzhenko et al. 1998; Pati et al. 2005). In the present study, embedding in thin alginate layer was used by applying only 300 μl of protoplast-alginate mixture to the surface of Ca-agar plates in 6 cm Petri dishes. Additionally, we minimized the layer thickness by a circular rotation of the protoplast-alginate suspension during the application and before its polymerization. That was technically easier than TAL and ETAF methods, in which reduction of the alginate gel thickness was achieved by inserting a polypropylene grid or coverglass, respectively. Besides the embedding system, source tissue and plant species are known factors influencing the frequency of cell divisions. Cultivation of mesophyll A. thaliana protoplasts in alginate beads resulted in plating efficiency of 0.6% (Damm and Willmitzer 1988) while in TAL system with cotyledon-derived protoplasts the values reached 30% (Dovzhenko et al. 2003). On the other hand, in such model species for in vitro culture as Nicotiana tabacum TAL technique applied for leaf protoplasts showed plating efficiency from 65 to 96% (Dovzhenko et al. 1998; Pati et al. 2005). The modified system for thin alginate layer formation, proposed in the protocol presented here allowed high plating efficiency both for leaf- (up to 60%) and hypocotyl-derived (over 70%) protoplasts. The results presented may suggest that this protocol enables higher efficiency than that described by Dirks et al. (1996) who used petiole-derived protoplasts (on average 43%). However, we used different carrot accessions and protoplast source tissues.
In contrast to the original composition proposed by Kao and Michayluk (1975), we simplified the CCP medium by removing all sugars (i.e. fructose, ribose, xylose, mannose, rhamnose, cellobiose, sorbitol and mannitol) except glucose. Our results demonstrate that these sugars are not essential for cell division and further growth of colonies, induction of somatic embryogenesis, and embryo development. The combination of 2,4-D and zeatin found by Dirks et al. (1996) as the best for the development of petiole-derived carrot protoplasts allowed to achieve very high plating efficiency for leaf and hypocotyl protoplasts as well. Additionally, we observed direct somatic embryogenesis before depolymerizing the alginate gel. It was not necessary to transfer the macrocolonies again to the culture medium supplemented with 2,4-D to induce somatic embryogenesis. That way, the total time required for the recovery of fully regenerated and rooted plantlets was significantly shortened and completed within 2 months. That period is not as short as in the efficient regeneration system reported for cotyledon-derived protoplasts in Arabidopsis thaliana (Dovzhenko et al. 2003) or for leaf-derived protoplasts in tobacco (Dovzhenko et al. 1998). However, to our best knowledge, this is the most productive protocol described for carrot. The regenerated plants were efficiently acclimatized to ex vitro conditions and for most of them their ploidy level, pollen viability, and seed set were not altered. That suggests that derived plants were genetically stable.
In conclusion, we developed a fast and efficient regeneration protocol for leaf- and hypocotyl-derived carrot protoplasts, which allowed plant production within 2 months after initiation of the culture. To our knowledge, this is the first report describing development of protoplast-to-plant system from hypocotyls as the donor tissue in this species. We demonstrated that high efficiency of direct somatic embryogenesis can be achieved in carrot protoplast culture using thin alginate layer system, simplified Kao and Michayluk medium (1975), and 2,4-D and zeatin as growth regulators. The presented protocol is simple in handling, it requires only two media for plant regeneration from protoplasts, thus is less laborious and less costly. The protocol can be used for micropropagation of valuable breeding materials (like DH lines) and easily applied for in vitro selection, protoplast fusion, and genetic transformation in carrot.
Abbreviations
- CPP:
-
Carrot petiole protoplast medium
- 2,4-D:
-
2,4-Dichlorophenoxyacetic acid
- ETAF:
-
Extra thin alginate film
- FDA:
-
Fluorescein diacetate
- MES:
-
2-(N-Morpholino)ethanesulfonic acid
- MS:
-
Murashige and Skoog medium (1962)
- NAA:
-
α-Naphthaleneacetic acid
- PEM:
-
Proembryonic mass
- TAL:
-
Thin alginate layer
References
Alexander MP (1969) Differential staining of aborted and nonaborted pollen. Stain Technol 44:117–122
Anthony P, Otoni W, Power JB, Lowe TC, Davey MR (1999) Protoplast isolation culture and plant regeneration from Passiflora. In: Hall RD (ed) Methods in molecular biology—plant cell culture protocols. Humana Press, Totowa, pp 169–179
Baranski R, Klocke E, Ryschka U (2007) Monitoring the expression of green fluorescent protein in carrot. Acta Physiol Plant 29:239–246
Borgato L, Pisani F, Furini A (2007) Plant regeneration from leaf protoplasts of Solanum virginianum L. (Solanaceae). Plant Cell Tiss Organ Cult 88:247–252
Castelblanque L, García-Sogo B, Pineda B, Moreno V (2010) Efficient plant regeneration from protoplasts of Kalanchoe blossfeldiana via organogenesis. Plant Cell Tiss Organ Cult 100:107–112
Damm B, Willmitzer L (1988) Regeneration of fertile plants from protoplasts of different Arabidopsis thaliana genotypes. Mol Gen Genet 213:15–20
Damm B, Schmidt R, Willmitzer L (1989) Efficient transformation of Arabidopsis thaliana using direct gene transfer to protoplasts. Mol Gen Genet 217:6–12
Davey MR, Anthony P, Power JB, Lowe KC (2005) Plant protoplast: status and biotechnological perspectives. Biotechnol Adv 23:131–171
Davey MR, Anthony P, Patel D, Power JB (2010) Plant protoplasts: isolation, culture and plant regeneration. In: Davey MR, Anthony P (eds) Plant cell culture essential methods. Wiley-Blackwell, New York, pp 153–173
Dudits D (1984) Isolation and culture of protoplasts from carrot cell suspension cultures. In: Vasil IK (ed) Cell culture and somatic cell genetics of plants. Academic Press, Orlando, pp 391–397
Dirks R, Sidorov V, Tulmans C (1996) A new protoplast culture system in Daucus carota L. and its applications for mutant selection and transformation. Theor Appl Genet 93:809–815
Dovzhenko A, Bergen U, Koop HU (1998) Thin-alginate-layer technique for protoplast culture of tobacco leaf protoplasts: shoot formation in less than two weeks. Protoplasma 204:114–118
Dovzhenko A, Dal Bosco C, Meurer J, Koop HU (2003) Efficient regeneration from cotyledon protoplasts in Arabidopsis thaliana. Protoplasma 222:107–111
Dudits D, Hadlaczky G, Lévi E, Fejér O, Haydu Z, Lázár G (1977) Somatic hybridisation of Daucus carota and D. capillifolius by protoplast fusion. Theor Appl Genet 51:127–132
Dudits D, Maroy E, Praznovszky T, Olah Z, Gyorgyey J, Cella R (1987) Transfer of resistance traits from carrot into tobacco by asymmetric somatic hybridization: regeneration of fertile plants. Proc Natl Acad Sci 84:8434–8438
Gamborg OL, Miller RA, Ojima K (1968) Nutrient requirements of suspension cultures of soybean root cells. Exp Cell Res 50:151–158
Gautheret RJ (1939) Sur la possibilité de realiser a culture indefinite des tissues de tubercules de carotte. C R Hebd Seances Acad Sc 208:118–120
Grambow HJ, Kao KN, Miller RA, Gamborg OL (1972) Cell division and plant development from protoplasts of carrot cell suspension cultures. Planta 103:348–355
Hall RD, Pedersen C, Krens FA (1993) Improvement of protoplast culture protocols for Beta vulgaris L. (sugar beet). Plant Cell Rep 12:339–342
Han L, Zhou Ch, Shi J, Zhi D, Xia G (2009) Ginsenoside Rb1 in asymmetric somatic hybrid calli of Daucus carota with Panax quinquefolius. Plant Cell Rep 28:627–638
Ichikawa H, Tanno-Suenaga L, Imamura J (1987) Selection of Daucus cybrids based on metabolic complementation between X-irradiated D. capillifolius and iodoacetamide-treated D. carota by somatic cell fusion. Theor Appl Genet 74:746–752
Kameya T, Uchimiya H (1972) Embryoids derived from isolated protoplasts of carrot. Planta 103:356–360
Kao KN, Michayluk MR (1975) Nutritional requirements for growth of Vicia hajastana cells and protoplasts at a very low population density in liquid media. Planta 126:105–110
Kiełkowska A, Adamus A (2010) In vitro culture of unfertilized ovules in carrot (Daucus carota L.). Plant Cell Tiss Organ Cult 102:309–319
Kisaka H, Kameya T (1994) Production of somatic hybrids between Daucus carota L. and Nicotiana tabacum. Theor Appl Genet 88:75–80
Kisaka H, Lee H, Kisaka M, Kanno A, Kang K, Kameya T (1994) Production and analysis of asymmetric hybrid plants between monocotyledon (Oryza sativa L.) and dicotyledon (Daucus carota L.). Theor Appl Genet 89:365–371
Kisaka H, Kisaka M, Kanno A, Kameya T (1997) Production and analysis of plants that are somatic hybrids of barley (Hordeum vulgare L.) and carrot (Daucus carota L.). Theor Appl Genet 94:221–226
Menczel L, Nagy F, Kiss Z, Maliga P (1981) Streptomycin resistant and sensitive somatic hybrids of Nicotiana tabacum + Nicotiana knightiana: correlation of resistance to N. tabacum plastids. Theor Appl Genet 70:590–594
Meyer L, Serek M, Winkelmann T (2009) Protoplast isolation and plant regeneration of different genotypes of Petunia and Calibrachoa. Plant Cell Tiss Organ Cult 99:27–34
Murashige T, Skoog F (1962) A revised medium for rapid growth and bioassayas with tobacco tissue culture. Physiol Plant 18:100–127
Orczyk W, Przetakiewicz J, Nadolska-Orczyk A (2003) Somatic hybrids of Solanum tuberosum - application to genetics and breeding. Plant Cell Tiss Organ Cult 74:1–13
Pati PK, Sharma M, Ahuja PS (2005) Extra thin alginate film: an efficient technique for protoplast culture. Protoplasma 226:217–221
Pati PK, Sharma M, Ahuja PS (2008) Rose protoplast isolation and culture and heterokaryon selection by immobilization in extra thin alginate film. Protoplasma 233:165–171
Prange ANS, Serek M, Bartsch M, Winkelmann T (2010) Efficient and stable regeneration from protoplasts of Cyclamen coum Miller via somatic embryogenesis. Plant Cell Tiss Organ Cult 101:171–182
Przetakiewicz J, Nadolska-Orczyk A, Kuć D, Orczyk W (2007) Tetraploid somatic hybrids of patato (Solanum tuberosum L.) obtained from diploid breeding lines. Cell Mol Biol Lett 12:253–267
Sheng X, Zhao Z, Yu H, Wang J, Xiaohui Z, Gu H (2010) Protoplast isolation and plant regeneration of different doubled haploid lines of cauliflower (Brassica oleracea var. botrytis). Plant Cell Tiss Organ Cult. doi:10.1007/s11240-011-0002-z
Sonntag K, Ruge-Wehling B, Wehling P (2009) Protoplast isolation and culture for somatic hybridization of Lupinus angustifolius and L. subcarnosus. Plant Cell Tiss Organ Cult 96:297–305
Steward FC (1958) Growth and organized development at cultured cells. II. Interpretation of the growth from free cell to carrot plant. Am J Bot 45:709–713
Tanno-Suenaga L, Ichikawa H, Imamura J (1988) Transfer of the CMS trait in Daucus carota L. by donor-recipient protoplast fusion. Theor Appl Genet 76:855–860
Wiszniewska A, Pindel A (2009) Improvement in Lupinus luteus (Fabaceae) protoplast culture – stimulatory effect of agarose embedding and chemical nursing on protoplast divisions. Aus J Bot 57:502–511
Yamamoto T, Nakajima Y, Oeda K (2000) Morphological changes in homeotic cytoplasmic male-sterile carrots combined with fertile cytoplasm by asymmetrical cell fusion. Plant Cell Rep 19:363–370
Acknowledgments
The authors thank Aleksandra Bemben for her excellent technical assistance. This work was supported by the Polish Ministry of Science and Higher Education (grant no. N310 047 32/2394).
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Grzebelus, E., Szklarczyk, M. & Baranski, R. An improved protocol for plant regeneration from leaf- and hypocotyl-derived protoplasts of carrot. Plant Cell Tiss Organ Cult 109, 101–109 (2012). https://doi.org/10.1007/s11240-011-0078-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11240-011-0078-5